

Abstract
The role of phospholipase C-coupled (group I) metabotropic glutamate receptors (mGluR1 and mGluR5) in post-traumatic neuronal injury was examined using rat in vivo and in vitro models. Traumatic injury to mixed neuronal/glial cultures induced phosphoinositide hydrolysis and caused neuronal death. Pharmacological blockade of group I receptors significantly reduced these effects in vitro and decreased neurological deficits as well as neuronal loss produced by traumatic brain injuryin vivo. In contrast, activation of group I receptors by a specific agonist in vitro exacerbated post-traumatic neuronal death in a dose-dependent manner. Antisense oligodeoxynucleotide directed to mGluR1, but not to mGluR5, was neuroprotective in vitro, although each oligodeoxynucleotide reduced the respective receptor-stimulated accumulation of inositol phosphates to a similar degree. Together, these findings suggest that activation of mGluR1 contributes to post-traumatic neuronal injury and that mGluR1 antagonists may have therapeutic potential in brain injury.
- antisense oligodeoxynucleotides
- brain trauma
- metabotropic glutamate receptors
- neuronal injury
- neuroprotection
- phosphoinositide hydrolysis
Increased glutamate release and activation of ionotropic glutamate receptors have been implicated in the pathophysiology of traumatic brain (Hayes et al., 1988; Faden et al., 1989; McIntosh et al., 1990) and spinal cord injuries (Faden and Simon, 1988; Wrathall et al., 1992). Metabotropic glutamate receptors participate broadly in the regulation of glutamate neurotransmission (Schoepp and Conn, 1993), yet their role in post-traumatic CNS injury has been largely unexplored.
Metabotropic glutamate receptors (mGluR) are coupled through G-proteins to second-messenger systems (Hollman and Heinemann, 1994; Pin and Duvoisin, 1995). Molecular cloning techniques have elucidated eight mGluR subtypes to date, some of which include alternately spliced variants (Hollman and Heinemann, 1994; Pin and Duvoisin, 1995). mGluR have been divided into three groups based on sequence homology: group I includes mGluR1 and mGluR5; group II includes mGluR2 and mGluR3; and group III includes mGluR4, mGluR6, mGluR7, and mGluR8 (Pin and Duvoisin, 1995). These three groups are further differentiated by their respective signal transduction mechanisms: group I receptors stimulate phospholipase C (PLC), leading to phosphoinositide (PI) hydrolysis and intracellular Ca2+ mobilization, whereas group II and group III receptors are coupled to the inhibition of adenylyl cyclase (Saugstad et al., 1994; Pin and Duvoisin, 1995).
There has been limited research to address a possible role for mGluR in modulating neuronal death after various nontraumaticinsults. To a certain extent, groups I and II mGluR appear to have contrasting actions. Group I receptors may potentiate neuronal excitation and excitotoxicity (Buisson and Choi, 1995; Pin and Duvoisin, 1995; Buisson et al., 1996), possibly through positive modulation of NMDA receptor activity (Fitzjohn et al., 1996). Group II receptors may exert a protective effect (Bruno et al., 1994; Buisson and Choi, 1995), possibly through presynaptic inhibition of glutamate release (Watkins and Collingridge, 1994; Pin and Duvoisin, 1995). Because of these apparently opposing actions, and the fact that more discriminating agonists/antagonists are not yet available, it is not surprising that data have been less than conclusive with regard to the role of mGluR in neuronal death. Indeed, the cyclic glutamate analog 1S,1R-1-aminocyclopentane-1,3-dicarboxylic acid (1S,1R-ACPD), which is an agonist at both group I and group II receptors, has been reported in various studies to either contribute to or protect against cell death (Koh et al., 1991; Chiamulera et al., 1992; McDonald and Schoepp, 1992; Sacaan and Schoepp, 1992; Siliprandi et al., 1992; Birrell et al., 1993;McDonald et al., 1993).
The synthesis of phenylglycine derivatives has permitted differentiation of the activity of various metabotropic receptor groups through comparison of the activities of selected compounds (Watkins and Collingridge, 1994). However, differentiation of receptor subtypes within groups has remained problematic (Saugstad et al., 1995).
In the present studies, rat in vitro and in vivotrauma model systems were both used to investigate the role of group I mGluR in mediating post-traumatic neuronal injury. We used (1) metabotropic receptor agonists and antagonists to elucidate the role of group I receptors in secondary injury, and (2) antisense oligodeoxynucleotides (AS ODN) directed to group I receptor subtypes to compare or contrast the neuroprotective roles of mGluR1 and mGluR5.
MATERIALS AND METHODS
Mixed neuronal/glial cultures. Sprague Dawley pregnant rats were obtained from Taconic Farms (Germantown, NY). Neocortex from 17- to 18-d-old embryos was dissociated, and cell suspension (5 × 105 cells/cm2) was seeded on top of confluent glial cultures 10 d in vitro (DIV). To prepare glial cultures, dissociated neonatal (1–2 d old) rat neocortex was seeded in 96-well Primeria plates (Falcon, Lincoln Park, NJ) at a density of 0.5 hemisphere/plate. In each case, the cortex was dissociated with a serological pipette in HBSS without magnesium or calcium, supplemented by 10 mm HEPES, pH 7.0, and 1 mm sodium pyruvate. The protocol for maintaining culture and media composition is detailed by Regan and Choi (1994). EBSS and HBSS were purchased from Mediatech (Herndon, VA); HEPES, glutamine, and glucose were from Biofluids (Rockville, MD); epidermal growth factor was from Life Technologies (Grand Island, NY); all other chemicals were procured from Sigma (St. Louis, MO).
