

Abstract
The neurotransmitters acetylcholine (ACh) and glutamate have been separately implicated in synaptic plasticity during development of sensory neocortex. Here we show that these neurotransmitters can, in fact, act synergistically via their actions at nicotinic ACh receptors (nAChRs) and NMDA receptors, respectively. To determine how activation of nAChRs modifies glutamatergic EPSPs, we made whole-cell recordings from visualized pyramidal neurons in slices of rat auditory cortex. Pulsed (pressure) ejection of nicotine onto apical dendrites selectively enhanced EPSPs mediated by NMDA receptors without affecting AMPA/kainate (AMPA/KA) receptor-mediated EPSPs. The enhancement occurred during a transient, postnatal period of heightened cholinergic function [neurons tested on postnatal day 8–16 (P8–16)], and not in the mature cortex (>P19). Three related findings indicated the mechanism of action: (1) The specific α7 nAChR antagonist methyllycaconitine citrate (MLA) blocked the effect of nicotine; (2) pulsed nicotine did not enhance postsynaptic depolarizations induced by iontophoretically applied NMDA; and (3) bath exposure to nicotine for several minutes produced apparent nAChR desensitization and precluded enhancement of EPSPs by pulsed nicotine. Together, the data suggest that nicotine acts at rapidly desensitizing, presynaptic α7 nAChRs to increase glutamate release onto postsynaptic NMDA receptors. The synergistic actions mediated by α7 nAChRs and NMDA receptors may contribute to experience-dependent synaptic plasticity in sensory neocortex during early postnatal life.
Patterns of sensory input can influence the development of sensory neocortex by way of activity-dependent synaptic plasticity (Stryker and Harris, 1986;Reiter and Stryker, 1988; Collingridge and Singer, 1990; Hata and Stryker, 1994; Scheetz and Constantine-Paton, 1994; Katz and Shatz, 1996). The cellular mechanisms of developmental plasticity involve the neurotransmitters ACh and glutamate, because plasticity is reduced by disrupting cortical pathways that use either ACh (Bear and Singer, 1986; Zhu and Waite, 1998) or glutamate (Kleinschmidt et al., 1987; Fox et al., 1996). It is not known if this reflects synergistic or separate actions by the two neurotransmitters.
During postnatal development of primary auditory, somatosensory, and visual neocortices, there is a dramatic increase in the expression of the cholinergic enzyme acetylcholinesterase (AChE) (Kristt, 1979;Prusky et al., 1988; Robertson et al., 1991). In auditory cortex, the increased expression occurs in layers III-IV beginning on postnatal day 3 (P3), reaches peak intensity at P8–10, and declines to low (adult) levels by P23 (Robertson et al., 1991). Recent studies have revealed a parallel increase in expression of the α7 nAChR in developing sensory cortex (Fuchs, 1989; Broide et al., 1995, 1996). The goal of this study was to determine the functional relevance of enhanced nicotinic receptor expression during development.
Despite the diversity and abundance of nAChRs in the CNS (for review, see Sargent, 1993; McGehee and Role, 1995; Wonnacott, 1997), there is little evidence for classical nicotinic synaptic transmission in the brain. Rather than mediating synaptic transmission as at the neuromuscular junction, nAChRs in the brain can act presynaptically to regulate the release of other neurotransmitters, including glutamate (for review, see McGehee and Role, 1996; Wonnacott, 1997). Because the enhanced expression of α7 nAChRs in developing sensory cortex occurs in a region of developing thalamocortical and corticocortical (glutamatergic) synapses, we sought to determine the effect of nicotine on glutamatergic EPSPs in developing auditory cortex.
We found that nicotine selectively enhances NMDA receptor-mediated synaptic transmission during the postnatal period of heightened cholinergic function. We propose that nicotinic modulation of NMDA receptor-mediated responses is important for experience-dependent maturation of thalamocortical and/or corticocortical synapses. Conversely, overexposure to nicotine may lead to nAChR desensitization, and hence disrupt synaptic plasticity during this critical period.
