

Abstract
For the mollusc Tritonia diomedea to generate its escape swim motor pattern, interneuron C2, a crucial member of the central pattern generator (CPG) for this rhythmic behavior, must fire repetitive bursts of action potentials. Yet, before swimming, repeated depolarizing current pulses injected into C2 at periods similar those in the swim motor program are incapable of mimicking the firing rate attained by C2 on each cycle of a swim motor program. This resting level of C2 inexcitability is attributable to its own inherent spike frequency adaptation (SFA). Clearly, this property must be altered for the swim behavior to occur. The pathway for initiation of the swimming behavior involves activation of the serotonergic dorsal swim interneurons (DSIs), which are also intrinsic members of the swim CPG. Physiologically appropriate DSI stimulation transiently decreases C2 SFA, allowing C2 to fire at higher rates even when repeatedly depolarized at short intervals. The increased C2 excitability caused by DSI stimulation is mimicked and occluded by serotonin application. Furthermore, the change in excitability is not caused by the depolarization associated with DSI stimulation or serotonin application but is correlated with a decrease in C2 spike afterhyperpolarization. This suggests that the DSIs use serotonin to evoke a neuromodulatory action on a conductance in C2 that regulates its firing rate. This modulatory action of one CPG neuron on another is likely to play a role in configuring the swim circuit into its rhythmic pattern-generating mode and maintaining it in that state.
- intrinsic neuromodulation
- serotonin
- nudibranch mollusc
- repetitive firing
- central pattern generator
- after- hyperpolarization
Extrinsic neuromodulatory inputs to central pattern generators (CPGs) alter the cellular and synaptic properties of CPG neurons, thereby reconfiguring a single circuit to produce multiple outputs (Katz and Harris-Warrick, 1990; Harris-Warrick and Marder, 1991; Katz, 1995b). We have described previously a preparation in which the neuromodulatory elements are themselves intrinsic to the CPG and thus are active whenever the CPG is activated (Katz et al., 1994; Katz and Frost, 1996b). We are interested in determining the function of such intrinsic neuromodulation and comparing it to the roles played by extrinsic neuromodulatory inputs to CPGs. Our results suggest that in this case intrinsic neuromodulation may unlock the oscillatory capability of the CPG circuit by altering key features of other CPG neurons and thereby providing the circuit with self-organizing capabilities.
The system under study is the escape swim network of the nudibranch mollusc, Tritonia diomedea (Getting et al., 1980; Getting and Dekin, 1985b). Tritonia is a bottom-dwelling sea slug that normally locomotes along the substrate via ciliary action of the foot (Audesirk, 1978). However, when contacted by a predatory starfish,Tritonia executes an escape swim response consisting of alternating ventral and dorsal whole-body flexions. The motor network mediating this response has been described as multifunctional; neurons participating in the generation of the swim response are thought also to mediate a nonrhythmic, reflexive withdrawal (Getting and Dekin, 1985a,b). Previous work had focused on the role of classical synaptic actions in mediating reconfiguration of the network from its nonrhythmic mode into a functional CPG (Getting and Dekin, 1985a,b). Our present results, however, suggest that neuromodulatory actions may play a significant role as well.
Sensory input for initiating the swim motor program is funneled through a bilateral pair of dorsal ramp interneurons (DRIs), which relay the excitation directly to one set of neurons intrinsic to the CPG, the dorsal swim interneurons (DSIs) (Frost and Katz, 1996b), causing them to fire intensely at the onset of the swim motor program (Fig.1A–C). The DSIs are serotonin-immunoreactive (Katz et al., 1994; McClellan et al., 1994) and appear to use serotonin both for neurotransmission and for heterosynaptic facilitation of transmitter release from another CPG neuron, C2 (Katz et al., 1994; Katz and Frost, 1995a,b). Interneuron C2 has a particularly prominent role within the CPG in the production of the swim motor program, in that C2 must fire throughout the swim motor program for the behavior to be produced (Getting, 1977; Taghert and Willows, 1978). If C2 is hyperpolarized during an ongoing motor program, then the motor pattern ceases, indicating that C2 activity is necessary for motor pattern generation.

Cerebral neuron 2 (C2) and the dorsal swim interneurons (DSI) produce repetitive bursts of action potentials during a swim motor program.A, The neuronal pathway underlying the generation of the swim motor program. Sensory input excites the dorsal ramp interneuron (DRI), which monosynaptically excites all three dorsal swim interneurons (DSI-A, DSI-B, DSI-C). The DSIs synapse withC2 and the ventral swim interneurons (VSI; VSI-A, VSI-B). Monosynaptic connections are shown as solid lines. Dashed lines indicate that the connection either is polysynaptic or has not been shown to be monosynaptic. Triangles depict excitatory synapses;circles represent inhibitory connections. Multicomponent connections are shown as combinations of circles andtriangles. The arrow from the DSIs represents the neuromodulatory actions of these cells onC2. All cell types are represented bilaterally.B, Simultaneous intracellular recordings fromC2 and a DSI. At thearrowhead, pedal nerve 3 was stimulated (Five 5 V, 2 msec pulses at 10 Hz). This resulted in an immediate excitation of the DSI, followed by an excitation of C2. Both neurons then produced repetitive busts of action po-tentials as part of the swim motor program. C, Instantaneous spike frequency profiles for C2 and the DSI. Inverse interspike interval for the two traces in B are plotted as a function of time. C2 fired four initial spikes at high frequency and then fired repetitive bursts with maximum spike frequencies, achieving 10–15 Hz. At the onset of the swim motor program, the DSI fired rapidly, approaching 40 Hz. TheDSI then fired repetitively at frequencies that declined to just under 10 Hz by the sixth cycle. D, Average instantaneous spike frequency exhibited by C2 in six cycle nerve-evoked swim motor programs (n = 4 preparations). Each point represents the mean and SE of the average instantaneous spike frequency during each cycle of the swim motor program.
Here, we report that, before swimming, C2 exhibits spike frequency adaptation (SFA) that prevents it from firing at the rates it attains during a swim. If not altered, this lack of excitability presumably would preclude the production of the swim behavior. It was noted previously that, after a swim, C2 excitability is much higher than before one (Frost et al., 1988). It also has been observed that the DSIs remain tonically active at higher rates after a swim as well (Katz et al., 1994), leading us to suspect that the DSIs might cause the observed enhancement of C2 excitability directly. We now find that the DSIs reduce spike frequency adaptation in C2, thereby enhancing its excitability. This neuromodulatory action of a neuron intrinsic to the CPG thus may enable the generation of the swim motor program.
Portions of this work have been reported in abstract form (Katz and Frost, 1994, 1996a).