In vitro trauma model. Our in vitro trauma model is based somewhat on the murine model developed previously by Choi and colleagues (Tecoma et al., 1989; Regan and Choi, 1994), which examined the role of ionotropic glutamate receptors in secondary injury using mixed neuronal/glial cultures. In the Regan and Choi model, injury is delivered by needle-scoring a grid of eight parallel cuts, crossed by eight perpendicular cuts in each well of 24-well tissue culture plates. In our model, trauma is delivered by a specially designed punch: this device consists of 28 stainless steel blades joined together, which produce 28 parallel cuts 1.2 mm in length uniformly distributed through the cell layer at 0.5 mm intervals in each well of a 96-well tissue culture plate. Cells that are located directly under the blades are damaged. Because the release of lactate dehydrogenase (LDH) from initially damaged cells is washed out after a 30 min incubation period, our obtained LDH values appear to reflect secondary injury. Sixteen to eighteen hours after injury, we observed death in up to 60 percent of total neurons. This paradigm was used in all of our studies, except those that examined the group I mGluR agonistR,S-3,5-dihydroxyphenylglycine (DHPG). For the latter experiments, a lower level of injury was required to assess possible exacerbation of trauma-induced injury. In these experiments, trauma was delivered by a circular punch that produced a circular cut with a 4 mm diameter in the cell layer. After 16 hr, this injury caused death in up to 25% of neurons.
The advantages of our model include a high reproducibility and consistency of injury across the different wells, and the convenience and ability to perform many replications over a short period of time (up to 20 per min). In our model, as in that described previously (Tecoma et al., 1989; Regan and Choi, 1994), secondary cell death can be largely prevented by the addition of the NMDA receptor antagonist 5R,10S-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (MK-801) (Mukhin and Faden, 1995), but 6-nitro-7-sulfamoylbenzo[f]quinoxaline-2,3-dione (NBQX), which is an antagonist of AMPA/kainate receptors, exerts no neuroprotective effect (our unpublished data).
Before injury, neuronal/glial cultures (17–19 DIV) were transferred to HEPES salt solution (120 mm NaCl, 5.4 mm KCl, 0.8 mm MgCl2, 1.8 mmCaCl2, 15 mm glucose, 15 mm HEPES, final pH 7.4) and incubated at 37°C for 30 min in the presence or absence of (+)-α-methyl-4-carboxyphenylglycine (MCPG), (S)-4-carboxyphenylglycine (4CPG), orR,S-3,5-dihydroxyphenylglycine (DHPG). MCPG, 4CPG, and DHPG were obtained from Tocris Cookson (St. Louis, MO). After injury, cells were incubated at 37°C for 30 min and washed five times (>1000-fold dilution) with growth medium (25 mm glucose, 1.0 mm glutamine, 25 mm HEPES, pH 7.2, 1% antibiotic–antimycotic in MEM EBSS). Cultures were then incubated in 5% CO2 for 16–18 hr at 37°C before assessment of injury. Control (uninjured) cultures (sister cultures from the same plate) were treated, i.e., washed, incubated (in the presence of agonist or antagonist, as required), and analyzed, using the same methods as for injured cultures.
Neuronal death was assessed by phase-contrast microscopy, trypan blue staining, and LDH assay. In our preliminary studies and those of Regan and Choi (1994), injury-induced LDH release 18 hr after injury is highly correlated to the number of nonviable neurons. Therefore, in our present experiments we quantitated neuronal death by measuring LDH efflux into the medium.
LDH measurement. LDH activity was measured at room temperature using a modification of the method described by Amador et al. (1963). A volume of 75 μl of culture medium was transferred to a 96-well microplate. A volume of 200 μl of LDH assay reagent (5 mm β-NAD, 25 mm lactic acid, 0.03% BSA, 100 mm Tris, final pH 8.45) was then rapidly added to each sample. Increases in optical density at 340 nm were measured at 10 sec intervals for 6 min using a Ceres 900 microplate reader (Biotek Instruments, Winooski, VT). Lyophilized serum (Accutrol) served as the LDH standard. Background LDH levels were estimated in the control (uninjured) cultures and subtracted from the values obtained after experimental injury. Results are expressed as percentage of LDH release observed after injury without any treatment. All reagents, including standard serum, were procured from Sigma.
Phosphoinositide (PI) hydrolysis. PI hydrolysis was measured in mixed neuronal/glial cultures (17–20 DIV), incubated overnight with 0.1 mCi/well myo-[3H]inositol (45 Ci/mmol, DuPont NEN, Boston, MA). Before injury, cells were washed three times with HEPES salt solution and incubated at 37°C for 30 min in the presence or absence of 500 μm MCPG. Five minutes after addition of 20 mm LiCl, cells were injured and incubated at 37°C. In 1S,3R-ACPD stimulation studies, cells were washed as above and preincubated at 37°C for 30 min, and 20 mmLiCl was added simultaneously with 500 μm1S,3R-ACPD (Tocris Cookson). Thirty minutes after injury or addition of 1S,3R-ACPD, incubation medium was aspirated and inositol phosphates were extracted by 0.1 m HCl containing 2 mm CaCl2. As detailed by Berridge et al. (1982), after separation on anion exchange columns (AG 1-X8, Bio-Rad, Hercules, CA), accumulated [3H]inositol phosphates were measured using a liquid scintillation counter (LS 6500, Beckman Instruments, Fullerton, CA).