MATERIALS AND METHODS
Slice preparation and maintenance. Sprague Dawley rats (8–24-d-old males or females) were anesthetized with barbiturate or halothane and decapitated. Brains were rapidly removed and blocked with a razor blade in cold artificial CSF (ACSF) containing (in mm): NaCl 125.0, KCl 2.5, NaHCO3 25.0, KH2PO4 1.25, MgSO4 1.2, CaCl2 2.0, and dextrose 10.0, bubbled with 95% O2 and 5% CO2, pH 7.4, osmolarity 298–302 mosmol/kg. Coronal sections (300 μm) containing auditory cortex (Paxinos and Watson, 1986; Sally and Kelly, 1988) were made using a Vibroslice (World Precision Instruments, Sarasota, FL). The location of auditory cortex was determined based on landmarks (e.g., the dorsoventral extent of the CA1–3 fields of the hippocampus in the coronal section and distance dorsal to the rhinal fissure) and AChE histochemistry (AChE delineates primary sensory cortex in juvenile rats; see below). Slices were placed in a holding flask containing oxygenated ACSF at room temperature for at least 30 min before use. Low Ca2+ experiments substituted 0.2 mm CaCl2 and 3.0 mmMgSO4 in the ACSF.
For recordings, a slice was submerged in a chamber located on the fixed stage of an upright microscope (Zeiss Axioskop) and maintained at 34°C (ACSF flow rate, 3.5–4.0 ml/min). To allow for accurate placement of recording, stimulating, and drug pipettes, neurons were visualized with infrared differential interference contrast (IR-DIC, Nomarski) optics with a video camera (Hamamatsu Photonics) and monitor (Stuart et al., 1993). Whole-cell recordings (WCRs) were made from layer II-V neurons that had an obvious pyramidal shaped soma and ascending apical dendrite. Neurons were routinely filled with biocytin to confirm morphology.
Electrophysiological recording and stimulation. WCR pipettes were made from filamented glass capillary tubes (1.5 mm outer diameter; AM Systems, Seattle, WA) with tip diameters of ∼2.5 μm and DC resistances of 4–6 MΩ using a horizontal micropipette puller (Sutter Instrument Co., Novato, CA). The pipettes were filled with (in mm): 125.0 K-MeS03, 0.05 CaCl2, 0.5 NaCl, 2 Mg-ATP, 0.5 Na-GTP, 0.16 EGTA, 10 HEPES, and 0.3–0.5% biocytin. The pH was adjusted to 7.3 with KOH (1 m), and final osmolality was 270–280 mosmol/kg.
ACSF-filled pipettes with tip diameters of ∼5 μm were used for electrical stimulation of afferent fibers. The electrode was placed 40–150 μm (mean, 77 ± 2 μm; n = 131) lateral to the recording site. Stimulation consisted of monophasic constant current pulses at intensities subthreshold for spike generation (200 μsec, 5–100 μA).
WCRs were obtained using an intracellular amplifier (Axoclamp 2B, Axon Instruments, Foster City, CA). Seal resistance was 5–12 GΩ. Series resistance ranged from 3 to 25 MΩ, but most often was <12 MΩ and was compensated using the bridge balance control. Membrane potential (Vm) was not corrected for liquid junction potentials. Vm was monitored on an oscilloscope and chart recorder, digitized at 5.5 kHz, and stored on computer. Software (AXODATA, AXOGRAPH, Axon Instruments) controlled data acquisition and analysis.
AChE histochemistry. Whole brains or hemispheres from 8 to 24-d-old rats were placed in 4% paraformaldehyde for at least 4 d. After fixation, brains were blocked and 50 or 100 μm coronal sections through auditory cortex were made using a Vibratome (Polysciences, Warrington, PA). AChE histochemistry followed a modifiedKoelle and Friedenwald (1949) method. Slices were rinsed in 0.1m sodium acetate buffer and incubated in medium containing the substrate acetylthiocholine iodide (1.0 × 10−4m) and the nonspecific cholinesterase inhibitor tetraisopropylpyrophosphoramide (1.14 × 10−4m). After 3 d of incubation, slices were rinsed in phosphate buffer and developed in 1% ammonium sulfide for ∼30 sec. Slices were then rinsed, mounted on slides, and allowed to dry overnight. The next day, slices were dehydrated, cleared, and coverslipped.
