

Abstract
Adenosine is a potent neuromodulator in the CNS, but the mechanisms that regulate adenosine concentrations in the extracellular space remain unclear. The present study demonstrates that increasing the intracellular concentration of adenosine in a single hippocampal CA1 pyramidal neuron selectively inhibits the excitatory postsynaptic potentials in that cell. Loading neurons with high concentrations of adenosine via the whole-cell patch-clamp technique did not affect the GABAA-mediated inhibitory postsynaptic potentials, the membrane resistance, or the holding current, whereas it significantly increased the adenosine receptor-mediated depression of excitatory postsynaptic currents. The effects of adenosine could not be mimicked by an agonist at the intracellular adenosine P-site, but the effects could be antagonized by a charged adenosine receptor antagonist and by adenosine deaminase, demonstrating that the effect was mediated via adenosine acting at extracellular adenosine receptors. The effect of adenosine loading was not blocked by BaCl2 and therefore was not caused by an adenosine-activated postsynaptic potassium conductance. Adenosine loading increased the paired-pulse facilitation ratio, demonstrating that the effect was mediated by presynaptic adenosine receptors. Finally, simultaneous extracellular field recordings demonstrated that the increase in extracellular adenosine was confined to excitatory synaptic inputs to the loaded cell. These data demonstrate that elevating the intracellular concentration of adenosine in a single CA1 pyramidal neuron induces the release of adenosine into the extracellular space in such a way that it selectively inhibits the excitatory inputs to that cell, and the data support the general conclusion that adenosine is a retrograde messenger used by pyramidal neurons to regulate their excitatory input.
- adenosine
- synaptic modulation
- hippocampus
- adenosine transport
- A1 receptors
- fEPSP
- electrophysiology
- whole-cell recording
Adenosine is a neuromodulator with potent inhibitory effects in the CNS that are mediated, in part, by the inhibition of neurotransmitter release (Dunwiddie, 1985). Low concentrations of adenosine are present continually in the extracellular space of the brain (Zetterstrom et al., 1982; Dunwiddie and Diao, 1994), and adenosine release from brain tissue can be evoked by various stimuli, including electrical (Pull and McIlwain, 1972;Daval and Barberis, 1981; Mitchell et al., 1993a) and chemical (Daval and Barberis, 1981) depolarization, hypoxia (Zetterstrom et al., 1982;Phillis et al., 1987), and ischemia (Berne et al., 1974; Phillis et al., 1987). Furthermore, these same types of stimuli have been shown to inhibit synaptic activity through the activation of adenosine receptors (Fowler, 1989, 1990; Gribkoff et al., 1990; Pedata et al., 1993).
The most likely source of adenosine released by these stimuli is the intracellular adenosine that is formed from the breakdown of ATP (Lipton and Whittingham, 1982; Yoneda and Okada, 1989; Lloyd et al., 1993). It is likely that this adenosine is released into the extracellular space via bidirectional, facilitated diffusion nucleoside transporters (Bender et al., 1980, 1981). Under normal conditions, adenosine kinase phosphorylates intracellular adenosine and keeps the intracellular concentrations low; thus the direction of adenosine transport is down the concentration gradient and into the cell. However, a rise in the intracellular concentration of adenosine would reverse the concentration gradient, allowing the transporters to mediate the release of adenosine into the extracellular space (Plagemann and Woffendin, 1989; Gu et al., 1995). Under various conditions, adenosine transport blockers can both decrease (Jonzon and Fredholm, 1985; Gu et al., 1995) and increase (Sanderson and Scholfield, 1986; Phillis et al., 1989; Dunwiddie and Diao, 1994) extracellular adenosine concentrations, depending on the extent to which they block either uptake or efflux of adenosine from brain tissue. Although previous experiments have implicated nucleoside transporters in the release of adenosine evoked by hypoxia and ischemia (Bender et al., 1980; Belloni et al., 1985; but see Phillis et al., 1989; Craig and White, 1993), several important questions remain unresolved. First, there is no clear evidence demonstrating that an increase in the intracellular concentration of adenosine is sufficient to increase extracellular adenosine levels. Second, the extent to which neurons (as opposed to glial cells or other cellular elements) can release adenosine is unclear. Finally, it is not known whether the amount of adenosine that can be released in this manner is sufficient to activate extracellular adenosine receptors. More specifically, it is not known whether a single cell can release enough adenosine to modulate synaptic activity or whether extracellular adenosine concentrations only rise during pathological conditions such as hypoxia or ischemia, when many cells would be expected to release adenosine in a concerted manner. If a single cell can use adenosine to modulate its synaptic inputs, then one role of adenosine might be to serve as a retrograde synaptic messenger, which could provide inhibitory feedback to excitatory synapses that impinge on that cell. To resolve these issues, experiments were designed to increase the intracellular concentration of adenosine in individual CA1 pyramidal neurons directly and to determine whether this increase modulates cellular physiology and synaptic activity in a way that would be consistent with increased extracellular concentrations of adenosine.
MATERIALS AND METHODS
Hippocampal slices 400 μm thick were prepared from male Sprague Dawley rats (100–140 gm) with a Sorvall TC-2 tissue chopper. Slices were incubated in artificial cerebral spinal fluid (aCSF) containing (in mm): NaCl 126, KCl 3, MgCl2 1.5, CaCl2 2.4, NaH2PO4 1.2, glucose 11, and NaHCO3 25.9 oxygenated with 95% O2/5% CO2 and kept at 33°C for at least 1 hr before experimentation. Experiments were conducted on slices submerged in oxygenated aCSF and superfused at 2 ml/min.
