

Abstract
To identify the molecular determinants underlying the pharmacological diversity of neuronal nicotinic acetylcholine receptors, we compared the α7 homo-oligomeric and α4β2 hetero-oligomeric receptors. Sets of residues from the regions initially identified within the agonist binding site of the α4 subunit were introduced into the α7 agonist binding site, carried by the homo-oligomeric α7-V201–5HT3 chimera. Introduction of the α4 residues 183–191 into α7 subunit sequence (chimera C2) selectively increased the apparent affinities for equilibrium binding and for ion channel activation by acetylcholine, resulting in a receptor that no longer displays differences in the responses to acetylcholine and nicotine. Introduction of the α4 residues 151–155 (chimera B) produced a ∼100-fold increase in the apparent affinity for both acetylcholine and nicotine in equilibrium binding measurements. In both cases electrophysiological recordings revealed a much smaller increase (three- to sevenfold) in the apparent affinity for activation, but the concentrations required to desensitize the mutant chimeras parallel the shifts in apparent binding affinity. The data were fitted by a two-state concerted model, and an alteration of the conformational isomerization constant leading to the desensitized state accounts for the chimera B phenotype, whereas alteration of the ligand binding site accounts for the chimera C2 phenotype. Point mutation analysis revealed that several residues in both fragments contribute to the phenotypes, with a critical effect of the G152K and T183N mutations. Transfer of α4 amino acids 151–155 and 183–191 into the α7-V201–5HT3chimera thus confers physiological and pharmacological properties typical of the α4β2 receptor.
The neuronal nicotinic acetylcholine receptors (nAChRs) are involved in cholinergic transmission in the peripheral nervous system as well as in the CNS (for review, seeBertrand and Changeux, 1995; Role and Berg, 1996). To date, 11 members of the neuronal nAChR family have been identified and cloned from vertebrate genomes (for review, see Lindstrom, 1996). Classified according to their sequences, these subunits have been named α2-α9 and β2-β4, and their genes are postulated to derive by duplications and mutations from a common ancestor (Le Novère and Changeux, 1995; Ortells and Lunt, 1995).
Reconstitution in host systems revealed that the physiological and pharmacological properties of the responses, including activation and desensitization, depend on both the α and the β subunits (Couturier et al., 1990; Gross et al., 1991; Luetje and Patrick, 1991; for review, see Bertrand and Changeux, 1995). This subunit diversity, which was also observed on native receptors using various techniques such as electrophysiological recordings and equilibrium binding experiments, most probably accounts for the specific pharmacology of physiological processes.
The nAChR channel opens in response to the binding of agonist (activation) but also becomes refractory to activation in the course of prolonged exposure to nicotinic agonists (desensitization). The two processes display different pharmacological specificities, typically illustrated by the Torpedo receptor for which concentrations required to desensitize the receptor are nearly 1000-fold lower than those required for activation (for review, see Changeux, 1990). This dual aspect of agonist pharmacology could play a predominant role in shaping synaptic currents and modulating the fraction of activatable receptor molecules (for review, see Heidmann and Changeux, 1982;Edelstein and Changeux, 1996; Jones and Westbrook, 1996), in particular in the mesolimbic system, a structure known to contribute to the reinforcing effects of nicotine and putatively to nicotine abuse in smokers (for review, see Dani and Heinemann, 1996). Subcutaneous injection of nicotine increases dopamine release in the nucleus accumbens, but these effects are antagonized by chronic administration of nicotine at lower concentrations (Benwell and Balfour, 1992). A comparable finding was obtained from rat striatum synaptosomes (Rowell and Hillebrand, 1994, and references therein). In addition, nicotine acts in vivo, as well as in vitro, as a positive reinforcer (Merlo Pich et al., 1997) and produces an upregulation of nicotinic receptors, possibly by stabilizing a desensitized state (Peng et al., 1994). Moreover, studies with β2 subunit knock-out mice show that the high-affinity β2 subunit containing nAChR contribute to the reinforcement by nicotine (Picciotto et al., 1997).
Little is known, however, about the molecular determinants underlying the pharmacological diversity observed between agonists such as ACh and nicotine, yet residues identified by affinity labeling experiments as contributing to agonist binding on Torpedo nAChR are highly conserved among neuronal nAChR. The aim of this work is to examine which portions of the binding sites determine differences in agonist pharmacology, including activation and desensitization, by performing parallel equilibrium binding and electrophysiological experiments with various recombinant subunits.
We used an approach based on two pharmacologically different receptors, the high-affinity hetero-oligomeric α4β2 receptor, widely distributed in the brain, and the low-affinity homo-oligomeric α7 receptor (see Table 1). We introduced into α7 subunit sequences the residues from α4 that surround homologs of the affinity-labeled amino acids in Torpedo, assuming that the residues determining agonist affinity and specificity are located within, or in close proximity to, the ligand binding pocket.
MATERIALS AND METHODS
Site-directed mutagenesis. The chimeric receptor (chick α7)-V201–5HT3 (named α7–5HT3) in the vector pMT3(Eiselé et al., 1993) was cloned as aNotI–XhoI fragment into the vector Bluescript KS to permit single-stranded DNA synthesis. For the introduction of chick α4 residues, oligonucleotide-directed specific mutagenesis was performed using the sculptor kit supplied by Amersham (Arlington Heights, IL). All constructs were then subcloned as aNotI–XhoI fragment back into pMT3(Swick et al., 1992) for expression both in human embryonic kidney (HEK) 293 cells and in Xenopus oocytes.