AS ODN treatment. After 14 DIV, mixed neuronal/glial cultures were maintained in serum-free medium (25 mmglucose, 1.0 mm glutamine, 25 mm HEPES, pH 7.2, antibiotic–antimycotic, and MEM EBSS), and supplemented by N2 supplement (Life Technologies) in the absence (control) or presence of 2 μm of appropriate ODN. Fresh ODN (up to 2 μm) was added every 24 hr for 5 d. Antisense ODN (5′-CCGGACCATTGTGGCGAA-3′) directed to mGluR1 is complementary to the sequence region that begins with −9 and ends with +9 nucleotide (Masu et al., 1991). Position +1 corresponds to the A nucleotide in the first ATG codon. Sense ODN is 5′-TTCGCCACAAT-GGTCCGG-3′, and missense ODN is 5′-CCGGAGCATAGTGGGG-AA-3′. Antisense ODN directed to mGluR5 (5′-AGAAGGACC-ATTTTAGGA-3′) is complementary to regions from −7 up to +11 (Abe et al., 1992). In this instance, sense ODN is 5′-TCCTAAAATG-GTCCTTCT-3′ and missense ODN is 5′-AGAAGCACCATATTACGA-3′. Oligodeoxynucleotides used in all our studies were obtained from Bio-Synthesis (Lewisville, TX) as phosphorothioate derivatives.
Immunoblot analysis. Neuronal/glial cultures (17–19 DIV) from 8 wells of the 96-well plate were collected in 1 ml of ice-cold 10 mm Tris-HCl buffer, pH 7.4, containing 100 μmphenylmethylsulphonyl fluoride (Sigma) and lysed by freezing and thawing. The membrane suspension was centrifuged at 14,000 × g for 10 min. The pellets were washed twice in the same buffer, and protein concentration was measured by Bio-Rad Protein Assay using bovine serum albumin as standard. Eight micrograms of membrane protein were separated by SDS-PAGE in 7.5% acrylamide and transferred onto Hybond-PVDF membrane (Amersham, Arlington Heights, IL). Immunostaining was done with primary affinity-purified rabbit polyclonal antibodies to the C terminus of mGluR1α (Chemicon, Temecula, CA; dilution 2.5 μg/ml) and to the C terminus of mGluR5 (Upstate Biotechnology, Lake Placid, NY; dilution 1 μg/ml). Signals were detected using enhanced chemoluminescence (ECL) Western blotting detection system (Amersham).
Traumatic brain injury (TBI) in vivo. Male Sprague Dawley rats (400 ± 25 gm, Harlan, Indianapolis, IN) were anesthetized with sodium pentobarbital (Abbott Laboratories, North Chicago, IL; 70 mg/kg, i.p.) and subjected to lateral fluid percussion-induced brain trauma. In this model, a fluid wave is delivered to the extradural space over the left parietal cortex, transiently deforming the underlying brain. Details are provided byFaden et al. (1989) and McIntosh et al. (1989). This model simulates many features of human concussive brain injury and has been well defined in terms of its behavioral, biochemical, and histological outcomes (Faden et al., 1989; McIntosh et al., 1989; Sun and Faden, 1995b).
In central administration studies, vehicle (10 μl of 25 mm Tris, pH 7.4, in normal saline, n = 12 animals) or R,S-MCPG (0.5 μmol in 10 μl of 25 mm Tris, pH 7.4, in normal saline, n = 9 animals) was injected into the lateral ventricle 15 min before and 1 hr after percussion-induced TBI of 2.8 atmospheres (atm). In systemic administration studies, vehicle (0.4 ml of 10 mm Tris, pH 7.4, in normal saline, n = 15 animals) orR,S-MCPG (48 μmol in 0.4 ml of 10 mm Tris, pH 7.4, in normal saline, n = 14 animals) was administered intravenously 15 min after TBI of 2.8 atm. In our laboratory, TBI of 2.8 atm results in moderately severe injury, as revealed by behavioral and/or histological changes (Sun and Faden, 1995a,b). R,S-MCPG was obtained from Tocris Cookson.
Neurological scoring. Neuroscore tests included evaluation of resistance to lateral pulsion (right and left), forelimb contraflexion upon suspension by the tail (right and left), and ability to maintain position on an incline plane (right, left, vertical). These tests show high interobserver reliability and have been used by us in a number of studies to discriminate treatment effects (Faden et al., 1989; Sun and Faden, 1995a,b). Each test was rated from 0 (no function) to 5 (normal function); therefore, maximum composite score was 35. Neuroscore tests were conducted by an individual unaware of treatment using procedures detailed previously (McIntosh et al., 1989; Sun and Faden, 1995a).
Cresyl violet staining. Two weeks after trauma, animals were anesthetized (100 mg/kg sodium pentobarbital, i.p.) and perfused intracardially with heparinized saline (1 U/ml, Sigma) followed by 10% buffered formalin (Fisher Scientific, Fair Lawn, NJ). Brains were harvested, fixed for an additional 24 hr, cryoprotected in 10% sucrose in PBS, and frozen at −80°C in embedding medium (Miles, Eckhart, IN). Coronal sections (8 μm) from the dorsal hippocampus (−2.2 to −3.8 relative to bregma) were thaw-mounted onto gelatin-rubbed slides and maintained at −80°C for histological study. Sections were stained with Cresylecht Violet (Cellpoint Scientifica, Rockville, MD) and differentiated in ethanol and xylenes. The number of surviving neurons in the ipsilateral hippocampus was quantitated using light microscopy (Model BH2, Olympus) under 400× magnification by counting cells in areas CA1 and CA3 showing distinct nuclei and nucleoli (Sun and Faden, 1995b).
Data analysis. Data reflecting continuous variables were analyzed by Student–Newman–Keuls test after ANOVA or by Student’st test for two-group comparisons. Neuroscore data were analyzed using the Mann–Whitney U test after Kruskal–Wallis nonparametric ANOVA. p < 0.05 was considered statistically significant.