All drugs were applied by injection into the superfusion line. Theophylline, adenosine, and adenosine deaminase were purchased from Sigma Chemical (St. Louis, MO), 8-p-sulfophenyltheophylline (8-pSPT) was purchased from Research Biochemicals (Natick, MA), BaCl2 was obtained from Aldrich Chemical Company (Milwaukee, WI), and 2′,5′-dideoxyadenosine (DDA) was purchased from ICN Pharmaceuticals (Costa Mesa, CA).
Experiments were performed by using the whole-cell patch-clamp technique and extracellular recording techniques. Patch pipettes were pulled from borosilicate glass [outer diameter (o.d.) 1.5 mm, inner diameter (i.d.) 0.86 mm, with filament; Sutter Instrument, Novato, CA] on a Flaming/Brown Micropipette puller Model P-87 (Sutter) and had a tip resistance of 4–10 MΩ when filled with a solution containing (in mm): K-gluconate 125, KCl 11, HEPES 10, CaCl20.1, K-EGTA 1, Mg-ATP 2, and Tris-GTP 0.3, pH-adjusted to 7.2–7.4 with KOH and osmolarity adjusted to 275–295 mOsm. Adenosine and other drugs were added to the stock electrode filling solution before the adjustment of pH or osmolarity. Whole-cell recording was performed by using the blind-patch technique as described previously (Blanton et al., 1989). Briefly, the patch pipettes were lowered into the stratum pyramidale layer of the CA1 region until the presence of a neuron cell body was detected by a change in the electrode resistance. Gentle suction was applied to the pipette, a >109 Ω seal was allowed to form, and the membrane patch was ruptured with further suction. Cells were voltage-clamped at −60 mV (−70 mV after correction for the electrode tip potential that exists when the voltage is zeroed with the electrode in the superfusion solution) with an Axoclamp-2A amplifier (Axon Instruments, Burlingame, CA) in the continuous single-electrode voltage-clamp mode.
In all experiments at least 15 min was allowed to pass between rupturing the patch and bath application of drugs, thus allowing responses to stabilize and the contents of the electrode to equilibrate with the cytoplasm of the cell. All responses were digitized with an R. C. Electronics ISC-16 analog-to-digital card and analyzed by computer with software developed in our laboratory. The membrane potential and holding current were monitored every 10 sec. The membrane resistance was determined from the current response to a −10 mV voltage command step every 20–30 sec. Synaptic responses were evoked every 20–30 sec by a bipolar-stimulating electrode placed on the Schaffer collateral and commissural afferent axons between the CA1 and CA3 regions. Excitatory postsynaptic currents (EPSCs) and the GABAAreceptor-mediated inhibitory postsynaptic currents (IPSCs) were measured as the peak amplitude of the evoked inward and outward currents, respectively. All measurements were taken from the average of 6–12 individual responses. Paired-pulse facilitation (PPF) ratios were determined by evoking a single EPSC 10 sec before evoking a pair of EPSCs 50 msec apart. The waveform recorded for the single EPSC was subtracted from the waveform for the pair of EPSCs to isolate the second EPSC in the pair. The PPF ratio was determined as the amplitude of the isolated second EPSC divided by the amplitude of the first EPSC of the pair. All PPF ratio measurements were conducted with 2 mm BaCl2 in the superfusing solution. Extracellular recordings were conducted simultaneously in the same slice as some of the intracellular recordings. Glass micropipettes were filled with 3 m NaCl and placed in the stratum radiatum in a region that would correspond to the dendritic field of the cell from which whole-cell recordings were obtained. Field excitatory postsynaptic potentials (fEPSPs) were recorded simultaneously with the whole-cell synaptic responses and were triggered by the same stimulus. Extracellular data were digitized as above, and the fEPSPs were measured as the amplitude of the average of 6–12 individual responses. All data were analyzed by using the two-tailed Student’s ttest with a p < 0.05 criterion for statistical significance.
Concentrations of adenosine at the presynaptic A1 receptors were estimated by using the equation:
 in which [Ado] is the concentration of adenosine,FI is the fractional increase of the EPSC after theophylline application (0.45 with adenosine-free electrodes, 3.43 with electrodes containing 5 mm adenosine), H is the Hill slope, which is assumed to be 1.52, and the EC50 is assumed to be 680 nm (Dunwiddie and Diao, 1994). This analysis is based on the assumption that the inhibition of the EPSC is mediated by a single class of receptors (Reddington et al., 1982; Dunwiddie and Fredholm, 1989; Alzheimer et al., 1991).
in which [Ado] is the concentration of adenosine,FI is the fractional increase of the EPSC after theophylline application (0.45 with adenosine-free electrodes, 3.43 with electrodes containing 5 mm adenosine), H is the Hill slope, which is assumed to be 1.52, and the EC50 is assumed to be 680 nm (Dunwiddie and Diao, 1994). This analysis is based on the assumption that the inhibition of the EPSC is mediated by a single class of receptors (Reddington et al., 1982; Dunwiddie and Fredholm, 1989; Alzheimer et al., 1991).