Expression in HEK 293 cells and [125I]α-bungarotoxin (Bgt) binding measurements. Chimeric cDNAs were transfected into HEK 293 cells by calcium phosphate precipitation (Chen and Okayama, 1987). All binding experiments were performed at 18°C as previously described (Weber and Changeux, 1974; Corringer et al., 1995). Briefly, HEK 293 cells expressing the α7–5HT3 and mutant chimeras [0.1–0.3 pmol of α-Bgt ([125I]α-Bgt, Amersham) binding sites] were diluted in 250 μl of HEPES buffer (10 mm HEPES, 2.5 mm CaCl2, 2.5 mm MgCl2, 82.5 mm NaCl, pH 7.2) and incubated for 10 min with various concentrations of cholinergic effectors. [125I]α-Bgt (final concentration 2.5 nm) was added, and after 5 min the sample was quickly diluted into 5 ml of PBS buffer, filtered through GF-C filter (Whatman, Maidstone, UK), and rinsed with 5 ml of PBS buffer. The amount of radioactivity remaining on the filter was determined by gamma counting. We verified that 3 μm and 30 nm nicotine equilibrated with the α7–5HT3chimera and chimera B, respectively, under the present conditions in <1 min (data not shown). Fitting the dose-inhibition curve to the empirical Hill equation yielded a protection constant,Kp, which gives a reasonable estimate of the apparent dissociation constant (Weber and Changeux, 1974).
Electrophysiology. Xenopus oocytes were prepared, injected, and recorded as described previously (Bertrand et al., 1991). Whole-cell recordings were performed in OR2 medium (containing 82.5 mm NaCl, 2.5 mm KCl, 1 mm Na2HPO4 2H2O, 2.5 mm CaCl2, 1 mmMgCl2, and 15 mm HEPES, pH 7.4) at 18°C.
Modeling. Dose-inhibition curves of desensitization were interpreted in terms of a simplified two-state model derived from the Monod-Wyman-Changeux scheme (Monod et al., 1965; Heidmann and Changeux, 1979, 1980; Edelstein et al., 1996), which postulates that the protein is in equilibrium between a basal B state, predominant in the absence of effectors, and a desensitized D state, and that during the isomerization process, all subunits undergo the conformational change, regardless of the occupation of the binding sites: B ⇋ D.
Each state displays an intrinsic affinity for a given agonist as expressed by the respective intrinsic dissociation constantsKB and KD, withc =KD/KB, and these states interconvert in the absence of effector with an isomerization constant L = [B]/[D]. Given the homomeric nature of the α7–5HT3 chimera, we postulated, in agreement with previous observations (Palma et al., 1996), that the protein carries five equivalent binding sites.
For nicotine desensitization experiments, we assumed that equilibrium was reached during the prepulse procedure (8 min), leading to fixedB̄pp and D̄pppopulations, where “pp” indicates prepulse. Activation was neglected under these conditions, because at equilibrium the current was always <5% of maximally evoked currents. Desensitization was assessed by application of a short pulse (2 sec) of agonist at its EC50 concentration. Assuming that this procedure does not further affect desensitization, the recorded response is proportional to the fraction of receptors remaining in an activatable conformation, and thus to B̄pp = (1 −D̄pp). We verified that the values of the parameters found with nicotine agreed reasonably with the data obtained with ACh.
Equations for 1 − D̄pp and of the Hill coefficient as a function of L were taken from previously published papers (Monod et al., 1965; Rubin and Changeux, 1966; Karlin, 1967; Edelstein et al., 1996), where X refers to the ligand concentration:
 Equation 1The apparent Hill coefficient nH at 50% desensitization is obtained from:
Equation 1The apparent Hill coefficient nH at 50% desensitization is obtained from:
 Equation 2In these equations:
Equation 2In these equations:
 Equation 3and with:
Equation 3and with:
 Equation 4where c =KD/KB andL = [B]/[D].
Equation 4where c =KD/KB andL = [B]/[D].
RESULTS
The α7–5HT3 chimera, a tool to investigate the nAChR binding site
Up to now, expression of neuronal nAChRs in quantities sufficient for biochemical experiments has remained difficult, and the only known systems in which successful expression of the α7 receptor has been obtained are stably transfected cell lines (Lukas et al., 1993;Gopalakrishnan et al., 1995). Although high yields of receptor expression have been achieved in these systems, they remain impractical for extensive studies of engineered cDNA constructs.
One way to overcome these technical difficulties is to use a chimera combining portions of chick α7 and 5HT3, the α7–5HT3 receptor chimera transiently expressed in HEK 293 cells or Xenopus oocytes. This chimera preserves the unmodified α7 binding site and the main pharmacological properties of agonists and competitive antagonists for the chick α7 wild-type receptor (Eiselé et al., 1993; Corringer et al., 1995) (Table1), with the exception of methyllycaconitine (Palma et al., 1996), probably because this large antagonist contacts distant parts of the protein. All experiments reported here were performed using α7–5HT3 chimeras with parallel electrophysiological recordings after cDNA expression inXenopus oocytes (Bertrand et al., 1991) and equilibrium binding measurements with transiently transfected HEK 293 cells (Corringer et al., 1995).
Apparent affinity constants of α4β2, α7, and α7–5HT3 and mutant chimeras
Construction of chimeras between α7 and α4 subunits
The chick α4β2 and α7 nAChRs display major differences in their respective apparent affinities for ACh and nicotine. First, both from electrophysiological recordings and equilibrium binding experiments, the α7 homo-oligomer displays a higher apparent affinity for nicotine than for ACh, whereas the α4β2 receptor does not discriminate between these two agonists. Second, the α4β2 receptor displays a 180-fold higher apparent binding affinity than EC50 obtained from electrophysiological recordings, whereas this difference is only two- to eightfold for the α7 receptor (summarized in Table 1).