RESULTS
Activation of group I mGluR after mechanical injury
To determine whether there was trauma-induced activation of PLC-coupled (group I) mGluR, we examined the effect of mechanical injury on accumulation of IP in the in vitro trauma model. In this model, mechanical injury to neuronal/glial cultures produced death in up to 60% of neurons 16–18 hr after trauma. As demonstrated in Figure 1, mechanical trauma markedly increased IP accumulation (p < 0.01); this increase was significantly reduced (p < 0.05) by addition of 500 μm MCPG. Because MCPG is an equipotential antagonist at mGluR groups I and II (Watkins and Collingridge, 1994; Pin and Duvoisin, 1995), and its effects on PLC are related to blockade of group I receptors (but not of group II receptors), these in vitro results suggest that traumatic injury activates group I mGluR.

Trauma to mixed neuronal/glial cultures induced significant PI hydrolysis that was attenuated by treatment with an mGluR antagonist (500 μm MCPG added 30 min before injury). MCPG alone in control (uninjured) cultures had no significant effect on PI hydrolysis. PI hydrolysis was measured as accumulation of [3H]inositol phosphates from 5 min before and up to 30 min after injury. Data represent mean ± SEM;n = 16–22 cultures per condition. **p < 0.01 versus control (uninjured culture);†p < 0.05 versus injury (untreated injury), using the Student–Newman–Keuls test after ANOVA.
Immunodetection of group I mGluR subtypes in neuronal/glial cultures
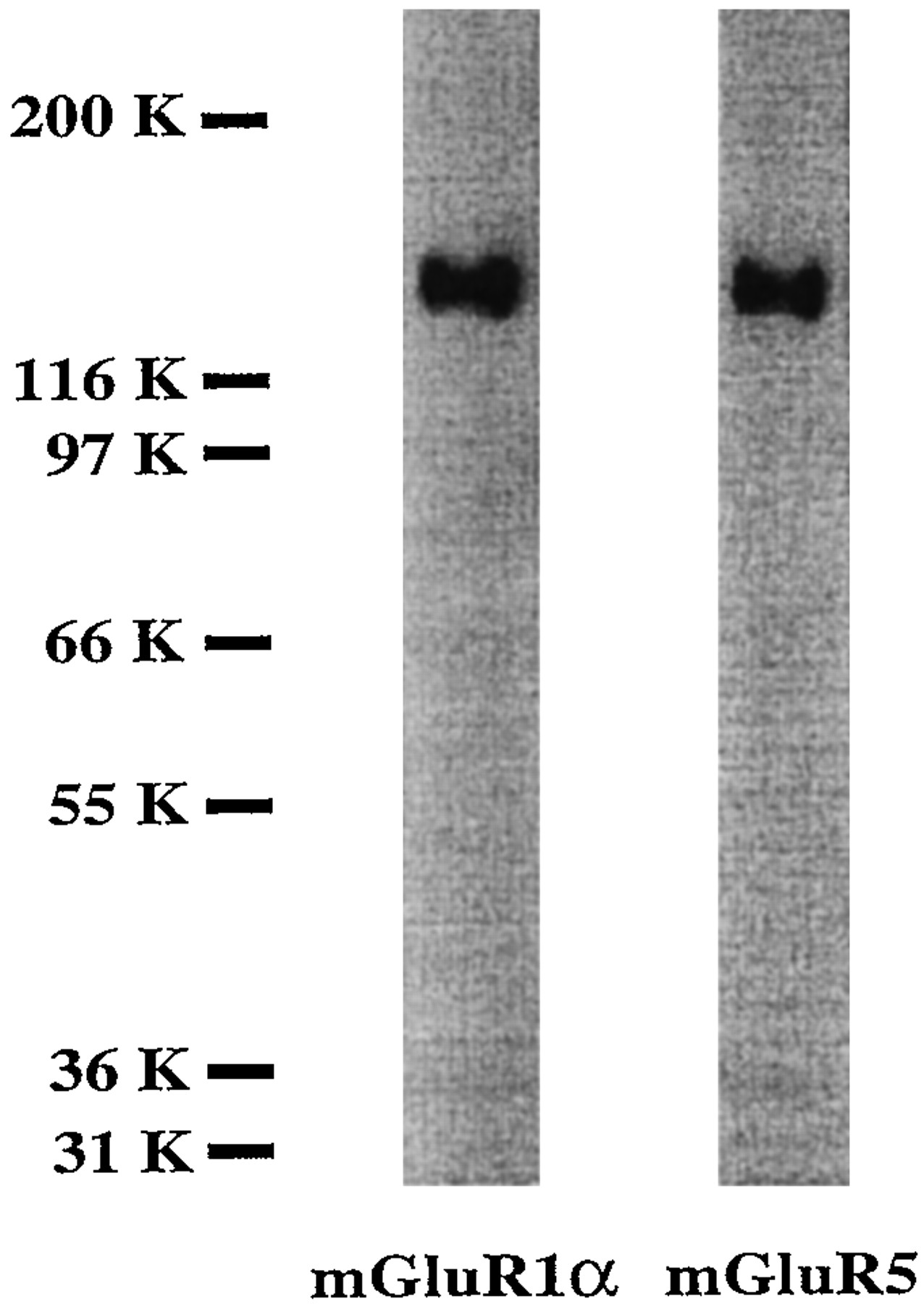
As noted above, group I mGluR includes two receptor subtypes: mGluR1 with four splicing variants and mGluR5 with two splicing variants. To evaluate the possible presence of group I mGluR specific subtypes in our neuronal/glial cultures, we used two available polyclonal antibodies, the first directed to the C terminus of one of the splicing variants of mGluR1 (mGluR1α), and the second directed to the C terminus of the splicing variants of mGluR5 (mGluR5α and mGluR5β) (Reid et al., 1995; Romano et al., 1995). As shown in Figure2, each antibody recognizes only one specific band with a molecular weight of ∼140 kDa.