RESULTS
Adenosine loading of pyramidal neurons inhibits synaptic activity
To determine whether changes in intracellular adenosine could affect the concentration of adenosine in the extracellular space, CA1 pyramidal neurons were “loaded” with adenosine by including adenosine in the electrode filling solution. Under these conditions, adenosine will diffuse into the pyramidal neurons and begin to elevate the intracellular concentration of adenosine as soon as the membrane patch is ruptured. If this treatment results in the release of adenosine into the extracellular space, the increased concentration of extracellular adenosine should alter electrophysiological responses via interactions with cell-surface adenosine receptors. To avoid any contamination from adenosine that might have leaked from the pipette into the extracellular space before formation of the gigaseal and to allow the concentration of adenosine within the cell and in the extracellular space to come to steady-state, all recordings were begun at least 15 min after the rupture of the membrane patch. To estimate the concentration of extracellular adenosine, we superfused slices with the competitive adenosine receptor antagonist theophylline. Theophylline blocks the action of adenosine at adenosine receptors and will therefore change the adenosine-sensitive electrophysiological responses in direct proportion to the amount of adenosine present in the extracellular space (Dunwiddie and Diao, 1994). Because EPSCs in the CA1 region are inhibited by adenosine profoundly, they should be enhanced by theophylline, and the magnitude of the increase produced by theophylline thus serves as an indirect measure of the amount of adenosine present. The initial experiments shown in Figure1 demonstrate that (1) theophylline produced a small increase in the EPSCs of control cells, (2) theophylline produced a larger increase in the EPSCs of adenosine-loaded cells, and (3) the increase in the EPSCs caused by theophylline was larger in the presence of exogenous adenosine.

The response to theophylline is proportional to the amount of adenosine in the extracellular space. A, The effect of theophylline on the excitatory postsynaptic current (EPSC) in a single neuron. Theophylline (THEO; 200 μm) was superfused during the time shown by thehorizontal bar and caused a small increase in the amplitude of the EPSC. The responses illustrated on theright were obtained during the baseline recording (a) and during the application of theophylline (b) at the time points indicated. Theophylline caused a 22% increase in the EPSC response, consistent with an 18% inhibition of the response by adenosine during the baseline recording.B, The effect of 200 μm theophylline (THEO; horizontal bar) on an adenosine-loaded cell (5 mm adenosine in the recording pipette). Theophylline caused a 395% increase in the amplitude of the EPSC, consistent with an 80% inhibition of the EPSC during the baseline. C, The effect of 200 μmtheophylline (THEO) on a cell recorded with normal filling solution in the recorded pipette (no adenosine inside) but with 30 μm exogenous adenosine (ADO) added to the bath. Exogenous adenosine inhibited the EPSC by 66% relative to the baseline, and this inhibition was reversed by theophylline. Thus, the increase in the EPSC produced by theophylline can be used to estimate the amount of adenosine-mediated inhibition of baseline responses. Each point on the graphs represents the average of five EPSCs evoked at 20 sec intervals, and each response on the right is averaged from seven consecutive sweeps.
In the subsequent experiments, four cellular parameters were measured: evoked excitatory postsynaptic currents (EPSCs), GABAAreceptor-mediated inhibitory postsynaptic currents (GABAAIPSCs), membrane resistance, and the holding current required to clamp the cell membrane potential at −70 mV. The effect of theophylline (200 μm) on these parameters was determined in control neurons and neurons recorded with an electrode filling solution containing 5 mm adenosine (Fig. 2). In control cells, theophylline increased EPSCs by 45 ± 13% (n = 11), which is consistent with previous reports indicating that there are small but significant amounts of endogenous extracellular adenosine even under control conditions (Dunwiddie, 1980; Dunwiddie and Diao, 1994). The effect of theophylline on EPSCs recorded from cells with adenosine-containing electrodes was approximately eightfold greater than in controls (343 ± 73% increase; n = 13;p < 0.002), demonstrating that the adenosine in the electrode caused a significant inhibition of the EPSC during the baseline recording. Theophylline caused a nonsignificant increase in the GABAA IPSC in control cells (70 ± 45% increase;n = 8; p > 0.16) and in adenosine-loaded cells (90 ± 62% increase; n = 10; p > 0.18), and there was no significant difference between the control and adenosine-loaded conditions (p > 0.8), which is consistent with previous reports that adenosine modulates excitatory but not inhibitory synaptic transmission in the hippocampus (Lambert and Teyler, 1991; Yoon and Rothman, 1991). Theophylline caused a small shift in the membrane resistance of control cells (−3.4 ± 1.5 MΩ; n = 11) and adenosine-loaded cells (2.2 ± 2.1 MΩ;n = 13), which was not significantly different between the two conditions (p > 0.075). However, theophylline caused a significant negative shift in the holding current in both control cells (−33.2 ± 6.7 pA; n = 11) and adenosine-loaded cells (−66.9 ± 17.5 pA; n = 13). Although there was a trend toward a greater effect of theophylline on the holding current in adenosine-loaded cells, this did not reach statistical significance (p > 0.19). In a second set of experiments, we investigated the dose–response relationship between the concentration of adenosine in the whole-cell electrode and the inhibition of the EPSCs. As shown in Figure3, the degree of adenosine-mediated inhibition of the EPSC, as determined by the increase in the responses after theophylline application, was dependent on the concentration of adenosine in the electrode filling solution, and the 5 mm concentration of adenosine used in the previous set of experiments gave a significant, reproducible response that was not maximal.