Photoaffinity labeling experiments performed on the muscle typeTorpedo nAChR revealed that the agonist/competitive antagonist binding pocket is composed of two main components of the N-terminal domain and overlaps the boundary between subunits (for review, see Bertrand and Changeux, 1995; Corringer et al., 1995; Karlin and Akabas, 1995). The “principal component” is carried by the α subunit and consists of three loops: loop A (Trp-86, Tyr-93), loop B (Trp-149, Tyr-151), and loop C (Tyr-190, Cys-192, and Cys-193), whereas the “complementary component” is carried by the non-α subunits and comprises at least two loops: loop D (Trp-55 and Trp-57 on the γ and δ subunits, respectively) and loop E (Asp 182 on the δ subunit) (Fig. 1). Recent experiments suggest the contribution of other regions of the γ or δ subunits to the complementary component (Prince and Sine, 1996). Site-directed mutagenesis experiments performed with muscle-type and neuronal homo-oligomeric α7 receptors established the functional role of loops A, B, C, D, and E in ACh and agonists binding. The labeled amino acid residues from loops A, B, and C are conserved in α1–α8 subunits (except in α5, which lacks the complete principal component of binding), whereas Trp from loop D is conserved in all neuronal β subunits, showing the overall conservation of the structure of the agonist binding pocket within the nAChR family (for review, see Galzi and Changeux, 1995; Karlin and Akabas, 1995).

Model of the ACh binding site of nAChRs, which includes loops A, B, and C from the principal binding component, and loops D and E from the complementary binding component. Sequences of fragments in the region of loops of the principal binding component from chick α4 and chick α7 are shown on the left. The residues homologous to those labeled by cholinergic effectors on the Torpedo receptor are shown in bold letters. The regions that differ between α4 and α7 areunderlined, and each of the fourunderlined α4 segments were grafted in the α7–5HT3 chimera. Light gray arrows correspond to the single mutations analyzed in this work. Thenumbers shown are related to α7 sequence. chimera A, α7–94-α4–104-α7–201-5HT3; chimera B, α7–151-α4–155-α7–201-5HT3; chimera C1, α7–174-α4–182-α7–201-5HT3; chimera C2, α7–183-α4–191-α7–201-5HT3.
To examine in greater detail the principal component of the binding site, we investigated whether discrete segments of the protein could determine specific pharmacological and functional features. Alignments of the regions surrounding the three loops of α4 and α7 are shown in Figure 1. Regions that are not conserved between α7 and α4, but are flanked by conserved motifs, are underlined. These α4 regions were introduced into the α7–5HT3 receptor using single-stranded site-directed mutagenesis. Two chimeras were constructed in the region of loop C (chimeras C1 and C2), one in the region of loop B (chimera B), and one in the region of loop A (chimera A).
All constructs displayed expression levels similar to those of the α7–5HT3 chimera in HEK 293 cells and yielded robust currents in the microampere range (with the exception of chimera C1, which gave very low expression levels in both systems). No major alterations of the time course of the currents evoked by ACh on these mutant receptors were observed, either during the rising phase of the responses, as illustrated in Figure2C, or at the level of the desensitization time course. Indeed, Figure 2A shows that in the absence of calcium ions, which may interfere with desensitization through a channel block mechanism, the decay of the responses and thus the desensitization kinetics are similar for the α7–5HT3, A, B, and C2 chimeras. In the presence of calcium, responses are larger because of potentiation through allosteric binding sites for calcium, as described previously for the α7–5HT3 chimera (Eiselé et al., 1993;Galzi et al., 1996a), and no significant differences are observed between the decay of the responses. For technical reasons, recordings were performed in the presence of calcium ions.

A, Time course of currents evoked by 100 μm ACh on Xenopus oocytes expressing the α7–5HT3 chimera and chimera A, B, and C2. Recordings were performed in control medium (2.5 mm Ca2+, thin lines) or in a medium from which Ca2+ was removed (thick lines). Note that no major differences are observed between the time courses of the currents and that all constructs are potentiated by Ca2+.B, Time course of currents elicited by three ACh concentrations on an oocyte expressing the chimera C1 are superimposed. All cells tested with this chimera displayed currents of comparable amplitude. C, Higher time resolution of the rising phase of the ACh evoked currents (at saturating concentrations of ACh, 100 μm, for all constructs except 320 μm for chimera C1). Currents have been normalized to their maximum value. From the right to theleft, traces correspond to constructs designated in the insert by a, b and c(which are superimposed), d and e.
EC50 values for the activation were determined by using brief agonist applications separated by adequate recovery intervals. Peak current may not exclusively reflect activation processes, because desensitization or channel block may start before the full onset of activation. However, because all constructs display similar time courses of their responses, a truncation of the response, if it occurs, would be comparable for all constructs. Thus the evaluation of the relative effects of the mutations, as compared with the α7–5HT3 chimera, would still be valid. Such features, however, could explain that for particular constructs, as seen for nicotine dose–response curves of chimeras A and B (Fig.3D), a slight decrease of the response is observed at submaximal concentrations.