Pharmacological agents and application. The following pharmacological agents were used: 6 cyano-7-nitroquinoxaline-2,3-dione (CNQX); (±)-2-amino-5-phosphonopentanoic acid (APV), methyllycaconitine citrate (MLA); NMDA, and (−)-nicotine di-d-tartrate (nicotine) (all from Research Biochemicals, Natick, MA), and picrotoxin (Sigma, St. Louis, MO). Drugs were prepared as a concentrated stock solution in distilled H2O or DMSO (for CNQX) and diluted to their final concentration with ACSF. Final concentration of DMSO in CNQX solution was 0.3%. Receptor antagonists were superfused for at least 10 min before initiating the experiment. When CNQX was applied by pressure pipette (only in Fig.1Aii), it was dissolved in ACSF alone (no DMSO).

Experimental design and pharmacological profile of EPSPs. Ai, Synaptic potentials were elicited by a single stimulus pulse every 10 sec. After control responses, pressure-ejected nicotine (N) was paired for several trials, with each pressure pulse beginning 5 msec before the stimulus. Responses for each condition depicted in figures are averages of three to five traces. Aii, Orthodromic stimulus (arrowhead) elicited a glutamatergic EPSP in a layer III pyramidal neuron. Pressure ejection of CNQX (100 μm, 50 msec beginning 5 msec before the stimulus) reduced the peak EPSP amplitude, demonstrating that pressure-ejected drugs reach the synapse sufficiently quickly to affect the peak EPSP. Receptor antagonists are bath-applied in all other figures. Vm, −70 mV.B, Glutamatergic EPSPs are produced by activation of AMPA/KA and NMDA receptors. Bi, The peak EPSP (i.e., early EPSP) was significantly reduced by CNQX (20 μm in bath), indicating the involvement of AMPA/KA receptors. The late EPSP was subsequently reduced by APV (50 μm; data not shown). Vm, −66 mV. Bii, In another cell, the late/descending slope of the EPSP (i.e., late EPSP) was reduced by APV (50 μm), indicating the involvement of NMDA receptors. Note that APV did not reduce the peak EPSP amplitude. Vm, −70 mV.
Nicotine was applied by either pressure ejection (0.5–25 μm, 10–200 msec pulses at 20 psi) from a pipette with a 5 μm tip, using a Picospritzer (General Valve, Fairfield, NJ), or superfused to induce receptor desensitization (see Fig. 5C). The nicotine-filled pipette was placed adjacent to the apical dendrite of the neuron, 7.6 ± 0.6 μm lateral and 24.6 ± 0.9 μm distal to the recording electrode (n = 119). The onset of the nicotine pulse generally preceded the afferent stimulus by 5 msec (Fig. 1Ai). This interval was not critical, and in control experiments nicotine administered 5–55 msec before the afferent stimulus produced similar increases in the late EPSP (similar increased amplitude at similar latencies) without affecting the peak (early) EPSP. In contrast, pressure ejection of CNQX pulsed 5 msec before the afferent stimulus reduced the peak EPSP by up to 50% (Fig.1Aii), confirming the presence of the drug at the neuron at the time of synaptic activation. The volume of nicotine ejected by a single pressure pulse (20–1500 pl) was estimated by ejecting nicotine into oil and measuring the resulting drop with a microscope reticule.
NMDA (50 mm, pH 8.0) was dissolved in distilled H2O and applied iontophoretically (10–30 nA; holding current, 5–10 nA). The iontophoretic pipette had a tip diameter of ∼1 μm and was typically placed adjacent to the apical dendrite of the neuron within 30 μm of the recording electrode at the soma.
Data analysis. Early EPSP amplitudes were measured at peak response. Late EPSP amplitudes were measured at the point of greatest deviation from control after nicotine pulses (range, 50–150 msec) or at 56 msec for neurons with no nicotine effect. All traces are averages of three to five responses taken at 10 sec intervals (Fig.1Ai). Mean data are presented ±1 SE. Differences between means were evaluated for statistical significance using thet test for paired samples. Correlation coefficients were calculated using the product-moment method. Differences were considered statistically significant if the probability of occurrence by chance was ≤0.05.