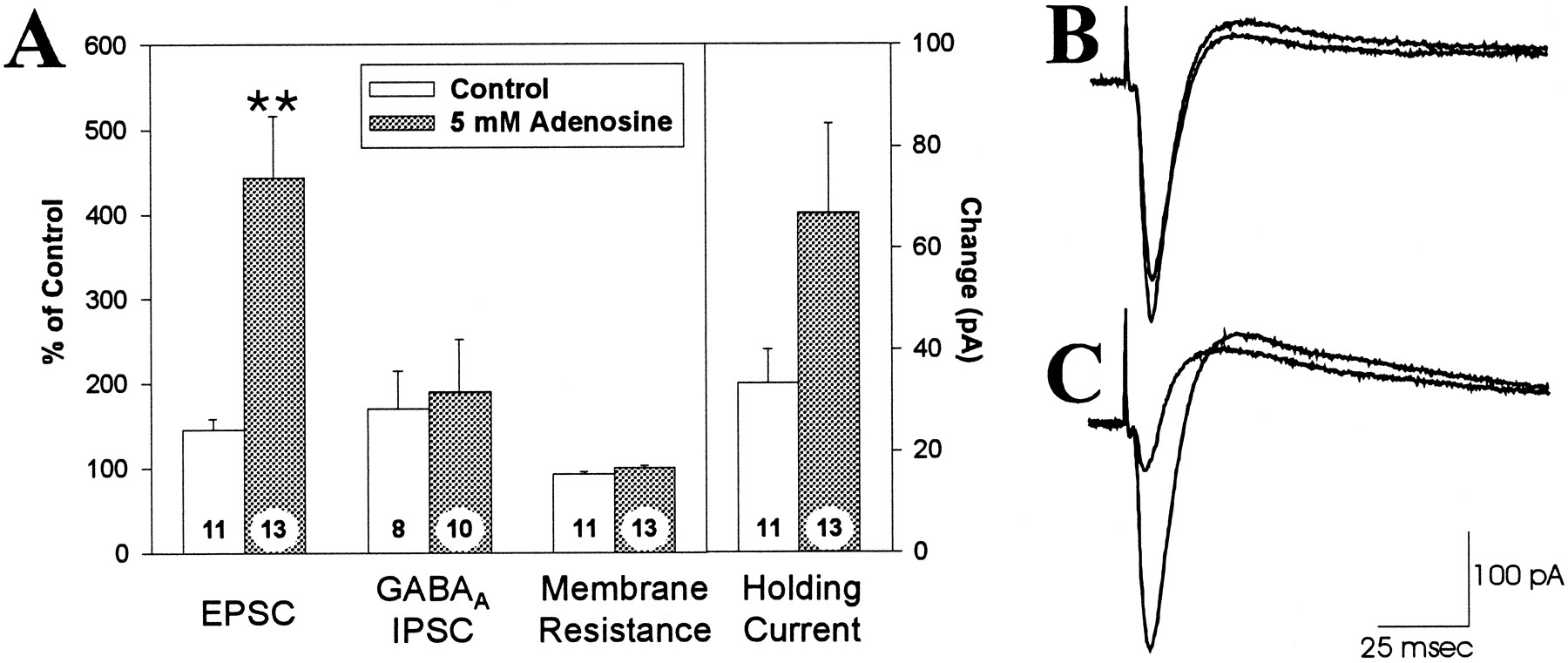
The response to 200 μm theophylline in neurons recorded with and without 5 mm adenosine in the electrode. A, The effects of theophylline on the amplitude of the EPSC, the GABAA receptor-mediated IPSC, membrane resistance (as determined by the current response to a fixed voltage step), and the holding current required to clamp the cell at −70 mV. Each parameter is expressed as a percentage of the pretheophylline control values, with the exception of the holding current, which is shown as the inward current response to theophylline in pA. Error bars represent SEM; **, denotes a significant difference (p < 0.01) relative to the control electrode solution. The numbers on each barindicate the number of cells recorded. B, Synaptically evoked currents from a cell recorded with a control electrode solution. The downward deflection (inward current) is the EPSC. The smaller EPSC response is the control response, and the larger response is the EPSC during application of 200 μm theophylline.C, Synaptic currents in a cell recorded with an electrode solution containing 5 mm adenosine; as inB, the larger response is the EPSC during superfusion with 200 μm theophylline. Each tracing is an average of 8–12 sweeps.

Concentration–response relationship for adenosine in the electrode filling solution. The data were transformed from the percentage of increase of the EPSC induced by theophylline to the percentage of inhibition of the EPSC by adenosine before theophylline application (assuming the response to 200 μm theophylline represents a response without any adenosine-mediated inhibition). This transformation facilitates the quantitative analysis of the data, because the maximal effect of adenosine is 100% inhibition of the EPSC response, whereas the maximal response to theophylline essentially could go to infinity. Under control conditions (no adenosine in electrode), endogenous adenosine inhibits the EPSC by ∼20%, whereas with the highest concentration of adenosine tested (15 mm), the EPSC is depressed by ∼80%. The line represents the computer-generated fit to the data according to the equation:Y = minresp + (maxresp − minresp)/(1 + 10[(logEC50− X) × H]), with an EC50 value of 2.12 mm and a Hill slope of 0.58. The numbers in parentheses indicate the number of cells recorded. In this and subsequent figures, *p < 0.05 and **p < 0.01 relative to 0 mm adenosine.