MATERIALS AND METHODS
Tritonia diomedea were obtained from the intracoastal waters of the state of Washington and British Columbia. All experiments were performed on the isolated CNS, consisting of the left and right pedal ganglia and the fused cerebral and pleural ganglia, along with associated nerves. The ganglia were removed from the animal as previously described (Willows et al., 1973; Katz and Frost, 1995a,b) and immediately placed in a 1 ml Sylgard-lined chamber superfused with saline cooled to 2°C. Normal saline composition was (in mm): 420 NaCl, 10 KCl, 10 CaCl2, 50 MgCl2, 11 d-glucose, and 10 HEPES, pH 7.6. The ganglia were pinned dorsal side up to the base of the chamber, and the connective tissue sheath was removed from the surface of the ganglia, exposing the cell bodies. Suction electrodes were placed on pedal nerves 3 and 6. Then the preparation was warmed to 10°C and left to rest for at least 3 hr and in most cases overnight. All measurements of excitability were taken from preparations that had not produced a swim motor program for at least 3 hr. This is defined as the rested, nonswimming condition. The swim motor program was evoked by stimulating pedal nerve 3 with 2 msec pulses (5–10 V at 10 Hz for 0.5–1 sec).
Neurons were impaled with glass microelectrodes filled with 4m K-acetate and inserted in chucks containing 3m KCl and having resistances of 5–20 MΩ. For most experiments in which C2 membrane potential was varied, C2 was impaled with two microelectrodes, one for current injection and the other for voltage recording. Alternatively, in some experiments single-electrode discontinuous (switching) current clamp was used for an accurate measure of C2 membrane potential. For other experiments and for DSI stimulation, current was injected through a single recording electrode, using a balanced bridge circuit.
Neurons were identified by soma location and coloration, synaptic connectivity, and activity pattern at rest and during the swim motor program (Taghert and Willows, 1978; Getting et al., 1980; Getting, 1981; Katz and Frost, 1995a,b). There are three DSIs (DSI-A, DSI-B, and DSI-C) on each side of the animal’s fused central ganglia. DSI-B and DSI-C are equivalent neurons but can be distinguished from DSI-A on the basis of synaptic interactions and activity pattern. No differences were observed in the neuromodulatory effects of any of the DSIs.
The DSIs were stimulated by trains of 10 or 20 msec current pulses that each elicited a single action potential. In this way we were able to control their spike frequency precisely. Serotonin (creatinine sulfate, Sigma, St. Louis, MO) was applied by switching perfusion lines, allowing a known concentration to replace the bath saline. Perfusion rate was ∼1 ml/min.
For experiments examining the effect of DSI stimulation and serotonin on the C2 afterhyperpolarization, the bathing medium was switched to high divalent cation saline, the composition of which was (in mm): 285 NaCl, 10 KCl, 25 CaCl2, 125 MgCl2, 11 d-glucose, and 10 HEPES, pH 7.6. In high divalent cation saline the threshold for eliciting C2 spikes increased to the point that C2 would not fire repetitively to constant current pulses. To observe the C2 afterhyperpolarization (AHP), we used trains of relatively short depolarizing current pulses (20 msec, >4 nA) in which each pulse evoked a single action potential.
Data were recorded onto videotape and digitized directly at 1000–1250 Hz with hardware and software from Cambridge Electronic Design (CED, Cambridge, UK). Data acquisition and analysis were performed with Spike2 software (CED) and SigmaPlot (Jandel Scientific, San Rafael, CA). Traces were smoothed by using a local averaging routine. Spikes were excluded from the local averaging.
RESULTS
Under rested conditions, spike frequency adaptation in C2 prevents it from mimicking its firing pattern during a swim
During a swim motor program C2 exhibits robust spiking behavior (Fig. 1B, top trace). It fires repetitive bursts of action potentials with a period between 6 and 10 sec. The peak firing frequency for each burst was above 10 Hz (Fig. 1C). For nerve-elicited swim motor programs consisting of six dorsal/ventral cycles, the average instantaneous spike frequency (inverse interspike interval) exhibited by C2 during the first cycle was >10 Hz and remained above 7 Hz for all six cycles (Fig. 1D).
In contrast to its firing during a swim motor program, C2 was relatively inexcitable before a swim under rested conditions (see Materials and Methods). C2 exhibited SFA such that, when repeatedly depolarized at intervals similar to the normal burst period of a swim motor program, C2 was unable to maintain an elevated spike frequency (Fig. 2A); rather, its responses to successive depolarizing current pulses became progressively weaker (Fig. 2C, open circles). Under control conditions the mean instantaneous spike frequency resulting from the first 3 sec, 2 nA current pulse was 4.7 ± 0.6 Hz (mean ± SE). This fell to 1.6 ± 0.4 Hz by the sixth pulse (n = 6).

DSI stimulation acts to remove C2 spike frequency adaptation. Under rested conditions, C2 exhibited an accumulation of spike frequency adaptation in response to sequential depolarizations. DSI stimulation allowed C2 to fire repeated bursts of action potentials at a maintained spike frequency. A, C2 was repetitively depolarized six times with 2 nA, 3 sec current pulses with 3 sec between pulses. The top trace is a plot of C2 instantaneous spike frequency. C2 fired rapidly at first and then adapted to a lower frequency of firing. It fired progressively less to each subsequent depolarization. A postburst hyperpolarization accumulated after each current pulse. The dotted line(in A and B) corresponds to the resting membrane potential before stimulation (−62 mV). B, DSI stimulation (5 Hz) enhanced C2 firing. Although C2 spike frequency still adapted within the first pulse, C2 now fired at a higher frequency to subsequent depolarizing current pulses. The postburst hyperpolarization was much smaller than control. DSI stimulation itself did not cause a substantial depolarization of C2. C, Average results from six preparations. The average C2 instantaneous spike frequency is plotted as a function of stimulus number when C2 was stimulated alone (Control, open circles) and during DSI stimulation at 5 Hz (filled circles). The data were analyzed by repeated measures ANOVA, with DSI stimulation and stimulus number as within-subject factors. There was a significant effect of DSI stimulation (F(1,5) = 35.30; p < 0.01) and stimulus number (F(5,25) = 14.82;p < 0.01), as well as a significant interaction between DSI stimulation and stimulus number (F(5,25) = 22.19; p < 0.001). DSI stimulation caused a significant change (p < 0.05; Tukey post hoctest) in the average instantaneous firing rate of C2 for all stimuli except the first two, as designated by the asterisks. In all preparations included here, C2 was stimulated as shown inA, and there was a 2–4 min interval between stimulus sets.