Both subtypes of PLC linked mGluR—mGluR1 (as mGluR1α splicing variant) and mGluR5—are found in neuronal/glial cultures used in these studies. Cell membranes (8 μg protein) were analyzed by SDS-PAGE and immunoblotting with mGluR1α- and mGluR5-specific antibodies, as described in Materials and Methods. When primary antibody was omitted from the incubation phase, the specific bands were not detected (data not shown). Data represent results of 18-DIV cell culture study; similar results were obtained in two additional experiments with 17- and 20-DIV cultures.
As was shown previously in an in vitro expression system, the mGluR1α antibody used recognizes a specific 140 kDa band in CHO cells transfected by mGluR1α cDNA but not in cells transfected by mGluR5α cDNA (J. Wroblewski, personal communication). Moreover,Romano et al. (1995) have reported that the mGluR5 antibody used by us does not cross-react with mGluR1α. Additional control experiments, in the absence of primary antibodies, showed a lack of cross-reactivity of the secondary antibody to cell culture membrane proteins (data not shown).
Exacerbation of injury-induced neuronal death in vitroby DHPG, an agonist of group I mGluR
To study the possible role of group I mGluR activation in exacerbating neuronal death caused by injury, we examined the effects of DHPG, a selective group I mGluR agonist (Schoeppet al., 1994) in thein vitro trauma system. As shown in Figure 3, DHPG produces a significant, dose-dependent increase of injury-induced LDH release that can be attenuated by MCPG. In control (uninjured) cultures, neither DHPG nor MCPG, nor a DHPG-MCPG combination, had any significant effect on LDH release (data not shown). These data indicate that activation of group I mGluR after trauma increases injury-induced neuronal death.
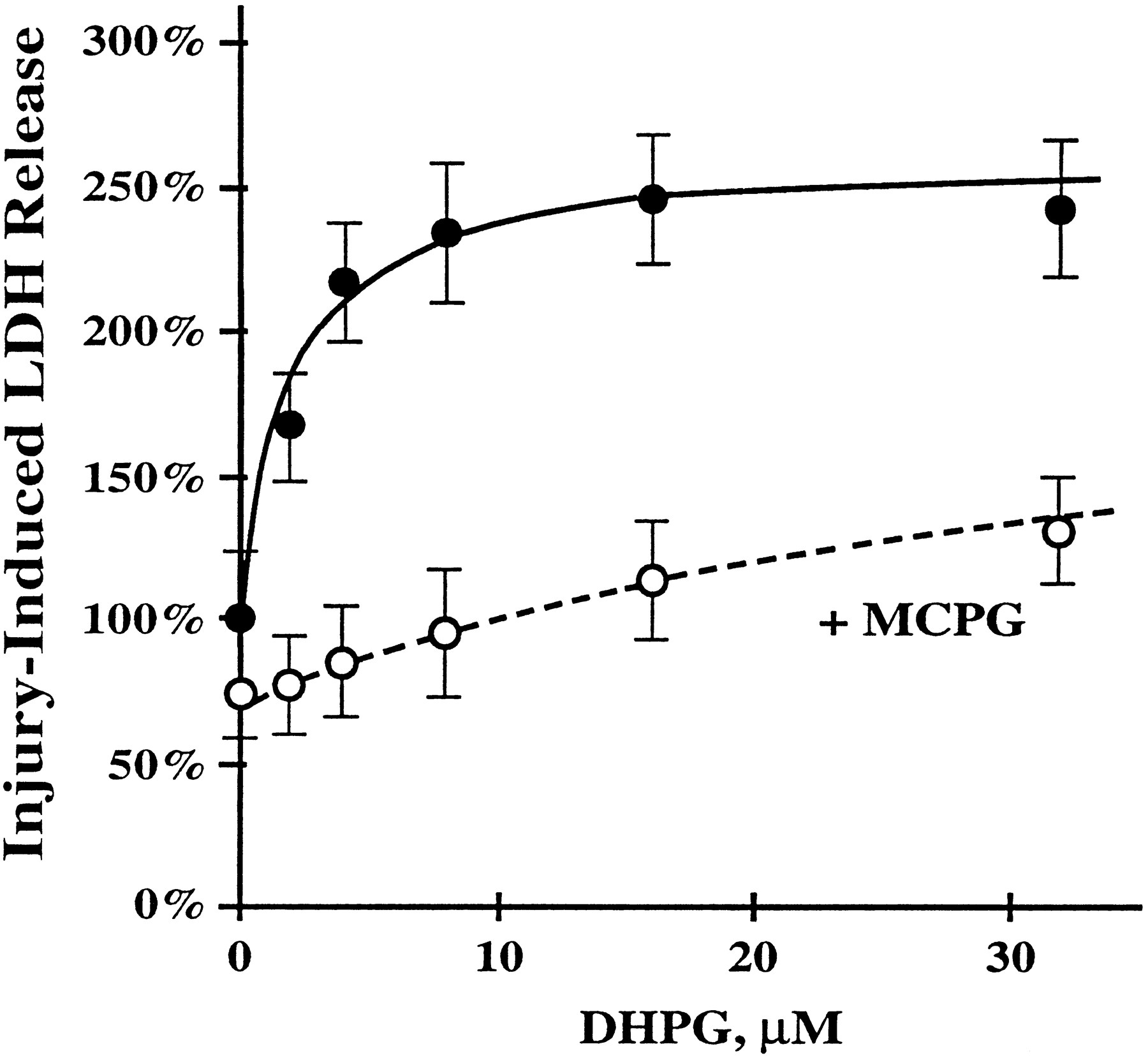
The agonist of group I mGluR DHPG in dose-dependent manner potentiates the injury-induced LDH release, which can be attenuated by the mGluR antagonist MCPG. Cultures were incubated in the presence of 2–32 μm DHPG (filled circles) or in the presence of 2–32 μm DHPG plus 1 mm MCPG (open circles) 30 min before and 30 min after injury. LDH levels were measured 16–18 hr after injury. Data represent mean ± SEM. DHPG alone: n = 19–20 cultures for each concentration; DHPG plus MCPG:n = 10 cultures for each concentration.