The effects of adenosine loading are mediated by extracellular adenosine
Adenosine loading of CA1 pyramidal neurons resulted in a theophylline-reversible depression of EPSCs that was consistent with mediation by cell-surface adenosine receptors. An alternative possibility is that adenosine reduced the EPSCs via an action at an intracellular site and that theophylline might reverse this effect via an intracellular mechanism. To test this possibility, we performed similar experiments with a charged adenosine receptor antagonist, 8-pSPT, which cannot cross the membrane barrier and enter cells. Like theophylline, 8-pSPT (100 μm) increased the EPSC response obtained with both control and adenosine-containing electrodes, but it caused a significantly greater increase in the amplitude of the EPSC in cells recorded with 5 mm adenosine in the electrode than in cells recorded with adenosine-free electrode solutions. The enhancement of the EPSC that was observed with 8-pSPT was not significantly different from that seen with theophylline (p > 0.87; Fig. 4). This result suggested that the effects of adenosine loading are mediated by extracellular adenosine receptors and not by an intracellular effect of adenosine.

Summary of the mean ± SEM inhibition of the EPSC as determined by application of adenosine antagonists under various conditions; the data were transformed, as in Figure 3.Open bars represent electrodes with 0 mmadenosine, filled bars with 5 mm adenosine, and the hatched bar is 5 mm dideoxyadenosine (DDA), a P-site agonist. The antagonists used were theophylline (Theo; 200 μm; these data are shown in Fig. 2 also and are included here for comparison), 8-p-sulfophenyltheophylline (8-pSPT; 100 μm), a charged adenosine receptor antagonist, and adenosine deaminase (ADA; 25 μg/ml). The fourth pair of bars demonstrates that including 5 mm DDA in the pipette had no significant effect on the baseline response. The last pair of bars shows that there was no effect of 2 mm BaCl2 on these responses (compare with first set of bars). The number on eachbar indicates the number of cells recorded; *p < 0.05; **p < 0.01 relative to 0 mm adenosine.
Another way to test whether the effects of adenosine loading are mediated by extracellular adenosine, rather than by some intracellular mechanism, is to antagonize the effects of extracellular adenosine with the enzyme adenosine deaminase. Adenosine deaminase catabolizes adenosine to inosine, which has little activity at adenosine receptors (Dunwiddie and Fredholm, 1984). Adenosine deaminase can thus antagonize the effects of adenosine (Dunwiddie and Hoffer, 1980; Haas and Greene, 1988), but because adenosine deaminase cannot cross cell membranes, it should antagonize an inhibitory effect of adenosine on the EPSC only if this effect is mediated by extracellular adenosine. As shown in Figure4, the baseline adenosine inhibition of the EPSC, as measured by application of 25 μg/ml adenosine deaminase, was significantly greater in cells loaded with adenosine than in cells recorded with adenosine-free solution. This was observed despite the apparent inability of adenosine deaminase to block all of the adenosine-mediated inhibition, as can be seen by the smaller amount of inhibition detected in control cells. The limited ability of adenosine deaminase to antagonize the effects of endogenous adenosine (relative to antagonists such as theophylline) probably reflects a more limited diffusion of this enzyme into the extracellular space of the slice and perhaps an inability to break down endogenous adenosine as rapidly as it is formed.
One potential mechanism by which adenosine could affect the EPSC magnitude via an intracellular site of action would be through an interaction with the P-site of adenylyl cyclase. Adenosine is capable of inhibiting cAMP formation by binding to an intracellular site on the adenylyl cyclase enzyme known as the P-site (Londos and Wolff, 1977). Thus, it is possible that the effects of adenosine loading were attributable to decreased levels of cAMP in the postsynaptic cell. To test whether P-site activation could mediate the effects of adenosine, recordings were made with patch electrodes containing 5 mmDDA, a selective P-site agonist (Londos and Wolff, 1977). The magnitude of adenosine-mediated baseline inhibition was almost identical in control and DDA-loaded cells (Fig. 4), and the inhibition in DDA-loaded cells was significantly smaller than that in adenosine-loaded cells (p < 0.0001). Thus, it is highly unlikely that the effects of adenosine loading are mediated by activation of the P-site.
The above experiments suggest that the effects of adenosine loading are attributable to the presence of elevated levels of adenosine in the extracellular space. To demonstrate that the adenosine in the extracellular space is released from the recorded cell and does not reflect leakage from the adenosine-filled electrode into the slice, we tested the stability of the effect of adenosine loading over time. It is possible that some of the adenosine that leaks out of the electrode before forming a tight seal with the cell membrane may still be present in the extracellular space during the baseline recording period. If this were the case, the adenosine-mediated inhibition should decay over time because of uptake and diffusion, whereas if the adenosine-mediated inhibition is attributable to the continuous release of adenosine from the cell, the response should be relatively stable over time. We have shown previously that an extracellular concentration of 20 mm adenosine can be cleared completely from the slice within 20 min (Mitchell et al., 1993b); therefore, the response to theophylline was compared 15 min after rupturing the patch (when some residual adenosine might be present) and 45 min after rupturing the patch. After a 45 min baseline, application of 200 μmtheophylline caused a 46 ± 10% increase in the amplitude of the EPSC with control electrodes (n = 4) and a 315 ± 48% increase in the amplitude of the EPSC with electrodes filled with 5 mm adenosine (n = 5). Neither of these values is significantly different from the effect of theophylline on the EPSC after a 15 min baseline (45 ± 13% increase with control electrodes, 343 ± 73% change with 5 mm adenosine; Fig. 2). These results are consistent with the conclusion that the increases in extracellular adenosine arise primarily from intracellular adenosine, that these increases are stable between 15 and 45 min, and that leakage of adenosine before seal formation does not account for the increased extracellular adenosine concentrations that are observed. As an additional control for leakage, we recorded from cells by using electrodes that contain adenosine and uridine in the filling solution. If adenosine is released by a specific transport mechanism, then uridine, which is a substrate for the nucleoside transporters (Paterson et al., 1991; Jones and Hammond, 1992), should compete with adenosine for transport out of the cell. If adenosine is leaking from the cells through a nonselective or nonsaturable mechanism, uridine will not be able to compete for adenosine release and will not decrease adenosine efflux. Cells recorded with 5 mm adenosine and 25 mm uridine in the electrode showed a 92 ± 16% increase in the EPSC after application of 200 μmtheophylline (n = 5). This is significantly less than the 343 ± 73% increase in the EPSC seen when electrodes contained 5 mm adenosine alone (p < 0.04). Thus, uridine significantly reduces the efflux of adenosine from adenosine-loaded cells, demonstrating that adenosine is being released from the cells by a saturable process.