RESULTS
Data were obtained from 159 pyramidal neurons in slices from rats ranging in age from 8–24 d. Neurons were located between 175 and 825 μm from the pia (mean, 433 ± 11 μm), corresponding to layers II-V of rat auditory cortex (Roger and Arnault, 1989). More than 80% of neurons were located in layers II/III and IV. For comparison purposes, we refer to P8–16 rats as juvenile and P19–24 rats as mature. Neurons obtained from juvenile rats had a resting Vm of −66.8 ± 1.0 mV (n = 141) and input resistance (Ri) of 222.6 ± 6.6 MΩ (n = 130; Ri measured within 5 min of establishing WCR). Neurons from mature rats had similar Vm(−70.4 ± 1.5 mV; n = 18; p > 0.05) and lesser Ri (97.9 ± 8.6 MΩ;n = 18; p < 0.01). The cells included in this report were regular-spiking pyramidal neurons as judged from examination of biocytin-filled neurons, i.e., filled neurons exhibited pyramidal shaped cell bodies with a large apical dendrite extending toward the pia and standard electrophysiological criteria, i.e., spike duration and spike frequency adaptation (Connors et al., 1982;McCormick et al., 1985). Fast-spiking cells were occasionally encountered and are not included in the present report.
Glutamate receptors mediate stimulus-evoked EPSPs
Electrical stimulation adjacent to the recorded neuron elicited a compound glutamatergic EPSP mediated by activation of AMPA/KA and NMDA receptors, similar to cortical EPSPs described previously (Jones and Baughman, 1988; Sutor and Hablitz, 1989a,b; Cox et al., 1992). The peak of the EPSP (i.e., the early EPSP) was reduced by the AMPA/KA receptor antagonist CNQX (Fig. 1Bi), and the residual slow EPSP was largely blocked by the NMDA receptor antagonist APV (data not shown; see Fig. 7). The late/descending phase of the EPSP (i.e., the late EPSP) was reduced by APV (Fig. 1Bii), and the residual response was largely blocked by CNQX (data not shown). Thus, AMPA/KA and NMDA receptors mediate separate, although overlapping, components of glutamatergic EPSPs in rat auditory cortex, as reported previously (Cox et al., 1992; Metherate and Ashe, 1994; Aramakis et al., 1997).
Developmental expression of AChE
AChE is the degradative enzyme for ACh. In rat auditory cortex, increased AChE expression in layers I and III-IV begins on P3, reaches peak intensity at P8–10, and declines to low (adult) levels by P23 (Robertson et al., 1991). We examined the pattern of AChE expression in coronal sections from P8–24 rats, the same age range used to study nicotinic effects on synaptic activity. In many cases, slices for physiological experiments and for AChE histochemistry were obtained from opposite hemispheres of the same brain (discussed below). Slices were rated with respect to the presence or absence of a band of AChE histochemical reaction product (dark brown product) in middle layers of auditory cortex. Confirming earlier reports, we found prominent bands of AChE reaction product in auditory cortex of rats aged P8–16 (Fig.2). There was an age-dependent decrease in the frequency of AChE expression (r = −0.91), such that the AChE band was observed in 100% of auditory cortical slices from P8–12 brains, 70% of slices at P13–14, and 60% of slices by age P15–16 d (Fig. 2B). The AChE band rarely appeared at P19–24 (Fig. 2). Thus, AChE is transiently overexpressed in the middle layers of auditory cortex during postnatal development.