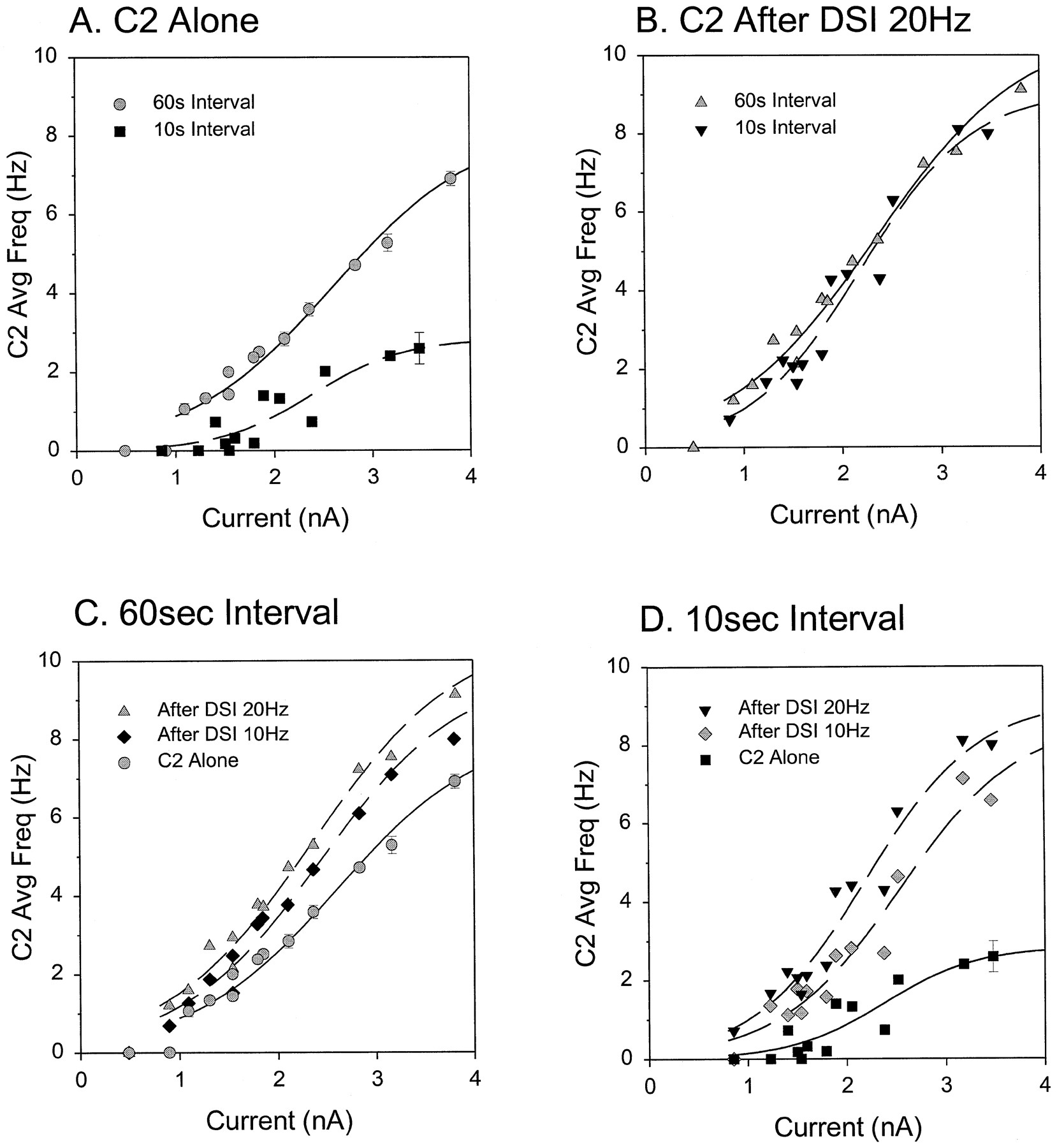
The excitability of C2 was critically dependent on how often it was stimulated. At long intervals (60 sec), C2 showed little or no decline in firing over repeated pulses (Fig.3A1). The average instantaneous spike frequency of C2 in response to a 2 sec, 2 nA current pulse was 7.2 ± 0.3 Hz (Fig. 3C), but when the interval was shortened to 10 sec, the cumulative spike frequency adaptation caused C2 to fire fewer spikes at a lower frequency in response to the same amount of current (Fig. 3B1). The average instantaneous spike frequency dropped to 3.9 ± 0.3 Hz (Fig. 3Ci), a significant change (p < 0.05; see figure legend for explanation of statistics). Figure 3D (circles) shows the effect of a wide range of intervals on C2 average firing frequency. Equivalent results were obtained in five preparations in which we varied the C2 test interval over a similar range. During a swim motor program the burst period varied from 6 to 10 sec, so the shorter intervals shown here are what is physiologically relevant for this behavior.

DSI stimulation reduced or eliminated accumulated spike frequency adaptation in C2.A, C2 was stimulated repeatedly with 2 nA, 2 sec current pulses every 60 sec. In the absence of DSI stimulation (A1, Control), C2 displayed a consistent level of excitability. The spiking response of C2 was enhanced when a DSI was stimulated at 20 Hz (A2, bottom trace) for 3 sec, ending 2 sec before the onset of a depolarizing current pulse. The resting membrane potential for C2 was −52 mV in A1 and −55 mV in A2.B, C2 was stimulated repeatedly with 2 nA, 2 sec current pulses every 10 sec. When stimulated alone (B1,Control), the C2 spiking response was significantly lower than the response to the same current pulses at a 60 sec interval (A1) because of accumulated spike frequency adaptation. At this lower level of C2 excitability, DSI stimulation at 20 Hz (B2) had a proportionally greater effect than it did when C2 was stimulated every 60 sec (A2). The resting membrane potential for C2 was −59 mV in B1 and −58 mV in B2.C, The average spike frequency response of C2 to 2 nA of current at 60 or 10 sec intervals when C2 was depolarized alone or after DSI stimulation (n = 24). A repeated measures ANOVA was performed, with DSI stimulation and interval as within-subject factors. There was a significant effect of interval (F(1,23) = 79.98; p < 0.001) and DSI stimulation (F(1,23) = 92.50;p < 0.001) and a significant interaction between the two factors (F(1,23) = 70.89;p < 0.001). Tukey post hoc tests indicated a significant difference (p < 0.05) between the average spike frequencies of the groups indicated (i, ii, and iii). D, For the preparation shown in A and B, the excitability of C2 was tested for a variety of intervals (8–120 sec). C2 was tested alone (circles) or after a 3 sec DSI stimulation at 5 Hz (upward triangles), 10 Hz (downward triangles), or 20 Hz (squares). The average spike frequency response of C2 varied with the test interval when C2 was tested alone, but it was invariant with respect to interval when C2 was depolarized after a DSI stimulus at 10 or 20 Hz. The effectiveness of DSI stimulation was dependent on the frequency at which it was made to spike.