Adenosine loading inhibits synaptic responses via a presynaptic mechanism
The reduction in evoked EPSCs produced by adenosine loading could occur by two possible mechanisms: adenosine could inhibit the release of glutamate via inhibitory A1 receptors on presynaptic terminals (Dolphin and Archer, 1983; Dunwiddie et al., 1984; Lupica et al., 1992;Wu and Saggau, 1994), or it could act on postsynaptic adenosine receptors and inhibit the EPSCs via a postsynaptic mechanism. A1 receptors on CA1 pyramidal neurons are coupled via a G protein to a Ba2+-sensitive potassium conductance, and the activation of this conductance could inhibit the EPSC by shunting the response (Siggins and Schubert, 1981; Segal, 1982; Dunwiddie and Fredholm, 1989). To test the latter hypothesis, we determined whether the effects of adenosine-containing electrode solutions on EPSCs were antagonized by 2 mm BaCl2 in the superfusion solution, which completely blocks the potassium conductance activated by postsynaptic adenosine receptors (Gerber et al., 1989). In the presence of Ba2+, theophylline increased the amplitude of the EPSC to a significantly greater extent in adenosine-loaded cells than in control cells, and neither of these responses was significantly different from those observed in control buffer without Ba2+ (Fig. 4). This suggests that adenosine loading of pyramidal neurons inhibits the EPSC by a mechanism independent of the Ba2+-sensitive postsynaptic potassium conductance.
Another approach to identifying the site of adenosine action took advantage of the phenomenon of PPF, in which the second of a pair of closely spaced evoked synaptic currents is enhanced relative to the first (Harris and Cotman, 1983). A distinguishing characteristic of modulators that inhibit release at hippocampal synapses is that they enhance PPF, as opposed to postsynaptically acting agents such as receptor antagonists, which have no effect on PPF (Creager et al., 1980; Dunwiddie and Haas, 1985). Therefore, the PPF ratio was characterized in pyramidal neurons recorded with adenosine-free and adenosine-containing electrode solutions (Fig. 5). During baseline recording, cells recorded with electrodes containing 5 mm adenosine had significantly greater PPF than did cells recorded with adenosine-free solutions (p < 0.02). Application of 200 μm theophylline reduced the PPF ratio of the adenosine-loaded cells so there was no difference between the adenosine-containing and control electrode solutions, which is consistent with the hypothesis that theophylline was blocking the actions of adenosine at a presynaptic site.

The paired-pulse facilitation (PPF) ratio of excitatory postsynaptic currents evoked in neurons recorded with control and adenosine-containing electrode solutions. The baseline magnitude of PPF in control neurons was significantly different (p < 0.05) from that in cells loaded with 5 mm adenosine (A), and this difference was abolished during superfusion with 200 μm theophylline. Individual points represent the mean ± SEM for the control (n = 5) or adenosine-loaded (n = 6) group of cells. B, A pair of EPSCs evoked 50 msec apart in a cell recorded with no adenosine added to the pipette. C, Comparable responses in an adenosine-loaded cell (5 mm adenosine); both are averages of seven evoked responses.
Adenosine loading selectively elevates extracellular adenosine near the loaded cell
The ability of adenosine loading to presynaptically inhibit excitatory synapses suggests the adenosine is being released into the extracellular space near these synapses. Alternatively, the adenosine could be released primarily from the cell body or other areas near the patch electrode in such large amounts that the adenosine could reach the presynaptic terminals after diffusion through theextracellular space. If this were the case, then extracellular adenosine would have to be elevated significantly over a relatively large area of the slice, in which event its effects would not be specific to the loaded cell. To examine this possibility, experiments were conducted with simultaneous whole-cell intracellular recording and extracellular recording of field potentials from the region of stratum radiatum corresponding to the dendritic field of the adenosine-loaded cell. The intracellular recordings confirmed the differential effects of theophylline on the EPSC in control and adenosine-loaded cells (Fig. 6A), while the simultaneous extracellular recordings demonstrated that there was no difference in the effects of theophylline on the corresponding fEPSPs (Fig. 6B), thus demonstrating that neighboring synapses in the same dendritic region were not affected by the adenosine-loading protocol.