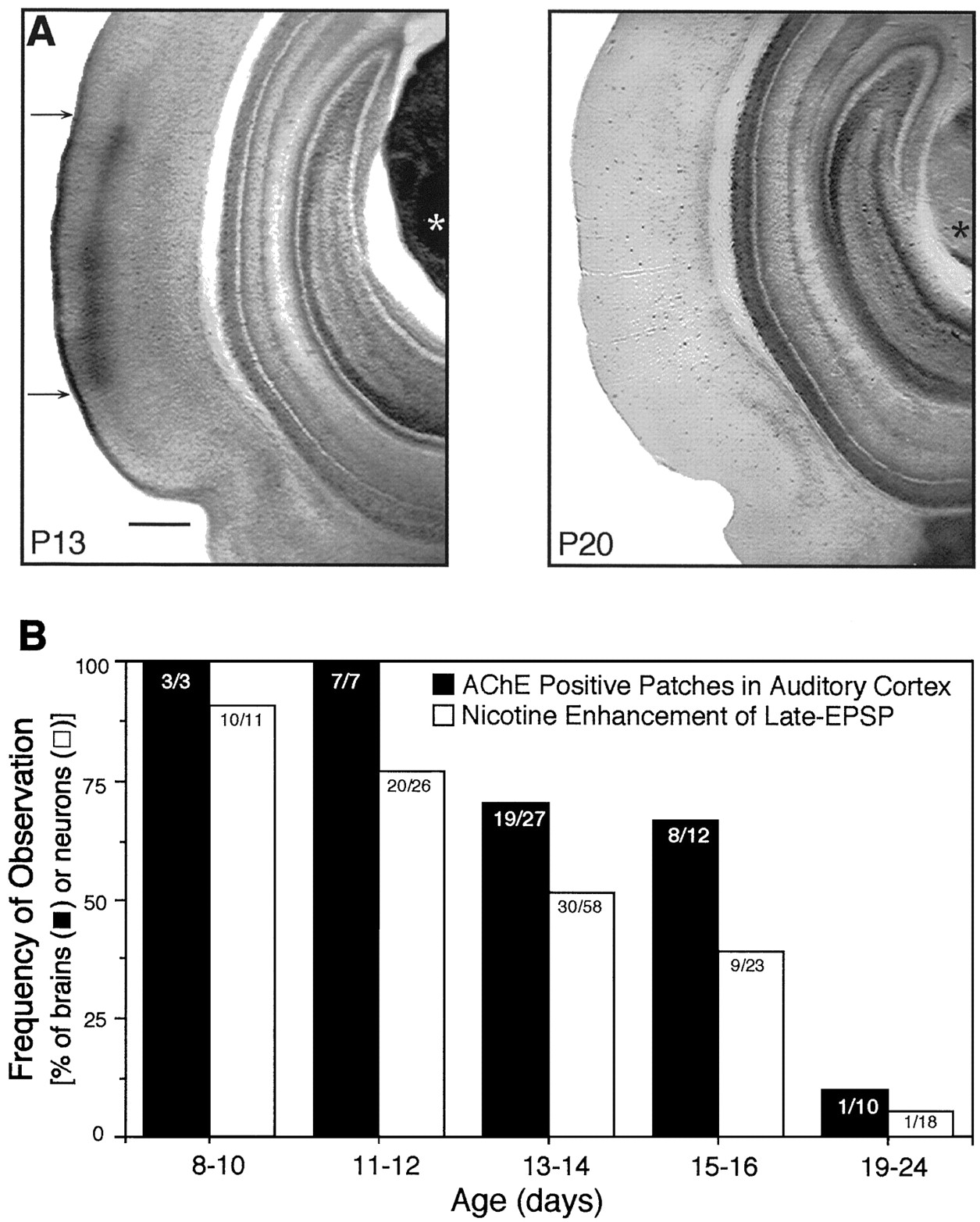
Age-dependent expression of AChE patches and responsiveness of neurons to nicotine. A, Dense AChE staining (between arrows) occurred in layers I and III/IV of primary auditory cortex and in the auditory thalamus (*) of a P13 rat but disappeared by P20. Scale bar, 500 μm. B, The frequency of slices with AChE-positive patches declined with increasing age (correlation coefficient r = −0.91), as did the frequency of neurons whose synaptic activity was modified by nicotine (r = −0.98). Correlation coefficients were calculated using the product-moment method. Number of observations indicated within each column.