The lack of C2 excitability at short intervals could not be overcome by injecting more current into C2. This can be seen by examining the relationship between injected current and spiking responses for long (60 sec) and short (10 sec) intervals (Fig.4A). When C2 was stimulated every 60 sec with depolarizing current pulses, its spike frequency increased sharply with the amount of current injected (Fig.4A, circles). However, when C2 was stimulated every 10 sec, its spike frequency was much lower for all levels of current injected (Fig. 4A, squares). Thus, at 10 sec the firing response did not exceed 3 Hz regardless of the amount of current injected.

DSI stimulation removed the effect of spike frequency adaptation from the current/frequency (I/F) relationship of C2. A, Inherent spike frequency adaptation decreased the slope of theI/F plot when C2 was stimulated at short intervals. C2 excitability was measured by stimulating C2 with 2 sec constant current pulses of varying amplitude every 60 sec (circles) or 10 sec (squares). The average instantaneous spike frequency of C2 during the current pulse is plotted as a function of the amplitude of the injected current. C2 spike frequency exhibited a sigmoidal relationship with current. There was a threshold for repetitive spiking at ∼1 nA. For the maximum current tested, C2 firing frequency decreased from ∼7 Hz at 60 sec intervals to less than 3 Hz when stimulated every 10 sec. B, When a DSI stimulus at 20 Hz preceded the C2 depolarization, there was no difference in the I/F plots for the two intervals tested (60 sec, upward triangles or 10 sec, downward triangles). C, At the 60 sec interval, the slope of the I/F plot increased progressively when a DSI was stimulated at 10 Hz (diamonds) or 20 Hz (triangles), as compared with when C2 was stimulated alone (circles). D, At the 10 sec test interval, C2 was not very excitable when stimulated alone (squares), but DSI stimulation at 10 Hz (diamonds) or 20 Hz (triangles) caused the I/F plots to resemble those measured at the 60 sec test interval. For all DSI trials a single DSI was stimulated for 3 sec at the designated spike frequency. The DSI stimulus train ended 2 sec before the onset of the C2 depolarizing current pulse. Control measurements of C2 spike frequency (C2 Alone) included only the steady-state firing response for that current, excluding those that occurred within 40 sec after either a DSI stimulus train or a change in current level. Variable numbers of C2 Alonetrials were averaged. In most cases the error bars were smaller than the symbol size. Data shown here are from many repetitions in a single preparation.
These results demonstrate that, before a swim, the intrinsic properties of C2 preclude it from firing repetitive, high-frequency bursts of action potentials to constant current pulses regardless of the amount of depolarizing current injected. This suggests that, for C2 spike frequency to remain elevated above 7 Hz as it does during a swim (Fig.1D), there must be a change in its firing properties, allowing it to fire in a repetitive manner without exhibiting cumulative SFA. Because maintained C2 firing is necessary for the production of the swim motor program, we hypothesized that the removal of SFA may be a necessary step for the reconfiguration of the resting motor network into a functional CPG. If so, then the mechanism for the removal of C2 SFA ought to be part of the pathway for initiating the motor pattern. We know that sensory input for initiating the swim motor program is funneled to the CPG through DRI (Frost and Katz, 1996b). DRI excites the serotonergic DSIs, which evoke a neuromodulatory effect on C2, namely an enhancement of transmitter release from C2 (Katz et al., 1994; Katz and Frost, 1995a,b). Here, we tested whether the DSIs also were responsible for enhancing C2 excitability.
Physiologically relevant DSI stimulation removes the effect of accumulated spike frequency adaptation
Although when repeatedly depolarized alone, C2 was unable to maintain an elevated spike rate as it does during a swim motor program (Fig. 2A), concurrent stimulation of a single DSI at 5 Hz changed the response of C2, allowing C2 to mimic more closely its firing pattern during a swim (Fig. 2B). There was no significant change in C2 firing during the first two bursts of the series; however, for all subsequent C2 depolarizations DSI stimulation caused a significant increase in the average instantaneous C2 spike frequency (n = 6; p < 0.05) (Fig.2C). Thus DSI stimulation decreased the expression of spike frequency adaptation in C2.
We sought to test the effectiveness of DSI stimulation at removing C2 SFA when the DSIs were stimulated at higher rates that more closely approximated their firing rate during a swim motor program. During the first 1 sec of a swim motor program, the mean firing rate of the DSIs was 22.3 ± 2.8 Hz, with a maximum rate during that time of 30.4 ± 4.1 Hz (n = 11). They maintained a firing rate above 10 Hz for the duration of the motor program. Stimulation of a single DSI for just 3 sec at 20 Hz resulted in a synaptic depolarization of C2 that occasionally caused C2 to fire (Fig.3A2, middle trace). Such a synaptically evoked depolarization would interfere with excitability measurements. Thus, to avoid the confounding effects of this synaptic depolarization, we tested C2 excitability by beginning our current pulse 2 sec after the end of DSI stimulation (Fig. 3A2).
Stimulating a single DSI at the same rate seen at the onset of the swim motor program (20 Hz) increased C2 excitability but had its largest effect when C2 SFA was already maximal (short intervals). DSI stimulation increased the spike frequency response of C2 to a 2 nA current pulse when C2 was stimulated every 60 sec by an average of 17 ± 4% (p < 0.05; Fig.3A2,Cii). However, DSI stimulation had a much stronger effect when C2 was depolarized every 10 sec (Fig. 3B2). When the accumulated effect of SFA was greatest, DSI stimulation increased C2 firing an average of 104 ± 13% (p < 0.05; Fig. 3Ciii). In five preparations we examined the effect of DSI stimulation on C2 excitability when C2 was tested over a wide range of intervals. The results of one such preparation show that DSI stimulation at 20 Hz caused C2 to fire equally well regardless of how often C2 was stimulated previously (Fig. 3D, squares). Similar results were obtained for all five preparations.
The extent to which DSI stimulation removed C2 SFA was a function of the frequency at which the DSI was fired. DSI stimulation at 5 Hz (three preparations tested) enhanced C2 excitability at all test intervals but did not remove the dependence of C2 firing on its test interval (Fig. 3D, upward triangles). In contrast, DSI stimulation at 10 Hz (four preparations tested) allowed C2 to fire equally well at all intervals (Fig. 3D, downward triangles) but at a lower rate than that produced by DSI stimulation at 20 Hz (Fig. 3D, squares).