The effects of 200 μm theophylline on simultaneously recorded intracellular EPSCs and extracellular field excitatory postsynaptic potentials (fEPSPs). A, The ensemble average response (mean ± SEM) induced by application of 200 μm theophylline for the EPSC recorded from control cells (•; n = 5) or 5 mmadenosine-loaded cells (○; n = 5). The duration of theophylline application is indicated by the horizontal bar at the bottom of the graph. B, The concurrent change in the fEPSP (mean ± SEM) when simultaneous whole-cell recording was conducted with control solution (•;n = 5 slices) or electrode solution containing 5 mm adenosine (○; n = 5 slices).C, D, The effect of 200 μmtheophylline on the EPSC (C) from a cell recorded with an adenosine-free electrode solution and the corresponding fEPSP responses (D). The smaller response in each case is the pre-drug control, and the larger response is during theophylline superfusion. The tracings are averages of 8–12 sweeps.E, F, Corresponding responses from a cell recorded with an electrode solution containing 5 mmadenosine (E) and the corresponding fEPSP responses (F). In both cases, the larger response was recorded in the presence of 200 μm theophylline.
Endogenous adenosine from a single cell can inhibit synaptic activity
To determine whether a single cell can generate and release enough endogenous adenosine to inhibit synaptic activity in the same manner as a cell loaded with exogenous adenosine, we filled a whole-cell recording electrode with 400 μm 5′-iodotubercidin, an inhibitor of adenosine kinase (Davies et al., 1984, 1986). When 5′-iodotubercidin was included in the electrode, theophylline caused a 514 ± 84% increase in the amplitude of the EPSC (p < 0.0001 vs control; n = 6), an effect comparable to that seen when a cell is loaded with 5–15 mm adenosine via the patch electrode. Thus, when adenosine kinase is inhibited in a single cell, the amounts of endogenous adenosine that are generated are comparable to the amounts that diffuse into the cell via adenosine-loaded patch electrodes.
DISCUSSION
The results of the present investigation demonstrate that a rise in the intracellular concentration of adenosine in a single CA1 pyramidal neuron leads to a corresponding increase in the activation of adenosine receptors via an increase in the concentration of adenosine in the extracellular space. Furthermore, the adenosine is released in such a way as to selectively inhibit evoked excitatory postsynaptic currents without markedly affecting the other electrophysiological parameters of the cell. This is the first demonstration that a hippocampal neuron is capable of regulating the strength of its excitatory synaptic inputs via this mechanism. The inhibition of the EPSC was mediated by extracellular adenosine acting at adenosine receptors, as shown by the ability of 8-pSPT and adenosine deaminase, neither of which can cross cell membranes and enter the cell, to block the inhibition. Furthermore, this inhibition was not attributable to activation of the intracellular P-site for adenosine on adenylyl cyclase, because the P-site agonist dideoxyadenosine did not mimic the effects of adenosine. Our results also demonstrate that the inhibition of the EPSC was not mediated by the activation of a postsynaptic potassium conductance by adenosine, because 2 mm BaCl2 did not block the effect and, instead, show that the inhibition was mediated by presynaptic adenosine receptors, as indicated by the change in the PPF ratio. Although it is possible that the sensitivity of postsynaptic adenosine receptors might be affected by intracellular adenosine, it is very unlikely that loading of the postsynaptic cell could affect the sensitivity of presynaptic receptors. Thus, the consequences of adenosine loading that we have reported are indicative of changes in extracellular adenosine concentrations and are not a reflection of changes in the sensitivity of adenosine receptors. Furthermore, the released adenosine is localized near the adenosine-loaded cell, because adenosine loading does not affect the population field EPSPs. A localized release of adenosine is also evident from the inability of adenosine loading to alter GABAA IPSCs. Although GABAergic nerve terminals do not contain adenosine receptors, exogenously applied adenosine inhibits the GABAA IPSC by decreasing glutamatergic stimulation of GABAergic interneurons (Fig. 1C). Adenosine loading does not seem to affect the excitatory inputs to the surrounding interneurons and only affects the excitatory inputs to the loaded cell. These data strongly support the hypothesis that including a high concentration of adenosine in the whole-cell electrode caused an increase in the concentration of adenosine within the pyramidal neuron, induced the release of adenosine into the extracellular space, and elevated the concentration of adenosine in the vicinity of Schaffer collateral/commissural afferent nerve terminals. This rise in extracellular adenosine was sufficient to activate presynaptic adenosine A1 receptors and inhibit the evoked release of glutamate.
The observation that increasing the intracellular concentration of adenosine can induce the release of adenosine into the extracellular space bears particular relevance to the mechanism by which metabolic disruptions might lead to the release of adenosine. One possibility that has been suggested is that an enzyme directly responsible for adenosine formation, such as 5′-nucleotidase, might couple the production of adenosine directly to its extrusion from the cell. The present experiments suggest that it is not necessary to invoke such a mechanism, because an increase in adenosine per se is sufficient to result in increased efflux from the cell. Previous experiments using ionophoretic injection of adenosine in medullary respiratory neurons of the cat also have reported results that are consistent with this hypothesis (Schmidt et al., 1995). It seems likely that adenosine release into the extracellular space is mediated by bidirectional purine nucleoside transporters, because loading the cell with a competing substrate for these transporters, such as uridine, can decrease the adenosine efflux from the cell. Previous evidence that nucleoside transporters are bidirectional and work by a facilitated diffusional mechanism (Bender et al., 1981; Plagemann and Woffendin, 1989; Gu et al., 1995), combined with evidence that simply increasing the intracellular concentration of adenosine is sufficient to evoke release, also supports the conclusion that the nucleoside transporters are involved.