A, C, Effect of increasing concentrations of ACh (A) and nicotine (C) on the initial rate of [125I]α-Bgt binding to the α7–5HT3 and mutant chimeras. Each pointcorresponds to the mean value of duplicate experiments, normalized to the maximum value. Lines represent fits to the empirical Hill equation, yielding apparent binding affinities (Kp) and Hill coefficients (nH). MeanKp and nH values, corresponding to the average of three separate experiments, are summarized in Table 1. B, D, ACh and nicotine dose–response relationships of α7–5HT3 and mutant chimeras. Peak responses evoked by 3 sec agonist application of increasing agonist concentrations were measured in three to five oocytes held at −100mV. Peak currents of each experiment are normalized to the maximum values, and the Figure represents the average of all experiments for each construct. Lines represent the mean of the curves resulting from the fit with the empirical Hill equation of each individual normalized experiment. The corresponding apparent affinities of activation (EC50) and Hill coefficients are summarized in Table 1.
Loop C contributes to the pharmacological specificity
Substitution of residues 183–191 of the α7 subunit by those of α4 yielded the construct called C2 (Fig. 1). Determination of the apparent binding affinity (Kp) and apparent affinity of activation (EC50) was derived from the empirical Hill equation for ACh and nicotine with this chimera. It showed a marked difference from the α7–5HT3 chimera. A 30-fold decrease inKp for ACh was observed, whereas electrophysiological recordings revealed only a sixfold decrease in EC50 for ACh (Fig. 3A,B, Table 1). Moreover, the apparent affinities for nicotine remained almost unchanged (Fig.3C,D, Table 1), resulting in similar apparent affinities for binding (Kp of 2.8 and 1.4 μm) and for activation (EC50 of 3.7 and 2.1 μm) for ACh and nicotine, respectively. Because recombinant α4β2 receptors do not discriminate in terms of pharmacological properties between ACh and nicotine, residues 183–191 thus contribute to the difference in agonist specificity observed between α7 and α4β2 nAChR.
Substitution of residues 174–182 of the α7 subunit by those of the α4 subunit yielded the construct designated C1 (Fig. 1). In contrast to C2, this construct yielded no detectable [125I]α-Bgt binding sites in transfected cells, and very small currents in the range of 46 ± 19 nA (n = 5, for 320 μm ACh) were measured in oocytes, therefore precluding a more extensive study. Nevertheless, EC50 for ACh of C1 resembles that of the α7–5HT3 chimera (∼60 μm) (Fig.2B). Taken together, these data indicate that these mutations did not alter the ligand binding site but most probably reduced the level of receptor expression. Inspection of the protein sequence revealed that a putative glycosylation site (NYT), not present in α4, had been created at position 181–183. To explore whether this element is responsible for the observed phenotype, another chimera in which the Asn 181 was replaced by an Ala was engineered, but this mutant also failed to express detectable levels of [125I]α-Bgt binding, suggesting that the side chain residues are critical for correct assembly.
Loop B determines agonist affinity at equilibrium
Substitution of residues 151–155 of the α7 receptor by the corresponding amino acids of the α4 subunit were used to construct chimera B. Determination of the Kp in HEK 293 cells revealed decreases of 75- and 120-fold for ACh and nicotine, respectively (Fig. 3A,C). In addition, a comparable reduction in Kp was observed for the agonist cytisine and the antagonist d-tubocurarine [for cytisine,Kp(wt) = 2.1 ± 0.6 μm,Kp(chimB) = 0.011 ± 0.004 μm; for d-tubocurarine,Kp(wt) = 1.0 ± 0.5 μm,Kp(chimB) = 0.035 ± 0.007 μm]. Interestingly, this chimera displayed only modest (although significant) 2.8- and 4.4-fold decreases in EC50for ACh and nicotine, respectively (Fig. 3B,D). By comparison, recombinant chick α4β2 receptor exhibits large differences (two orders of magnitude) between the apparent affinities measured in binding experiments at equilibrium or in activation experiments by electrophysiological recordings for ACh and nicotine (Table 1). Thus, the mutations conferred a high apparent affinity for agonists on the α7 binding site in equilibrium binding experiments, thereby mimicking α4β2 receptors.
A minor contribution from loop A
Substitution of the α4 loop A residues into α7 yielded chimera A. The construct was characterized by a slight decrease inKp for ACh (twofold), with almost no effect on the Kp of nicotine and on the EC50in electrophysiological experiments. Mutations of loop A thus do not significantly alter the apparent equilibrium binding and activation affinities for both agonists.
Analysis of chimeras B and C2 by point mutations
To investigate the contribution of individual amino acid residues in the portions exchanged in the chimeras, a series of point mutations was generated. All constructs yielded normal expression levels in both HEK cells and oocytes.
In the case of loop B, mutations G151D, G152K, W153A, S154K, and L155I were introduced individually into the α7–5HT3 chimera. As demonstrated by the results presented in Table2, parallel effects were observed for ACh and nicotine apparent binding affinities. Mutations at positions 151, 152, and 153 produced an increase in apparent binding affinity for both agonists, in the range of 2.4- to 7.5-fold for the G151D and W153A, and of 22- and 50-fold in G152K for ACh and nicotine, respectively. On the other hand, the mutation L155I had almost no effect on the AChKp and produced a small (fourfold) decrease in nicotine Kp, whereas S154K resulted in a large decrease in apparent affinity for both agonists (20- and 7-fold for ACh and nicotine, respectively). In contrast to the observations with the chimera B, these point mutations produced similar effects in binding and activation experiments. Indeed, mutations G152K and W153A resulted in 11- and 5.7-fold decreases in EC50 for ACh, and S154K resulted in a 8.7-fold increase in EC50 for ACh.