Nicotine enhances the late EPSP in slices from juvenile rats
To investigate the role of nAChRs during the period of enhanced expression of cholinergic markers (AChE and α7 nAChR), we paired microapplication of nicotine with synaptic activation in slices from juvenile rats. Pulses of nicotine immediately preceding the afferent stimulus selectively enhanced the amplitude and duration of the late EPSP without affecting the peak amplitude of the early EPSP (Fig.3A, n = 69 of 118 neurons). The effect of nicotine was dose-dependent (Fig.3A,B), dissipated quickly (an afferent stimulus 10 sec later elicited a recovered EPSP), and in general was easily repeatable at 10 sec intervals. Membrane depolarization increased the enhancement of the late EPSP by nicotine (Fig. 3C, n = 14), which, given the voltage dependence of the NMDA receptor (Mayer et al., 1984), could reflect an increased contribution of NMDA receptors to the EPSP. In a minority of neurons (13 of 69), the late EPSP was greatly enhanced to the point of generating multiple action potentials. In these neurons replication required longer periods between applications (>5 min), which may reflect recovery from nAChR desensitization.

Nicotine selectively enhanced the late EPSP.A, Afferent stimulation elicited an EPSP (black traces) in a P16 pyramidal neuron. Pressure ejection of nicotine (25 μm, 20–40 msec pulse) produced a dose-dependent increase in the magnitude of the late EPSP without affecting the amplitude of the early EPSP (gray traces). Afferent stimuli delivered at 10 sec intervals; control traces were obtained between nicotine doses. Vm, −66 mV. B, Nicotine produced a dose-dependent increase in the late EPSP (•; n = 47; p < 0.05), but did not affect the amplitude of the early EPSP (○; p > 0.05). Average Vm, −67.8 ± 0.8 mV; neurons from P8–16 animals. C, Membrane depolarization increased the amplitude of the late EPSP (•, n = 14) and the degree to which nicotine enhanced the late EPSP (○).
Effect of nicotine on pharmacologically isolated glutamatergic EPSPs
Because nicotine appeared to differentially modify the early and late EPSP, we determined if the effect could be differentially affected by selective antagonists of NMDA or AMPA/KA receptors. In a subset of nine neurons, nicotine enhanced the late EPSP to 210.9 ± 20.9% of its control amplitude without affecting the amplitude of the early EPSP. These cells were further tested for nicotinic effects in the presence of APV or CNQX. APV (50 μm) reduced the amplitude of the late EPSP and prevented enhancement of the late EPSP by nicotine (Fig. 4A,C; area of EPSP in APV + nicotine condition was 102.6 ± 7.3% of response in APV alone; p > 0.10; n = 5). Conversely, CNQX (20 μm) reduced the amplitude of the early EPSP but did not prevent the effect of nicotine (Fig.4B,C; area of EPSP in CNQX + nicotine was 214.5 ± 35.9% of response in CNQX alone; p < 0.01;n = 4). Thus, nicotine selectively enhanced the NMDA receptor-mediated synaptic response.

Effect of nicotine on pharmacologically isolated EPSPs. A, Nicotine (25 μm, 20–30 msec pulse) produced a dose-dependent increase in late EPSP magnitude (Control; P13 pyramidal neuron). Pharmacological isolation of the AMPA/KA receptor-mediated early EPSP with APV (50 μm) revealed no effect of nicotine. Recovery followed wash-out of APV. Vm, −70 mV. B, The NMDA receptor-mediated late EPSP was isolated with CNQX (20 μm) and was enhanced by nicotine (25 μm, 30 msec). Recovery from CNQX was not attempted. Neuron from P16 rat; Vm, −64 mV. C, In nine cells with nicotine-induced enhancement of the late EPSP, the effects of nicotine were subsequently tested on pharmacologically isolated EPSPs (neurons from P8–16 animals). Nicotine had no effect on the isolated early EPSP (102.6 ± 7.3% of EPSP area in APV alone; p> 0.10; n = 5) but significantly increased the isolated late EPSP (to 214.5 ± 35.9% of EPSP area in CNQX alone;p < 0.01; n = 4).
Involvement of presynaptic α7 nAChRs
The transient increase of cholinergic markers in developing sensory neocortex includes heightened expression of the α7 nAChR subunit (Broide et al., 1996), which confers high Ca2+ permeability (Séguéla et al., 1993) and can mediate presynaptic enhancement of transmitter release (for review, see Role and Berg, 1996; Wonnacott, 1997). The α7 nAChRs can also mediate postsynaptic depolarizations produced by the influx of Na+ and Ca2+ (Zhang et al., 1994). Several findings indicate that nicotine enhances the NMDA receptor-mediated EPSP by acting presynaptically at α7 nAChRs.