An interesting aspect of our data is that we can use it to make a rough estimate of the concentration of adenosine at the presynaptic A1 receptors. Dunwiddie and Diao (1994) used a modified Hill equation to relate the percentage of inhibition of excitatory synaptic responses to the actual concentration of adenosine at presynaptic receptors in a hippocampal slice preparation. On the basis of their data and the inhibition of the EPSC in Figure 1, the basal concentration of adenosine at presynaptic A1 receptors in the present experiments was estimated to be 400 nm (see Materials and Methods). The concentration of adenosine at the presynaptic receptors when 5 mm adenosine is included in the electrode was estimated to be 1.5 μm. These results suggest two conclusions; first, it is clear that, despite the high concentrations of adenosine in the pipette, very little actually reaches the presynaptic receptors. However, given the limitations of dialysis into the cell, the relatively long diffusion path out to the dendrites, and the ability of intracellular enzymes such as adenosine kinase and adenosine deaminase to metabolize adenosine, this is not surprising. A second point is that, with the millimolar concentrations of ATP that are normally present in cells, only a very small fraction needs to be converted to adenosine to generate a physiologically significant amount of adenosine, particularly if it is generated close to the site of efflux from the cell.
An unexpected finding was that adenosine loading produced only a small, nonsignificant change on the holding current. Adenosine receptors on CA1 pyramidal neurons are coupled to inwardly rectifying K+channels that hyperpolarize these cells (Siggins and Schubert, 1981;Haas and Greene, 1984; Dunwiddie and Fredholm, 1989), and previous studies have reported changes in the membrane potential and membrane resistance when adenosine was applied exogenously, although these effects are typically small (Siggins and Schubert, 1981; Segal, 1982). There are several possible explanations for why we observed such a small effect on postsynaptic adenosine receptors. First, the postsynaptic response to adenosine is less sensitive and less robust than the presynaptic response, and the amount of adenosine that is released may be difficult to detect postsynaptically. Second, it is possible that the nucleoside transporters are localized relatively far from the postsynaptic receptors, so the extracellular concentration of adenosine does not rise near these receptors. A third possibility is that adenosine does reach postsynaptic receptors, but unlike the presynaptic response, the postsynaptic response desensitizes before superfusion with the adenosine receptor antagonists. In any case, the protocol we use seems to evoke a predominantly presynaptic adenosine response.
One novel finding of these studies is that they clearly establish that CA1 pyramidal neurons can release physiologically significant amounts of adenosine. Although numerous studies have shown that adenosine can be released from brain slices, there has been little evidence that neurons, as opposed to glia or other types of cells, can release adenosine. Manzoni et al. (1994) demonstrated a release of adenosine in the hippocampus induced by application of NMDA and by electrical stimulation. Although they suggested the adenosine might be coming from interneurons, their experiments did not rule out indirect mechanisms that might involve mediation by other cell types. Our experiments do not provide any evidence about the ability of interneurons to release adenosine, but they clearly suggest that CA1 pyramidal neurons are capable of releasing significant amounts of adenosine at their own synaptic inputs. Because CA1 pyramidal neurons contain NMDA receptors and are stimulated synaptically by the protocols used by Manzoni et al. (1994), they are also good candidates for the source of adenosine released in their experiments.
The release of adenosine near the excitatory inputs of the loaded cell has some interesting implications. Presumably, the intracellular concentration of adenosine needs to be elevated only near the site of release for adenosine release to occur. Thus, elevating the concentration of adenosine throughout the entire cell may not be necessary to modulate synaptic activity. A localized elevation of adenosine in one small part of a cell, such as a dendritic spine, may be sufficient to affect synaptic activity. There is good evidence that, in some cells, adenosine is formed rapidly and then rephosphorylated as a part of normal purine metabolism and that disrupting this pathway by inhibiting adenosine kinase can induce the release of adenosine from cells (Pak et al., 1994; Lloyd and Fredholm, 1995; J. M. Brundege and T. V. Dunwiddie, unpublished observations). Indeed, we have shown that inhibiting adenosine kinase in a single cell produces an adenosine-mediated inhibition of its excitatory inputs. It may be possible for neurons to release adenosine during normal physiological activity by modifying the activity of the enzymes adenosine kinase or 5′-nucleotidase. It is interesting to note that biochemical assays have shown that adenosine kinase is subject to substrate inhibition by adenosine (Fisher and Newsholme, 1984), suggesting that if adenosine concentrations within the cell rise past a certain point, a rapid, dramatic elevation of adenosine levels may ensue. It is possible that such a mechanism may play a role in the release of adenosine during our adenosine-loading protocol and that this mechanism could amplify greatly the effects of an otherwise small, transitory rise in adenosine levels in a subregion of a neuron. Hence, a large-scale metabolic disruption, such as occurs during ischemia, may not be necessary to evoke the release of adenosine, and a small, local release could occur in response to normal physiological activity.
In summary, these experiments support several conclusions. First, a simple rise in the intracellular concentration of adenosine leads to an increase in extracellular adenosine. This provides a mechanism that might account for the release of adenosine and the corresponding inhibition of synaptic activity, which is induced by hypoxia and ischemia. Second, the release of adenosine from a single neuron is sufficient to inhibit excitatory synaptic responses onto that cell by >80%. This suggests that individual cells have a powerful mechanism by which they can regulate their afferent synaptic activity, and it raises the possibility that pyramidal neurons use adenosine as a retrograde messenger to regulate their own excitatory inputs.
Footnotes
This work was supported by Grant R01 NS29173 from the National Institute of Neurological Disorders and Stroke, and by the Veterans Administration Medical Research Service.
Correspondence should be addressed to James M. Brundege, Department of Pharmacology, Box C-236, University of Colorado Health Sciences Center, Denver, CO 80262.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}