Affinity constants of α7–5HT3 mutants from loop B and C2 for ACh and nicotine
To analyze in greater details the S154K mutation, which unexpectedly produced a large decrease in ACh apparent binding affinity, the double mutant G151D/S154K was generated. This construct displayed an apparent ACh binding affinity (Kp = 73 ± 16 μm with nH= 1.6 ± 0.1) identical to the one of the α7–5HT3 chimera, whereas adding the effects of the two single mutations would result in an eightfold decrease in apparent affinity for ACh. This suggests that the side chains of the two residues interact when incorporated together in chimera B.
Concerning the C2 chimera, mutants T183N, E184S, S185K, F186K, and K191T were constructed and tested for equilibrium binding of ACh and nicotine. These mutations produced no significant effects on apparent affinity for nicotine, except for T183N, which results in a threefold decrease in Kp. For ACh, however, the mutants E184S, S185K, and F186K all resulted in small decreases inKp (2.1- to 3.6-fold) and T183N produced a 7.5-fold decrease in Kp, whereas K191T had no effect.
In conclusion, several residues from loops B and C contribute to the phenotypes, with a critical role for the mutations G152K and T183N.
Increase in equilibrium binding apparent affinity reflects an increase in sensitivity to desensitization
Chimeras B and C2 display high apparent affinities for agonists at equilibrium, which presumably reflects the population of a desensitized state of the receptor. As a result, a prolonged application of a low agonist concentration would be expected to elicit desensitization of the responses recorded electrophysiologically to a greater extent than for the α7–5HT3 chimera. To evaluate this prediction, we monitored the amount of activatable α7–5HT3 or B and C2 chimeras before and after continuous application of agonist, using the following protocol. In an agonist-free medium, oocytes displaying large currents were tested at 98 sec intervals with short pulses of agonist (2 sec, at a concentration near the EC50; named “test pulse”). After a recording time of 5 min under these control conditions, a low concentration of the same agonist was added to the perfusion medium (named “prepulse”), and test responses were monitored for at least 8 additional minutes (Fig.4). The control perfusion was then reestablished, and another prepulse at a different concentration of agonist was applied. These recordings were made with both ACh and nicotine. A 5 min prepulse period was sufficient to reach a steady-state level of desensitization for the three constructs and for all agonist concentrations used.
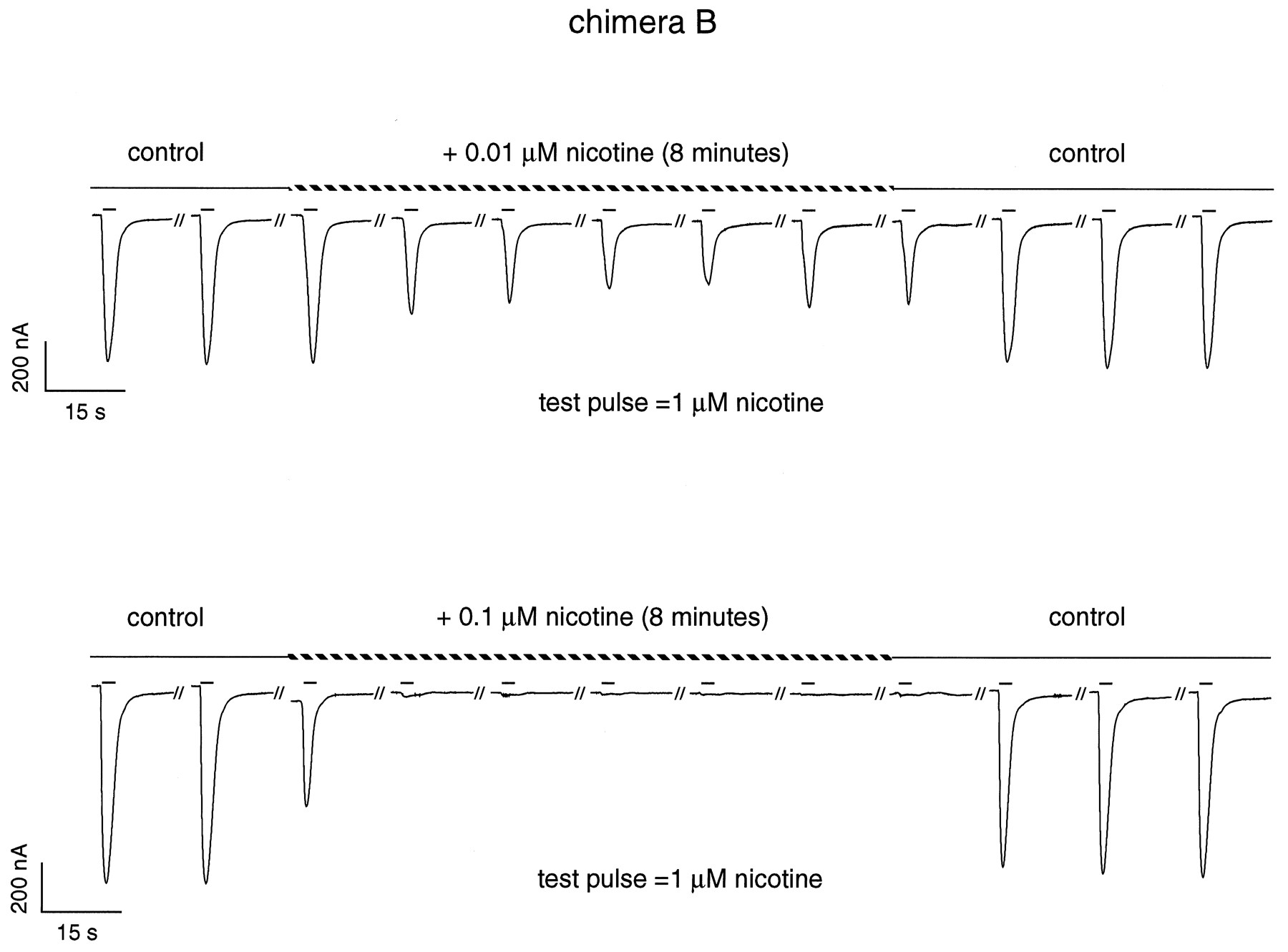
Typical desensitization recordings of the chimera B. The oocyte was challenged at regular intervals (98 sec) by a short nicotine test pulse (2 sec) at a concentration near the EC50 (1 μm). Control conditions show that the test pulses do not produce significant desensitization. Addition of low concentrations of nicotine to the perfusion medium (prepulse,dashed lines) results in a decrease of the test pulse responses. Equilibrium is reached within a few minutes, and full recovery was always observed when going back to control conditions.