DSI stimulation removes spike frequency adaptation for all levels of injected current
Instead of testing C2 with a single level of current, we next tested whether DSI removed C2 spike frequency adaptation for a range of injected currents. As shown above, the current/frequency (I/F) relationship for C2 was strongly dependent on the test interval when C2 was stimulated alone (Fig.4A). DSI stimulation removed the effect of the test interval so that C2 spiking was dependent only on how much current was injected; after DSI stimulation at 20 Hz the I/Frelationship for C2 was the same for both test intervals (Fig.4B).
DSI stimulation increased the excitability of C2 at both the longer and the shorter intervals. At the 60 sec interval DSI stimulation at 10 Hz (diamonds) or 20 Hz (triangles) increased the slope of the I/F relationship, causing C2 to fire more spikes at a higher frequency for all levels of injected current (Fig.4C). Once again, the stronger DSI stimulation produced a larger effect. The increase in C2 firing caused by DSI stimulation was consistent in each of the nine preparations examined.
As was seen with just the 2 nA current pulses, DSI stimulation had a larger effect on C2 when C2 was stimulated at the shorter interval (10 sec) because of the removal of more accumulated SFA (Fig.4D). Although C2 fired at lower rates for all currents when tested alone every 10 sec, DSI stimulation at 10 or 20 Hz before C2 depolarization increased C2 firing to almost the same levels as when C2 was stimulated every 60 sec (compare Fig. 4C andD). Similar results were obtained in all three preparations tested. Thus, DSI stimulation removed the effect of cumulative spike frequency adaptation in C2 for the entire range of current levels tested. This caused C2 to maintain the same I/F relationship regardless of how often it was stimulated.
The time course of the DSI modulatory action is appropriate for playing a role in generation of the swim motor program
For the DSI-induced enhancement of C2 excitability to be important for the initiation of the swim motor program, it must begin within a few seconds of the DSI firing onset. The DSIs began firing maximally almost immediately after a swim-initiating nerve stimulus (Fig.1B). The delay until the first C2 spike in the first burst of a swim motor program was 2.0 ± 0.6 sec, and the delay until peak C2 firing for that burst was 4.0 ± 0.8 sec (n = 11) (Fig. 1B). The onset of the modulatory effect of DSI stimulation was difficult to ascertain because of the confounding effects of the DSI-evoked synaptic depolarization in C2. However, it is clear from Figure 3, A2 andB2, that there was a large effect within 5 sec after the start of DSI stimulation. Thus, the modulation certainly would be effective at approximately the time C2 fires maximally during its first burst of a swim motor program.
The enhancement of C2 excitability caused by DSI stimulation at 20 Hz lasted longer than the period of a single swim cycle (6–10 sec). For example, when C2 was stimulated every 10 sec, the enhancement caused by a single DSI stimulus at 20 Hz was apparent even 12 sec after the end of the DSI train (Fig. 3B2). Thus, the modulatory effect of the DSIs likely would carry over from cycle to cycle during the swim motor program and possibly accumulate over the course of a swim motor program.
Serotonin mimics and occludes the enhancement of C2 excitability by DSI
The DSIs are serotonin-immunoreactive (Katz et al., 1994;McClellan et al., 1994) and serotonin has been implicated as the transmitter responsible for the other DSI-evoked effects (Katz and Frost, 1995b); exogenous serotonin mimics both of the DSI synaptic actions and the DSI-induced enhancement of C2-evoked synaptic potentials and serotonin antagonists block these effects (Katz et al., 1994; Katz and Frost, 1995b). We wanted to know whether serotonin also mimicked and occluded the enhancement of C2 excitability caused by DSI stimulation.
High concentrations of serotonin (100 μm) increased C2 excitability and prevented DSI stimulation from increasing C2 excitability any further. Serotonin application increased C2 excitability at the 60 sec interval (p < 0.05; Fig. 5A,B,Ei). Although C2 excitability still decreased when C2 was depolarized every 10 sec in serotonin, it was significantly higher than control at this test interval (p < 0.05; Fig.5C,D,Eii). The effectiveness of serotonin at increasing C2 excitability wore off over the course of 2 hr of continuous exposure, presumably because of receptor desensitization.

Serotonin mimics and occludes the enhancement of C2 excitability by DSI. A, The response of C2 to a 2 nA, 2 sec current pulse when applied every 60 sec. The resting membrane potential before the depolarizing test was −46 mV. B, The response of the same neuron to the same current pulse in the presence of 100 μm serotonin. Serotonin caused an increase in the number and frequency of spikes produced. The neuron was impaled with two electrodes. Current was injected through the second electrode to counteract the depolarizing action of serotonin. The resting membrane potential before the depolarizing test was −54 mV.C, As was shown previously, depolarizing C2 every 10 sec with a 2 nA, 2 sec current pulse substantially decreased its spiking response (leftmost stimulus). DSI stimulation (20 Hz, 3 sec) briefly depolarized C2 and then increased its spiking response for a number of seconds (right two stimuli).D, In the presence of 100 μm serotonin, the firing response of C2 when it was depolarized every 10 sec was elevated (compare leftmost stimulus with its counterpart in C), mimicking the effect of DSI stimulation (middle, C). In addition, the effect of DSI stimulation was occluded; DSI stimulation did not cause a depolarization of C2 and did not increase C2 firing further.E, Average responses of 10 C2 neurons from six preparations. In normal saline, when stimulated every 60 sec, C2 fired at an average spike frequency of 7.9 ± 0.9 Hz when stimulated alone and 8.5 ± 1.0 Hz when stimulated after a DSI. This decreased to 4.2 ± 0.7 when C2 was stimulated alone every 10 sec. DSI stimulation at this interval increased C2 spiking to 7.0 ± 0.6 Hz. In the presence of 100 μm serotonin, C2 firing response at the 60 sec interval increased to 9.2 ± 0.8 Hz when stimulated alone. There was no change when C2 was stimulated after a DSI at this interval (9.2 ± 0.9 Hz). When stimulated every 10 sec, there was less of a decrease than in control saline (C2 fired at 6.4 ± 0.5 Hz). In the continued presence of serotonin, DSI stimulation did not cause a substantial increase in C2 firing (7.2 ± 0.6 Hz). Data were analyzed with repeated measures ANOVA, with DSI stimulation, interval, and serotonin as within-subject factors. Although the ANOVA for the overall effect of serotonin was not significant (F(1,9) = 3.19;p = 0.11), there was a significant effect of interval (F(1,9) = 56.33;p < 0.001) and DSI stimulation (F(1,9) = 35.19; p < 0.001) and a significant interaction among DSI stimulation, interval, and serotonin (F(1,9) = 8.74;p < 0.05). Tukey post hoc tests indicated that serotonin caused a significant change in the average spike frequency of C2 at either the 60 or 10 sec interval when C2 was stimulated alone (i, ii). In contrast, in the presence of serotonin there was no significant difference between the spike frequency of C2 when stimulated alone or after a DSI at either interval. This indicates that serotonin effectively occluded the actions of DSI stimulation.