Neuroprotective effects of group I mGluR antagonistsin vitro
In the same in vitro model, MCPG significantly decreased (p < 0.01) trauma-induced LDH release (Fig. 4A), with EC50 = 50–100 μm (Fig. 4B). As noted above, MCPG is an equipotential antagonist at mGluR groups I and II (Watkins and Collingridge, 1994; Pin and Duvoisin, 1995). To evaluate further the neuroprotective effect of group I mGluR blockade in traumatic injury, we also studied 4CPG, which is a similar but more potent antagonist than MCPG at group I mGluR (IC50 ∼ 30–40 μm) and, only at much higher concentrations (EC50 ∼ 500 μm), is an agonist at group II mGluR (Watkins and Collingridge, 1994; Pin and Duvoisin, 1995). As shown in Figure 5, 4CPG—which is somewhat selective at group I receptors at the concentration used (30 μm vs EC50 on group II of 500 μm)—also significantly decreased trauma-induced LDH release (p < 0.001). The fact that MCPG and 4CPG have identical effects on group I receptors, yet opposite effects on group II receptors, suggests that each exerts neuroprotective action at group I receptors.

Treatment with MCPG attenuated neuronal injury after mechanical trauma to mixed neuronal/glial cultures, as reflected by changes in LDH release. Cultures were incubated in the absence or presence of 500 μm MCPG (A) or 12–1000 μm MCPG (B) 30 min before and 30 min after injury. LDH levels were measured 16–18 hr after injury. Data represent mean ± SEM. A, n = 25 cultures per condition; B, n = 18–20 cultures for each concentration; **p < 0.01 versus control (untreated injury) using Student’s ttest.

Treatment with 4CPG, a somewhat selective antagonist at group I mGluR at the concentration used (30 μm), also showed neuronal protection in vitro. Culture conditions, LDH measurements, and methods of drug administration were similar to those described in Figure 4. Data represent mean ± SEM; n = 46–50 cultures per condition. ***p < 0.001 versus control (untreated injury) using Student’s t test.
Neuroprotective effects of MCPG in vivo
To assess the role of mGluR in traumatic injury in vivo, we examined the effects of R,S-MCPG in an in vivo injury model using rats subjected to lateral fluid percussion-induced brain trauma. R,S-MCPG is an antagonist of group I and group II mGluR; other available group I antagonists show agonist activity at group II receptors. Because group II agonists show neuroprotective action (Bruno et al., 1994; Buisson and Choi, 1995; Buisson et al., 1996), we chose MCPG to exclude the possibility that suggested neuroprotective effects could be related to agonist action at group II receptors.R,S-MCPG administered intracerebroventricularly 15 min before and 1 hr after trauma not only significantly improved neurological recovery (p < 0.01; Fig.6) but also attenuated post-traumatic CA1 pyramidal cell loss (p < 0.01, Student’s t test) in ipsilateral hippocampus (Fig. 7). Similar attenuation of cell loss was noted in CA3 (Fig. 7); however, it did not quite reach statistical significance (p < 0.06, Student’st test).

Intracerebroventricular administration ofR,S-MCPG (0.5 μmol/injection) at 15 min before and 1 hr after injury significantly improved neurological recovery after lateral fluid percussion-induced TBI in rats. Histograms represent median scores at different days post-trauma. Eachcircle represents individual animal cumulative neuroscore reflecting performance on a battery of motor tests.Filled circles, Untreated injury (vehicle),n = 12 animals; open circles, injury treated by R,S-MCPG,n = 9 animals. **p < 0.01 versus untreated injury (vehicle), using the Mann–WhitneyU test after Kruskal–Wallis nonparametric ANOVA.

Intracerebroventricular administration ofR,S-MCPG (0.5 μmol/injection) at 15 min before and 1 hr after injury attenuated post-traumatic cell loss in ipsilateral hippocampus measured 2 weeks after fluid percussion-induced TBI. Cells were counted after staining 8 μm coronal brain sections with cresyl violet. Data represent mean number of cells ± SEM per 0.25 × 0.25 mm field. Control group, n = 8 animals; R,S-MCPG-treated group,n = 5 animals. **p < 0.01 versus control (untreated injury, vehicle) using Student’st test.
In separate experiments designed to simulate more clinically relevant conditions, R,S-MCPG was given intravenously 15 min after trauma. MCPG-treated animals showed markedly enhanced neurological recovery compared with vehicle-treated controls at 1 week (p < 0.001) and at 2 weeks (p < 0.01) after TBI (Fig. 8).

Intravenous administration of 48 μmR,S-MCPG 15 min after injury significantly improved neurological recovery at 1 and 2 weeks after lateral fluid percussion-induced TBI in rats. Histograms represent median scores at different days post-trauma. Each circlerepresents individual animal cumulative neuroscore. Filled circles, Untreated injury (vehicle), n = 15 animals: open circles, injury treated byR,S-MCPG, n = 14 animals. **p < 0.01; ***p < 0.001 versus untreated injury (vehicle), using the Mann–WhitneyU test after Kruskal–Wallis nonparametric ANOVA.