As revealed by the experiments described in Figure5, exposure of oocytes expressing the α7–5HT3 chimera to increasing concentrations of nicotine and ACh resulted in a decreased fraction of activatable receptors. For both agonists, desensitization took place only in concentration ranges in which they elicited a current. Fitting the dose-inhibition curves by the Hill equation yielded an IC50 of 0.48 μmand 12.5 μm for nicotine and ACh, respectively, with corresponding nH values of 4.0 and 3.0 (Table1).

Activation dose–response (light gray symbols) and desensitization dose-inhibition (dark gray symbols) curves of the α7–5HT3, B and C2 chimeras, for nicotine (A) and ACh (B). Dose–response curves for activation and desensitization were performed successively on the same oocyte, and these data were normalized according to the maximum value of the activation curve. Desensitization was measured using the protocol illustrated in Figure 4, after an 8 min prepulse incubation, and with a test pulse at a concentration near the EC50, which had been determined on the same cell. Steady-state test pulse currents were plotted as a function of prepulse agonist concentration. The mean values of three separate experiments are shown in each case, except for the α7–5HT3 and B chimeras with ACh, in which two experiments were performed. Dashed lines represent the mean of the curves resulting from the fit of each individual curve using the empirical Hill equation, for both activation and desensitization. The corresponding apparent affinity of desensitization (IC50) and Hill coefficient are summarized in Table1. Solid lines represent the fit of the desensitization data using the two-state allosteric model (Eq. 1), normalized to the maximum test pulse currents.
In contrast, much lower and nonactivating concentrations of nicotine were sufficient to desensitize the chimera B. This construct is characterized by a 19- and 48-fold decrease in IC50 for ACh (Fig. 5B) and nicotine (Fig. 5A), respectively, which is associated with a large decrease in Hill coefficient. In the case of chimera C2, a 27-fold decrease in IC50 is observed for ACh, with a comparatively weak effect on nicotine IC50 (fourfold decrease). Interestingly, these changes are associated with no significant modifications of the Hill coefficients.
With chimera B and C2 for both nicotine and ACh and with the α7–5HT3 chimera for nicotine, the level of desensitization was at least 95% at the highest agonist concentration of the desensitization curves (Fig. 5). However, in the case of the α7–5HT3 chimera for ACh, the midpoint of the desensitization curve was close to the EC50 for activation, and at the highest prepulse concentration that could be tested (EC50), the desensitization was only 70%. Therefore, the full desensitization curve could not be measured.
In conclusion, the specific increase in apparent binding affinity reflects a specific increase in desensitization sensitivity, with a parallel effect on ACh and nicotine for chimera B and a specific effect on ACh for chimera C2.
Modeling of activation and desensitization data of mutant chimeras
Interpretation of these unusual phenotypes requires fitting of the available electrophysiological recordings with a model that includes the conformational transitions of the receptor molecule. Such a model is aimed at providing an interpretation in terms of the intrinsic properties of the receptor (intrinsic affinity for a given state and agonist, intrinsic isomerization constant between two states) of the apparent affinity constants (such as IC50, EC50, and Kp) and of the apparent cooperativity of the corresponding curves. We applied the simple two-state allosteric model (see Material and Methods), which takes into account only a basal and one desensitized state (with intrinsic dissociation constants KB andKD). Thus, activation was not included in the model because no major effects were observed at this level. Similar models have been currently used for the study of the desensitization of muscle-type (Sine et al., 1995) and neuronal (Galzi et al., 1996b) nAChR.
Our strategy was to constrain the parameter of the α7–5HT3 chimera first, using the unusually highnH value of the dose-inhibition curve of desensitization for nicotine, which depends only on the Land c parameters, and then to chose theKB to obtain the correct IC50. In Equations 1 and 2, various values for L and c are compatible with a nH of 4. Because the difference between the EC50 of activation and the IC50 of desensitization is small, our working hypothesis was to find a value of KB as close as possible to that of KD, i.e., the maximal value ofc consistent with the experimental data. Constructing from Equation 2 the curves of nH as a function ofL for various c values shows that the maximalc compatible with the observed nH isc = 0.01. This requires that L = 105 and results in KB = 5 μm to give the correct IC50. To allow comparison with experimental data in Figure 5 (also see Table3), this theoretical curve was normalized to the maximal current evoked by the test pulse of agonist. In the case of ACh, the nH of 3 was obtained withc = 0.04, leading to a KB of 20 μm. These KB values are in the range of the EC50 of activation (4.3 and 25 μm for nicotine and ACh, respectively) and thus are compatible with the activation data.