In the presence of serotonin, DSI stimulation before C2 stimulation did not cause any further enhancement even at a 10 sec C2 test interval (Fig. 5D,E). In the presence of serotonin the DSI-evoked synaptic potentials in C2 also were occluded (compare Fig.5C and D). The occlusion of DSI-evoked actions did not change over the course of 2 hr of continuous exposure to serotonin although the increased excitability caused by serotonin decreased during this time. Thus, prolonged serotonin application appeared to have desensitized the DSI-evoked effects.
The enhancement of excitability by DSI and 5HT is not caused by a simple depolarization
We were interested in determining whether the enhancement of C2 excitability resulted from a neuromodulatory action of the DSIs or whether it was simply a consequence of the conventional excitatory synaptic potentials that these cells evoke in C2 (Getting, 1981), depolarizing C2 toward threshold. Although measurements in the soma of C2 showed that low-frequency DSI stimulation increased C2 excitability without causing much depolarization of the membrane potential (Fig.2B) and although previous work suggested that synaptic inputs were transmitted faithfully to the soma (Getting, 1983a), it still might have been the case that local depolarization in the neuropil resulting from DSI-evoked EPSPs caused the apparent increase in C2 excitability. To test this, we looked at the relative effects of membrane potential changes and DSI stimulation on C2 excitability.
We tested the effect of C2 membrane potential on the steady-state level of SFA at short intervals (Fig. 6). When C2 was stimulated alone every 10 sec with a 2 sec, 2 nA current pulse, it reached a steady-state level of adaptation and fired only approximately five spikes per pulse (Fig. 6A). A 20 Hz, 3 sec DSI stimulus, ending 4 sec before C2 depolarization, greatly enhanced C2 excitability (Fig. 6B). Associated with the increase in excitability was a small synaptically evoked depolarization. We next injected hyperpolarizing current into C2 to counteract this DSI-evoked depolarization (Fig. 6C,D). C2 now fired only three spikes when stimulated alone (Fig. 6C). With C2 hyperpolarized, DSI stimulation still increased C2 excitability (Fig. 6D). Although the effect of DSI stimulation was reduced by hyperpolarizing C2, the resulting spiking response of C2 was nonetheless higher than that of the control at resting potential (compare Fig. 6A and D). Depolarizing C2 above the level caused by DSI stimulation did not increase C2 excitability as much as DSI stimulation did (compare Fig.6B and E).

The enhancement of C2 excitability is not related to simple depolarization. When C2 was stimulated at a 10 sec interval, DSI stimulation was more effective than subthreshold depolarization at decreasing the effect of steady-state SFA. C2 was stimulated with a 2 nA, 2 sec current pulse every 10 sec. A DSI was stimulated at 20 Hz for 3 sec, ending 4 sec before the C2 current pulse. A, At resting membrane potential (Vmem = −31 mV), C2 displayed a low level of excitability. B, Previous DSI stimulation greatly enhanced the spiking response of C2. The DSI stimulus evoked rapid EPSPs in C2 and caused a prolonged depolarization of C2 membrane potential. C, C2 was hyperpolarized through constant current injection (Vmem = −35 mV). This decreased the basal response of C2 to a 2 nA current pulse. D, After DSI stimulation the membrane potential of C2 at the onset of depolarization was less than control (dotted line), yet the C2 firing response was still higher than when C2 was stimulated alone from its resting potential (A). E, C2 was depolarized by ∼7 mV, using constant current (Vmem = −24 mV). Despite the large depolarization, its spiking response to an additional 2 nA of current was still less than when C2 was tested after DSI stimulation at rest (B). F, Plot of average C2 instantaneous spike frequency versus C2Vmem when C2 was stimulated alone (open circles) and after DSI stimulation (filled circles) for this example.Vmem was measured immediately before the depolarizing test pulse. Lines through data are least-squares best fits. Tonic depolarization of C2 increased C2 excitability, but DSI stimulation had an even greater effect, causing an increased slope of the linear regression. Thus, simple depolarization of C2 was unable to mimic the large increase in excitability caused by DSI stimulation. In this example single-electrode discontinuous current clamp was used to measure membrane potential while current was injected. Similar results were obtained in five other preparations, using separate electrodes for current injection and voltage recording.
It took large imposed changes in the resting membrane potential of C2 to cause small changes in its average spike frequency response to a current pulse (Fig. 6F, open circles). DSI stimulation increased the slope of the relationship between imposed C2 membrane potential and average frequency in response to a 2 nA, 2 sec depolarizing current pulse (Fig. 6F, filled circles). If the DSI modulatory effect were attributable to a depolarization of C2, then the two lines would be parallel. In each of six preparations in which it was tested, the maximal C2 spike frequency associated with DSI stimulation was greater than that produced by depolarization to any level in the absence of DSI stimulation. From these tests we conclude that, although membrane depolarization contributes to increasing C2 excitability, the effects of DSI stimulation on C2 excitability do not appear to be attributable solely to simple synaptically evoked depolarization; therefore, neuromodulation plays a role.
Although serotonin application also caused a depolarization of C2, the voltage change was not responsible for the concurrent enhancement of C2 excitability. In each case the rise in C2 excitability did not occur at the same time as the depolarization of C2 membrane potential but was separated temporally by as much as 8 min. Furthermore, in four preparations we varied C2 membrane potential by injecting current either through the use of discontinuous current clamp or through a second electrode. Counteracting the serotonin-induced C2 depolarizations did not prevent serotonin from increasing C2 excitability (Fig. 5A,B).
DSI stimulation decreases the C2 spike afterhyperpolarization
When C2 was depolarized repeatedly, its resting membrane potential gradually hyperpolarized (Fig. 2A). This change in membrane potential is indicative of a change in membrane conductance that may underlie the spike frequency adaptation. We observed that, when stimulated concurrently with a DSI, C2 exhibited markedly less postburst hyperpolarization than when stimulated alone (Fig.2B).
To examine directly the effect that DSI stimulation has on the C2 AHP, we stimulated C2 with a defined number of relatively brief (20 msec) depolarizations that each evoked a single spike. In this way we could control the number of spikes in the C2 train. Thus, if DSI stimulation changed the amplitude of the C2 AHP, it would not be attributable to an alteration in the number of spikes fired by C2. To evaluate further whether the DSI stimulation was having a direct effect on the C2 AHP, we conducted these experiments in high divalent cation saline. High divalent cation saline raises spike thresholds (Getting, 1981), thereby decreasing the likelihood that the DSI-evoked modulatory action is attributable to recruitment of another modulatory neuron (see Materials and Methods). In 15 of 18 preparations, DSI stimulation (5 Hz, 10 sec) reduced the AHP when C2 was stimulated with four pulses at 20 Hz (Fig.7A). In the remaining three preparations there was no observable change.