First, we tested the effects of the selective α7 nAChR antagonist MLA on nicotinic enhancement of EPSPs. MLA (5 nm) did not affect evoked EPSPs but did block nicotine-induced enhancement of the late EPSP (Fig. 5A,B;n = 5). Because an additional characteristic of α7 nAChRs is their rapid desensitization on exposure to agonist (decay time constants range from several milliseconds to several seconds) (Castro and Albuquerque, 1993; Zhang et al., 1994), we also evaluated the effect of prolonged exposure to nicotine on glutamatergic synaptic transmission. Bath application of nicotine (0.3–10 μm) had no effect on membrane potential or on evoked EPSPs (n = 9 of 10). Further, application of even low nicotine doses (0.3–0.5 μm) for several minutes in the bath largely prevented the effects of pressure-ejected nicotine pulses (Fig. 5C,D; n = 5). Thus, exposure to superfused nicotine for several minutes, which likely produced significant receptor desensitization (Castro and Albuquerque, 1993;Zhang et al., 1994), precluded enhancement of the late EPSP by pulsed nicotine. These findings further implicate the α7 nAChR subunit in the actions of nicotine.

The effect of nicotine was blocked by α7 nAChR antagonism and by persistent exposure to nicotine. A, Nicotine-induced (25 μm, 40 msec) enhancement of the late EPSP in a P8 neuron (top traces) was blocked by superfusion of the α7 receptor antagonist MLA (5 nm,bottom traces). Vm, −53 mV.B, In five neurons (age range, P8–13), nicotine enhancement of the late EPSP (to 188.1 ± 27.9% of control amplitude; p < 0.01) was prevented by MLA (5 nm; 95.5 ± 6.0% of control; p > 0.10). C, Enhancement of the late EPSP by pressure-pulse application of nicotine (25 μm, 30 msec; top traces) was prevented by bath application of a low concentration of nicotine (0.3 μm for 3 min,bottom traces). Vm, −68 mV.D, In five neurons (age range, P8–12), pulsed nicotine enhancement of the late EPSP (to 210.8 ± 37.1% of control;p < 0.05) was prevented by exposure to superfused nicotine (0.3–0.5 μm; 112.5 ± 8.5% of control;p > 0.10). Nicotine-induced enhancement was restored on washing for 6–8 min (220.5 ± 61.2%;n = 3).
Additional findings indicate a presynaptic locus for the action of nicotine. Because nicotine selectively enhanced the NMDA receptor-mediated late EPSP, we determined its effects on postsynaptic depolarizations elicited in response to iontophoretic application of the agonist NMDA. These experiments were performed with synaptic transmission blocked in low Ca2+/high Mg2+ ACSF. The results showed that nicotine, whether pulsed at the beginning of the NMDA application or at the peak of the NMDA-mediated depolarization, had no affect on the postsynaptic depolarization (Fig.6A,B, n= 6; p > 0.10). Thus, nicotinic enhancement of the NMDA receptor-mediated late EPSP is not likely to involve a postsynaptic interaction.

Nicotine did not exert direct postsynaptic effects. A, Iontophoretic application of NMDA (15 nA, 3 sec; 50 mm) to a neuron from a P10 rat produced repeatable membrane depolarizations in low Ca2+/high Mg2+ ACSF. Concomitant pulse application of nicotine (25 μm; 20–100 msec) did not alter the magnitude of NMDA-induced responses. Vm, −70 mV.B, In six neurons (age range, P8–10), nicotine did not affect the amplitude of NMDA-induced membrane depolarizations, which averaged 11.7 ± 3.0 mV (peak depolarization) in the control condition and 10.7 ± 3.0 mV with a 50 msec pulse of nicotine (p > 0.10). C, Pulsed nicotine (marked by rectangle under baseline) in the absence of electrical afferent stimulation resulted in a small amplitude membrane depolarization. The nicotine-induced depolarization was prevented by superfusion of i, APV (50 μm, Vm, −61 mV); or ii, MLA (5 nm, Vm, −53 mV); but notiii, CNQX (20 μm, Vm, −61 mV). D, The nicotine-induced depolarization of Vm was voltage-sensitive and of greatest amplitude at depolarized potentials (n = 10 neurons; age P10–13).