Neuroprotective effects of AS ODN directed to group I mGluR subtypes
Recent in vitro expression studies (Brabet et al., 1995; Kingston et al., 1995) have shown that although 4CPG is an antagonist to both mGluR1 subtypes, its potency is greater at mGluR1 receptor sites (IC50 = 40–70 μm) than at mGluR5 receptor sites (IC50 = 150–2000 μm). The neuroprotective effect of 4CPG in our trauma model suggests the involvement of mGluR1 without definitively excluding the involvement of mGluR5. To address this point more directly, we examined the effects of pretreatment with AS ODN directed to mGluR1 or mGluR5 in vitro. Controls included cells treated with mismatched (missense) oligodeoxynucleotides (MS ODN) and sense oligodeoxynucleotides (S ODN). As shown in Figure 9, treatment with AS ODN directed to mGluR1 for 5 d significantly reduced post-traumatic neuronal death and associated LDH release (p < 0.01). In contrast, identical AS ODN treatment directed to mGluR5 had no appreciable effect on neuronal survival (Fig. 9). Injury-induced LDH release was nearly identical in cultures treated with MS ODN, S ODN, and untreated control cultures. Pretreatment alone with any of the ODN (in the absence of injury) caused no significant changes (data not shown). To determine the presence of both mGluR1 and mGluR5 subtypes in our cultures, and to demonstrate the efficacy of AS ODN treatment, we measured agonist-induced IP accumulation in uninjured cultures. AS ODN treatment directed to both mGluR1 and mGluR5 produced virtually identical reduction in 1S,3R-ACPD-induced IP accumulation (p < 0.01; Fig.10). Therefore, the lack of neuroprotection with AS ODN treatment directed to mGluR5 did not appear to result from insufficient expression of mGluR5 or from failure of AS ODN to suppress translation of this receptor subtype.

Pretreatment with AS ODN directed to mGluR1, but not AS ODN directed to mGluR5, significantly reduced neuronal death after trauma in vitro. Treatment with mismatched (missense) MS ODN or with sense ODN (S ODN) had no significant effect on neuronal survival. Neuronal/glial cultures were treated with 2 μm appropriate ODN for 5 d. Neuronal death was evaluated as injury-induced LDH release 16–18 hr after injury. Data represent mean ± SEM; n = 30–40 cultures per condition. Multiple comparisons were performed using ANOVA and Student–Newman–Keuls test: **p < 0.01 versus control (untreated injury);††p < 0.01 versus MS ODN treatment;##p < 0.01 versus S ODN treatment.

Pretreatment with AS ODN directed to mGluR1 as well as pretreatment with AS ODN directed to mGluR5 significantly reduced 1S,3R-ACPD-induced PI hydrolysis. Treatment with mismatched (missense) ODN had no significant effect on 1S,3R-ACPD-induced PI hydrolysis. PI hydrolysis was measured as inositol phosphate (IP) accumulation within 30 min after addition of 500 μm1S,3R-ACPD. Data represent mean ± SEM; n = 30–40 cultures per condition. Multiple comparisons were performed using ANOVA and Student–Newman–Keuls test. **p < 0.01 versus control (untreated injury);††p < 0.01 versus MS ODN treatment.
DISCUSSION
Traumatic injuries to the CNS induce biochemical changes that contribute to irreversible tissue damage (Faden, 1993). This delayed autodestructive response, often referred to as “secondary injury,” appears to involve a cascade of events that involves alterations in blood flow and metabolism, membrane damage with lipid hydrolysis, modifications in neurotransmitters/neuromodulators and their receptors, changes in tissue content of monovalent and divalent cations, activation of free radical reactions and inflammatory/immune responses, among others (Faden, 1993; McIntosh, 1993; Panter and Faden, 1994).
TBI causes early (within 10 min) release of glutamate into the extracellular space, which is highly correlated to injury severity (Faden et al., 1989; Katayama et al., 1990; Nilsson et al., 1990;Palmer et al., 1993). Treatment with NMDA antagonists reduces neuronal loss (Toulmond et al., 1993; Hicks et al., 1995) and improves behavioral recovery (Hayes et al., 1988; Faden et al., 1989; Sharpira et al., 1990; McIntosh et al., 1993; Smith et al., 1993). Sun and Faden (1995a) have also shown that central administration of AS ODN directed to the NMDA-R1 subunit is neuroprotective. In addition, glutamate-release inhibitors, such as the sodium channel blockers BW1003C87 and 619C89, improve both behavioral and histological outcome after experimental brain trauma (Okiyama et al., 1995; Sun and Faden, 1995b).
Although the existence of metabotropic glutamate receptors was suggested in 1985 (Sladeczek et al., 1985), only the recent availability of somewhat selective agonists and antagonists has made it possible to address the potential pathophysiological roles for these receptors. Recent experimental studies have also indicated a possible, albeit inconsistent, action for metabotropic glutamate receptors in modulating neuronal death (Koh et al., 1991; Chiamulera et al., 1992;McDonald and Schoepp, 1992; Sacaan and Schoepp, 1992; Siliprandi et al., 1992; Birrell et al., 1993; McDonald et al., 1993; Bruno et al., 1994; Opitz et al., 1994; Gong et al., 1995; Buisson et al., 1996).