DSI stimulation and serotonin application both decrease the C2 afterhyperpolarization. A, In the presence of high divalent cation saline, C2 was made to fire four action potentials (clipped on top trace) at 20 Hz by injecting four 20-msec-duration current pulses (bottom trace). Higher amplification of the top traceshows that DSI stimulation decreased the afterhyperpolarization that followed the C2 spikes (middle trace). B, Application of 100 μm serotonin (5HT) also decreased the amplitude of the afterhyperpolarization and caused a slight afterdepolarization. All traces are averages of eight trials. Traces were aligned vertically to aid in seeing the differences in afterhyperpolarizations. The resting membrane potentials of C2 after DSI stimulation and in the presence of 5HT were depolarized slightly with respect to controls. However, depolarization of C2 via current injection caused an increase rather than a decrease in the size of the afterhyperpolarization (n = 5; data not shown), indicating that the decrease in AHP produced here was not attributable to the depolarization of C2 by DSI.
Serotonin application (100 μm) also caused a decrease in the AHP after C2 spikes in all five preparations that were tested (Fig.7B). In fact, serotonin often converted the late component of the AHP into an afterdepolarization. The decrease in AHP was not attributable to a simple depolarization of C2 by serotonin, because depolarization of C2 through current injection led to an increase, not a decrease, in the amplitude of the AHP, consistent with the AHP being attributable to an increased potassium conductance (n = 5; data not shown).
DISCUSSION
C2 must fire repetitively at a high frequency for the swim motor program to be generated (Getting, 1977; Taghert and Willows, 1978). Our results show that, before the generation of a swim motor program, interneuron C2 exhibits SFA, rendering it incapable of firing in such a manner. The six DSIs fire strongly at the onset of a swim motor program. Activation of just a single DSI enhances the excitability of interneuron C2 by decreasing SFA and thereby allowing C2 to fire more strongly to repetitive depolarizations. Thus, DSI-evoked reduction of SFA in C2 may be a necessary step in the generation of the swim motor program.
Potential mechanisms underlying modulation of spike frequency adaptation
Our evidence suggests that the DSI-evoked decrease in C2 SFA is attributable to a change in an active conductance in C2 rather than through synaptic summation or a change in passive integrative properties. Although the DSIs monosynaptically excite C2 via conventional synaptic connections (Getting, 1981), the enhancement of C2 excitability cannot be attributed completely to a simple depolarization of C2; direct subthreshold depolarization of C2 did not enhance C2 excitability as much as DSI stimulation did, nor is the enhanced excitability associated with a measurable decrease in the resting conductance of C2; previous work showed that DSI stimulation does not increase C2 resting input resistance as measured by hyperpolarizing current pulses in the soma (Katz and Frost, 1995a). Furthermore, DSI stimulation does not alter the degree of electrical coupling between the left and right C2s, indicating that, even in the neuropil regions of the cell, resting input resistance is not changed (Katz and Frost, 1995a).
There are numerous examples of enhanced cellular excitability caused by a decrease in the spike afterhyperpolarization (Fung and Barnes, 1987;McCormick et al., 1991; Shepard and Bunney, 1991; Womble and Moises, 1993; Cox et al., 1994; Pineda et al., 1995; Gorelova and Reiner, 1996). Furthermore, calcium-activated potassium conductances have been shown to underlie afterhyperpolarizations in a number of other systems, including sympathetic neurons in mammals and amphibians (McAfee and Yarowsky, 1979; MacDermott and Weight, 1982), lamprey spinal neurons (El Manira et al., 1994), vagal motor neurons (Sah, 1995), and hippocampal interneurons (Zhang and McBain, 1995). DSI stimulation decreased the amplitude of the C2 spike afterhyperpolarization. This effect was mimicked by serotonin. Serotonin has been shown to reduce afterhyperpolarizations by decreasing calcium-activated or voltage-dependent potassium conductances in many systems, including lamprey spinal neurons (Wallén et al., 1989), Aplysiasensory neurons (Baxter and Byrne, 1990; Sugita et al., 1994), and rat neostriatal neurons (Stefani et al., 1990), motor neurons (Berger et al., 1992; Bayliss et al., 1995), Purkinje cells (Wang et al., 1992), and hippocampal neurons (Pedarzani and Storm, 1993). Our data are consistent with the hypothesis that the DSIs enhance C2 excitability by releasing serotonin, which decreases a calcium-activated potassium conductance. However, other mechanisms are also feasible (Jakobsson, 1978; Partridge, 1980; Gean and Shinnick-Gallagher, 1989; Catarsi et al., 1995), and more experiments are needed to determine which are in use here.
The effects of serotonin and DSI stimulation on C2 inTritonia are remarkably similar to the effects of serotonin and serotonergic neurons on sensory neurons in Aplysia. In each system, serotonin application or stimulation of serotonergic neurons causes both an increase in excitability and a presynaptic enhancement of transmitter release (Kandel and Schwartz, 1982; Walters et al., 1983; Klein et al., 1986; Mackey et al., 1989). It will be interesting to compare the physiological mechanisms underlying the neuromodulatory actions in Tritonia with the well studied mechanisms in Aplysia.
Is the neuromodulatory effect direct?
It is difficult to determine whether a neuromodulatory action is “monosynaptic” in nature because many of the traditional tests of monosynapticity (Berry and Pentreath, 1976; Getting, 1981) do not apply to the slower neuromodulatory actions, and many neuromodulatory effects are attributable to the release of neurotransmitter from nonsynaptic sites (Vizi, 1984; Golding, 1994; Agnati et al., 1995). Still, there are some indications that the DSIs directly enhance C2 excitability. (1) The effect was observed with low levels of DSI stimulation (such as 5 Hz for 3 sec) that would be unlikely to recruit a second modulatory neuron in a reliable manner. (2) The strength of the DSI modulatory action increased monotonically with the strength of DSI stimulation. (3) The decrease in C2 AHP was observed in high divalent cation saline, which decreases the recruitment of polysynaptic pathways by raising the threshold for spiking (Getting, 1981). (4) The DSIs are serotonin-immunoreactive, and serotonin mimics the effect of the DSIs in both normal saline and high divalent cation saline.