Parameters of the two-state model yielding best fits of the dose-inhibition curves of desensitization (shown in Fig. 5)
Mutations introduced into chimera B result in an increase in apparent affinity and a decrease in cooperativity. A decrease in cooperativity can be obtained either by decreasing L or by increasingc. However, the latter alternative is unlikely, because a value of c = 0.1, for example, would require a 10-fold decrease in KB to produce the correct IC50, a feature inconsistent with the weak effect of the mutations observed in activation experiments. In contrast, a decrease in L from 105 to 10, with minor modification of the intrinsic affinities for nicotine, appeared sufficient to account for the phenotype observed for chimera B, giving the correct IC50 and a value of nHclose to the experimental one, for both ACh and nicotine, as shown in Figure 5 and Table 3. The value L = 10 predicted for chimera B also implies that in the absence of effector, 9% of the population is in the desensitized conformation. Electrophysiological recordings do not permit direct measurement of this basal level of desensitization, but this L value gives the normalized theoretical curve that best represents the normalized experimental curve.
Mutations introduced in chimera C2 result in a specific increase in apparent affinity for ACh, with small modification of thenH. Thus an alteration of the Lconstant is unlikely, whereas simply decreasing theKD values (5-fold and 20-fold decrease for nicotine and ACh, respectively) fully accounts for the phenotype. To account for the small but significant shifts in EC50, KB constants were slightly reduced. In all cases, the calculated average deviation was <5% of the amplitude of the curves, except for chimera B with nicotine, in which case this value reached 9% because of the difference in cooperativity of both curves (Table 3). Parameters from chimeras B and C2 also provided a semiquantitative description for the shifts in Kp for binding experiments. A more precise agreement is unlikely, because binding and desensitization experiments were performed in different expression systems.
DISCUSSION
Establishing the molecular determinants of the physiological and pharmacological diversity of nAChR for agonists such as ACh and nicotine has remained a difficult challenge. This issue, attributable to the high conservation of residues known (to date) to contribute to agonist binding of nAChR, is complicated by the multiple-loop nature of the agonist binding site. Such organization could imply that multiple mutations located in different parts of the sequence may contribute simultaneously to the modulation of receptor pharmacology. For example, it has been found previously that residues located in segments 1–84 and 195–215 were responsible for the differences in sensitivity of ACh versus nicotine observed between α2β2 and α3β2 receptors (Luetje et al., 1993).
The experiments reported here using chick α4/α7 chimeras of the N-terminal domain were designed to identify the segments that individually contribute to the marked pharmacological differences existing between the α4β2 and α7 receptors. The experimental strategy was to transfer amino acid residues from the chick α4 subunit that lie in the proximity of already identified points of ligand binding into the α7–5HT3 chimera. From the present study we cannot exclude the possibility that other regions of the N-terminal domain, either from the principal α4 or complementary β2 components, may also contribute to such differences. In addition, results with subunits from other species may also differ. However, the major pharmacological features of selectivity and apparent affinity at equilibrium with respect to ACh and nicotine for α4β2 receptors have been transferred to the α7–5HT3 chimera. We show here that incorporation of residues near loop B, in chimera B, results in a ∼100-fold increase in apparent binding affinity for agonist, whereas incorporation of residues near loop C, in chimera C2, results in a specific increase in apparent binding affinity for ACh compared with nicotine, with weak effects on activation in both cases. On the other hand, mutations near loop A, or in the region of loop C (chimeras A and C1), did not alter the apparent affinities of agonists, but the level of receptor expression was lower in the case of loop C1.
The major portion of the binding site is currently thought to be contributed by the loop C region. The Cys doublet motif is characteristic of the α subunit binding domain, and small synthetic peptide fragments from this region bind the competitive antagonist α-Bgt (Basus et al., 1993, and references therein). In the muscle-type receptor, mutations of the residues Y190 and Y198 identified by affinity labeling of Torpedo receptor (for review, see Bertrand and Changeux, 1995) resulted in large decreases of the apparent affinity for ACh (Tomaselli et al., 1991; Aylwin and White, 1994a,b; McLaughlin et al., 1995; Nowak et al., 1995; Kearney et al., 1996). However, the Y190F mutant has also been reported to display an altered “gating process,” at both the level of activation and desensitization (O’Leary and White, 1992; Sine et al., 1994; Chen et al., 1995).
Simulations using a concerted model suggest that mutations yielding the C2 chimera produced their effect at the level of the ligand binding site, consistent with their location in the loop C region. In agreement with this notion, neither the chimera C2 nor the corresponding single mutations produced large effects on nicotine binding, showing that this segment does not contain determinants causing major alterations in the receptor intrinsic properties and in particular of its intrinsic isomerization constants. Mutation of α7Y187 also specifically alters ACh apparent affinity as compared with nicotine (Galzi et al., 1991), which shows a different mode of interaction of the two agonists with the loop C. Mutagenesis studies onTorpedo receptor suggest that Y190 interacts with the quaternary ammonium portion of ACh (Sine et al., 1994; Dougherty, 1996). The interaction between tyrosines and quaternary ammonium could thus account for the weak effects of the α7 mutations on nicotine binding.