In the present studies, we demonstrated that DHPG, a specificagonist for group I mGluR (Schoepp et al., 1994), exacerbated trauma-induced neuronal cell death in tissue culture in a dose-dependent manner. Effects of DHPG were markedly attenuated by MCPG, an antagonist of group I/II mGluR. These results are in agreement with the findings of Buisson and Choi (1995), who showed that the group I receptor agonist 3HPG promoted neuronal death in culture after oxygen–glucose deprivation or after brief NMDA exposure. Like other investigators using in vitro models to examine injury (Koh and Choi, 1987; Regan and Choi, 1994), we used LDH release as a marker of cell death, based on studies showing that LDH increase is highly correlated with the percentage of nonviable neurons, as evidenced by trypan blue staining and other methods (Regan and Choi, 1994) (our unpublished data). Although the mechanism leading to LDH release after injury is not fully understood, it may reflect damage to cell membranes (Koh and Choi, 1987). In our experiments, treatment of cultures with DHPG alone, at doses that exacerbate trauma-induced cell loss (up to 32 mm), did not appear to be associated with neuronal death. These observations suggest that activation of group I mGluR, rather than exerting a direct action, may in fact trigger secondary injury mechanisms. For example, group I mGluR activation enhances the activity of NMDA receptors (Fitzjohn et al., 1996), thereby potentiating post-traumatic excitotoxicity mediated by those receptors (Faden et al., 1989). In this respect, it should be noted that in our in vitro model, treatment with the NMDA receptor antagonist MK-801 reduced cell death by up to 60% (Mukhin and Faden, 1995).
The fact that pharmacological blockade of group I mGluR decreases trauma-induced PI hydrolysis and neuronal loss in vitroindicates that traumatic injury, in addition to activating NMDA receptors in this model, also activates phospholipase C-linked mGluR. Recent studies have demonstrated that fluid-percussion TBI in rats causes PI hydrolysis (Prasad et al., 1994). Another laboratory (Delahunty et al., 1992) studied muscarinic or metabotropic agonist-stimulated PI hydrolysis in rat hippocampus and found a greater response in hippocampi of traumatized animals. These data are consistent with our findings regarding the participation of PLC, particularly mGluR-linked PLC, in TBI.
In our in vitro model, MCPG and 4CPG produced similar neuroprotective actions. These data are compatible with the observation of Opitz et al. (1994) that MCPG is neuroprotective in hippocampal slices subjected to hypoxia/hypoglycemia. Whereas MCPG is an equipotential antagonist of both group I and group II mGluR, 4CPG is a relatively selective group I antagonist at the concentrations used, and only at higher concentrations does it exert agonist activity at group II receptors (Pin and Duvoisin, 1994; Watkins and Collingridge, 1994). Taken together, these data support our hypothesis that the neuroprotective actions of these compounds result from blockade of group I mGluR. A role for group I mGluR in post-traumatic neuronal death is also strongly supported by our findings that AS ODN directed to the mGluR1 receptor subtype has a neuroprotective effect. This conclusion is reinforced further by results of our experiments using the group I receptor agonist DHPG.
Beneficial effects were also observed forR,S-MCPG after in vivo trauma; this compound, given centrally (i.c.v.), improved neurological recovery and survival of hippocampal neurons. These findings agree with recent work by Gong et al. (1995), which showed that pretreatment with MCPG, administered intracerebroventricularly in rats before fluid percussion-induced TBI, improved certain motor and memory tasks. By themselves, these in vivo results do not permit a definitive conclusion about the participation of group I mGluR in post-traumatic neuronal death because of the mixed pharmacological profile of MCPG. However, in vitro studies indicate that activation of group II mGluR is neuroprotective (Bruno et al., 1992), and that this neuroprotective effect is diminished by MCPG (Choi, 1996). Thus, studies using both in vivo and in vitro models appear to implicate group I mGluRs. Additional in vivostudies with more selective mGluR antagonists or with AS ODN are needed to confirm this conclusion. It should be noted that, in separatein vivo studies, we demonstrated protective effects for MCPG, even when administered systemically (i.v.), aftertrauma. These results indicate that group I mGluR antagonists may have therapeutic potential in the treatment of brain trauma.
In our in vitro studies, treatment with AS-ODN directed to mGluR1, but not treatment with AS-ODN directed to mGluR5, improved postinjury neuronal survival in our cultures. As we have shown, both mGluR1 and mGluR5 receptor subtypes can be detected by immunoblotting in our cultures. We have also found that AS ODN directed to either mGluR1 or mGluR5 decreased 1S,3R-ACPD-induced PI hydrolysis to similar levels. These findings suggest that the lack of neuroprotective effect of AS-ODN treatment directed to mGluR5 results neither from insufficient expression of mGluR5 nor from failure of AS ODN to suppress translation of this receptor subtype.
There are several possible explanations for the lack of neuroprotective effect of AS ODN at mGluR5 receptors. mGluR1 and mGluR5 may not be activated to the same degree by injury. For example, mGluR5 may not be the predominant receptor subtype in synapses activated after injury, or mGluR5 may be expressed by cell types, such as glia, which are less susceptible to trauma-induced cell death. Another explanation may be the differences in the signal transduction pathways activated by each receptor subtype. Whereas activation of both receptors causes stimulation of PLC activity, mGluR1 may additionally mediate increases in phospholipase A2 and/or adenylyl cyclase activity (Pin and Duvoisin, 1995). More studies will be necessary to address this issue. Taken together, our in vivo and in vitro studies show that activation of group I mGluR, particularly mGluR1, contributes to the pathophysiology of post-traumatic neuronal death. These observations further suggest that more selective mGluR1 antagonists may prove to have therapeutic utility.
Footnotes
This work was supported in part by grants from the Centers for Disease Control (CCR306634) and National Institutes of Health (NS27849) to A.I.F. We thank Randyll Goodnight for technical assistance, and Linda Powell and Elizabeth Wellner for editorial assistance in the preparation of this manuscript.
Correspondence should be addressed to Dr. Alan I. Faden, Georgetown University Medical Center, NW103 Med. Dent. Bldg., 3900 Reservoir Road NW, Washington, DC 20007-2197.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}