State dependence of cellular properties
SFA is common to many other systems (Gean and Shinnick-Gallagher, 1989; Sawczuk et al., 1995), including other pattern-generating networks (El Manira et al., 1994). Hypotheses of its function in motor systems sometimes have assumed that the amount of SFA observed in the quiescent state is reflective of the level to be found in the active state while the motor pattern is being generated (Jodkowski et al., 1988; Sawczuk et al., 1995). This was the case for a previous examination of the role of SFA in the Tritonia swim behavior (Getting, 1983a, 1989b). Our results indicate that the properties of a neuron in a quiescent state may not reflect the properties of that neuron while it is participating in the generation of behavior. During the initial moments of a swim motor program when all six DSIs are firing at frequencies of 20 Hz or more, C2 excitability would be much higher than at rest and perhaps even higher than we measured in this study when only a single DSI was stimulated to fire at a maximum of 20 Hz. This would lead to the removal of C2 SFA at the onset of the swim motor program. During the course of the swim motor program the DSI firing progressively decreases, presumably causing C2 SFA to increase again to some extent. The alteration of cellular properties observed inTritonia is consistent with findings from other systems, which show that synaptic strength and cellular properties can change dramatically during activation or alteration of a motor pattern (Mangan et al., 1994a,b; Johnson et al., 1995; Katz, 1995a,b).
Current view of the reconfiguration process in Tritonia
Previous work had suggested an hypothesis for the steps involved in initiating the Tritonia swim motor program (Getting and Dekin, 1985a,b; Getting, 1989a). In this scheme a brief sensory input is transformed into a long-lasting “ramp” depolarization of the DSIs. The DSIs then synaptically excite C2, bringing it above threshold. C2 inhibits an unidentified neuron (the I-cell) that mediates mutual inhibition among the DSIs, thereby changing the functional connectivity of the DSIs from inhibitory to excitatory and causing them to become self-excitatory. C2 has a delayed excitatory effect on the ventral swim interneurons (VSIs) (Getting, 1983b). When the VSIs fire, they then inhibit C2 and the DSIs, momentarily interrupting the self-excitation among the DSIs. This scenario for reconfiguring the network from a nonoscillatory state into a CPG does not involve any neuromodulatory changes in cellular or synaptic properties but, rather, depends exclusively on synaptic integration to switch from one state to the other.
A number of subsequent observations lead us here to revise this formulation (see also Frost et al., 1997). First, we have identified the source of the long-lasting input to the DSIs as DRI and found that DRI receives excitatory feedback from C2 (Frost and Katz, 1996b). Second, we have found that C2 synapses are enhanced strongly and rapidly by DSI stimulation (Katz et al., 1994; Katz and Frost, 1995a,b). Last, this paper shows that, because of its inherent SFA, in its resting state C2 is incapable of firing in the manner observed during a swim and that DSI stimulation alleviates this refractoriness.
Thus, dynamic neuromodulatory actions appear to act in concert with the previously hypothesized reconfiguration mechanism. The sequence of events underlying the generation of a swim motor program, as we currently envision them, is the following: sensory input excites DRI (Fig. 1A). DRI then monosynaptically excites all of the DSIs. The DSIs synaptically excite C2 while at the same time rapidly enhancing the excitability of C2 and evoking presynaptic facilitation of C2 synapses. The excited C2 then fires action potentials that both silence the I-cell, thereby removing the DSI–DSI inhibitory interactions, and polysynaptically excite DRI, perhaps because of the enhancement of C2 synaptic strength. Thus, in addition to the previously hypothesized recurrent excitation among the DSIs after removal of I-cell inhibition (Getting and Dekin, 1985b), we envision that a positive feedback loop is formed from the CPG (C2) to its own input neurons (DRI). Then the positive feedback loop is inhibited momentarily by the VSIs on each cycle. The modulatory actions of the DSIs seem to be a necessary component in this process, because they endow C2 with properties required for participation in the production of the motor pattern. Further work is needed to test this suggested scenario.
The role of intrinsic neuromodulation in gating its own pathway
The DSI-induced enhancement of C2 excitability may further act as a safeguard to prevent “unintended” swims. For example,Tritonia rarely swims in response to tactile stimulation but readily swims in response to contact with starfish tube feet or concentrated salt solutions. In semi-intact preparations, tactile skin stimulation causes C2 to fire action potentials but does not trigger the swim motor program (T. Hoppe and W. Frost, unpublished observations). In isolated brain preparations C2 receives excitatory inputs from unidentified neurons that are not active at times when the swim motor program is produced (P. Katz, unpublished observations). Were C2 able to generate high-frequency bursts easily in its unmodulated state, tactile stimuli and these other nonswim-related inputs also might initiate swims readily. By having C2 SFA removal and enhancement of C2 synaptic strength as necessary steps in the initiation and maintenance of the swim motor program, C2 may be prevented from triggering a swim motor program in response to inputs other than the DSIs, thereby establishing a high threshold for the response and ensuring that the animal does not produce spurious escape swims (Fig. 8).

Hypothetical mechanism for limiting access to the swim behavior while still allowing C2 to participate in other behaviors. Stimuli that do not modulate C2 synaptic strength and excitability will be incapable of evoking a swim motor program. However, C2 still will be able to participate in pathways that do not require it to fire at high rates for prolonged periods of time.
Previous work in Tritonia had shown that, on rare occasions, strong depolarization of C2 is, by itself, sufficient to elicit a swim motor program (Getting, 1977; Taghert and Willows, 1978). Our observations (unpublished) indicate that the times when C2 depolarization can by itself trigger a swim coincides with a high rate of spontaneous DSI firing, such as occurs in the time period after the generation of a swim motor program (Katz et al., 1994). Thus, after a swim motor program C2 excitability and synaptic strength may remain high as a consequence of elevated background DSI activity. This may act as a means of dynamically adjusting the threshold for triggering the behavior, such as occurs after sensitizing stimuli (Frost et al., 1988;Brown et al., 1996; Frost and Katz, 1996a).
There are now a number of examples in which extrinsic modulatory inputs have been shown to enhance cellular excitability via removal of SFA (Fung and Barnes, 1987; McCormick et al., 1991, 1993; Cox et al., 1994;Spain, 1994). In many of these cases such a modulatory action is presumed to play a role in controlling the ability of other inputs to excite the target neurons. In contrast, the DSIs are gating the output of their own target because they are the immediate source of the excitatory drive to which C2 is responding. This type of self-gating mechanism may have general relevance for other systems in which neurons need to control the outputs of the followers in their own pathway.
Footnotes
This work was supported by National Institutes of Health Grants R01-NS35371 and R01-NS36500. We thank Lian Ming Tian for participating in some of the experiments and Donna Mongeluzi for assistance with statistical analyses.
Correspondence should be addressed to Dr. Paul S. Katz, Department of Biology, Georgia State University, P.O. Box 4010, Atlanta, GA 30302.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}