Overall, the entire loop, amino acids 183–186, contributes to the increase in ACh apparent affinities. It is likely that these residues do not contact directly the agonists but rather act indirectly in shaping the ligand binding pocket, because (1) up to 10 residues, and no single residue belonging to this segment, have been shown by affinity labeling to reside in close proximity to the agonists, and (2) single mutation analysis, although it introduced very different side chain residues (T183N, E184S, S185K, and F186K), produced at most a sevenfold increase in binding affinity. It is noteworthy that these residues are not conserved among the α subunits sequenced to date (Fig. 6), suggesting that this highly variable region plays a specific role in determining the affinity and pharmacology according to subunit combination and species. A second consequence, considering that the effects were much stronger in binding than in activation experiments, is that the mutations specifically alter the intrinsic properties of the binding site of the desensitized conformation of the receptor, at least when they are introduced together in chimera C2. This point suggests a structural reorganization of the agonist binding site in the course of the desensitization of the receptor protein.

Comparison of the amino acid sequences in the region of loop B and C of several nicotinic subunits (Cockcroft et al., 1992). Numbers correspond to the chick α7 sequence.
Among the few studies concerning the loop B region that have been reported so far, changing Trp 148 to Phe in homo-oligomeric α7 receptors was found to decrease the apparent affinities of activation for nicotine and ACh by 100-fold (Galzi et al., 1991). In contrast to chimera C2, fitting the electrophysiological data of chimera B using a concerted model supported the notion that an alteration of the isomerization constants of the protein is the major cause of the increase in binding affinity and desensitization sensitivity, possibly in conjunction with an alteration of the ligand binding domain, attributable to the location of the mutation in the vicinity of Trp 148 (Sugiyama et al., 1996). This conclusion is consistent with the parallel increase in apparent binding affinity observed for all the agonists tested, and for the desensitizing antagonist d-tubocurarine (Bertrand et al., 1992), independent of their chemical structure, which argues in favor of a modification of the intrinsic properties of the protein. In agreement with this hypothesis is the report that in one case of genetically transmissible myasthenia gravis, the disease phenotype is caused by a mutation at the site homologous to α7G152 (mutation α1G153S) (Sine et al., 1995). This mutation causes a 50-fold increase in the apparent affinity for ACh in equilibrium binding experiments, and exposure to meproadifen, a noncompetitive blocker that stabilizes the desensitized state, results in a much smaller difference in apparent affinity between wild-type and mutant receptor. These data were interpreted in terms of a change in the intrinsic isomerization constant L of the receptor toward the desensitized state (Sine et al., 1995). So far, such regions of conformational control of desensitization have been found in the transmembrane portion of the receptor, in particular at the level of the M2 (Revah et al., 1991;Devillers-Thiéry et al., 1992) and M4 (Lee et al., 1994) segments.
The five mutated residues are well conserved within the α subunit phylogenetic subfamilies (α2/α3/α4, α7/α8, and muscle-type α1) but not in the β neuronal subunits (Fig. 6), an observation consistent with their critical role at the level of the principal component of the ACh binding site. Single mutation analysis revealed that the G152K mutation accounts for most of the observed phenotypic changes of chimera B, with a weaker yet significant effect of G151D and W153A. In contrast, the S154K mutation results in a large decrease in apparent affinities. Our results also suggest that these amino acids interact with each other when incorporated into the α7–5HT3 chimera. Indeed, adding the effects of each single mutation would result in a sixfold increase in binding affinity for ACh, whereas a 75-fold increase is observed when the mutations are incorporated together in chimera B. An interesting hypothesis would be that the S154K mutation, when incorporated alone, produces a decrease in apparent affinity either through repulsive interaction with the agonist or through an increase of the L constant, whereas this effect would be eliminated in chimera B where a D is present at position 151 through a side chain ionic interaction between D151 and K154. In agreement with this hypothesis, we show here that the double mutant G151D/S154K displays a binding affinity for ACh identical to the one of the α7–5HT3 chimera. Thus, addition of the effects of this double mutant, G152K, W153A, and L155I, would now result in a 53-fold increase in ACh apparent affinity, consistent with what is observed in chimera B. Finally, alterations in activation constants are more pronounced in single mutants than in the full chimera. Although difficult to interpret, these results further indicate that the region is involved in a complex process that could possibly be explained by side chain interactions as proposed above.
We also demonstrate that the increases in equilibrium binding affinity have major functional consequences. Indeed, the chimera B is desensitized by 20- to 50-fold lower concentrations of ACh and nicotine than the α7–5HT3 chimera, whereas chimera C2is desensitized by 27- and 4-fold lower concentrations of ACh and nicotine, respectively (Table 1). It is noteworthy that most of the mutations introduced in our investigation produced increases in apparent affinity of binding and desensitization. High-affinity nicotine binding to the α4β2 receptor has been related to the high-affinity reinforcement mechanism; in contrast, a low ACh binding affinity seems to be required in vivo for proper α7 function. This work thus opens the way to an analysis in molecular terms of the physiological and pharmacological aspects of nicotine abuse on ACh transmission by specifically modulating in vivothe pharmacology of desensitization of native receptors through homologous recombination experiments (Picciotto et al., 1995).
Footnotes
This work was supported by research grants from the Association Française contre les Myopathies, the Centre National de la Recherche Scientifique, the Collège de France, the Direction de la Recherche Etudes et Techniques, the European Economic Community Biotech Program, the Human Frontier Science Program, the Council for Tobacco Research, the Office Fédéral de l’Éducation et des Sciences, and the Swiss National Science Foundation.
Correspondence should be addressed to Dr. J. P. Changeux, Neurobiologie Moléculaire, Institut Pasteur, 25 rue du Docteur Roux, 75724 Paris Cedex 15, France.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}