Further evidence for a presynaptic locus of action came from the effects of nicotine pulses administered alone, without afferent stimulation. Nicotine pulsed alone (at doses higher than those required to modify the late EPSP) often produced a slow, small amplitude postsynaptic depolarization (0.5–3.5 mV; Fig. 6C, control traces; n = 31). The amplitude of the depolarization increased upon steady membrane depolarization (Fig.6D; n = 10), and the response was blocked by APV (Fig. 6Ci; n = 2), indicating the involvement of NMDA receptors. The depolarization did not occur in low Ca2+/high Mg2+ ACSF (n = 2), suggesting that it was not a direct postsynaptic response to nicotine. Finally, the depolarization was also blocked by MLA (Fig. 6Cii; n = 2) but not CNQX (Fig. 6Ciii; n = 2).
Taken together, the data thus far indicate that nicotine acts presynaptically at α7 nAChRs to enhance, or even elicit, glutamate release and that the released glutamate acts at postsynaptic NMDA receptors but not at AMPA/KA receptors.
Nicotine does not enhance EPSPs in slices from mature rats
The percentage of neurons that were sensitive to nicotine declined with increasing age (Fig. 2B). Nicotine enhanced the late EPSP in 91% of neurons recorded from P8–10 rats but, in contrast, did not modify either the early or late EPSP in 94% of neurons from P19–24 animals (Fig.7A,B). Although maturation of cortical inhibition could potentially suppress NMDA receptor-mediated activity (Luhmann and Prince, 1991; Agmon and O’Dowd, 1992) and mask the effects of nicotine, pharmacological blockade of inhibition and isolation of NMDA receptor-mediated EPSPs in slices from mature animals revealed no effect of nicotine (Fig.7A; n = 3).

Nicotine did not enhance EPSPs in the mature neocortex. A, Nicotine (10 μm, 50 msec) did not modify the EPSP in a P20 neuron. Superfusion of CNQX (20 μm) and picrotoxin (10 μm) isolated the late EPSP, which remained unaffected by nicotine (middle trace) and was blocked by APV (50 μm;lower trace). Increasing the stimulus intensity resulted in a larger (11 mV) isolated late EPSP in CNQX and picrotoxin that also was not affected by nicotine (data not shown). Vm, −77 mV. B, Nicotine did not affect EPSPs in P19–24 neurons (n = 18; p values > 0.10). Average Vm, −70.4 ± 1.5 mV.
The decrease in the efficacy of nicotine therefore paralleled the decrease in anatomical markers of cholinergic function (Fig.2B). In particular, when measurements of AChE expression and nicotine actions were obtained from opposite hemispheres of the same brain, 94% (17 of 18) of brains with AChE patches had positive nicotine effects, and 86% (6 of 7) of brains without AChE patches also lacked nicotine effects. Thus, nicotinic enhancement of synaptic activity correlates with the heightened expression of cholinergic markers in the juvenile auditory cortex.
DISCUSSION
We have investigated the influence of nAChRs on glutamatergic transmission in developing auditory neocortex. Nicotine enhanced the late component of glutamatergic EPSPs that is largely mediated by NMDA receptors but did not affect the early component mediated by AMPA/KA receptors. Furthermore, the effect of nicotine appears to involve presynaptic α7 nAChRs, implying enhanced release of glutamate at synapses in which NMDA, but not AMPA/KA, receptors are found postsynaptically. Finally, the effect occurred during a postnatal period of enhanced expression of cholinergic markers and not in the mature cortex. The synergistic actions involving nAChRs and NMDA receptors during early postnatal life may contribute to experience-dependent synaptic plasticity in sensory neocortex.
Nicotinic AChRs regulate synaptic transmission in the CNS
Although there is little evidence for the involvement of nAChRs in direct mediation of synaptic transmission in the brain (but see Zhang et al., 1993; Feller et al., 1996; Roerig et al., 1997), a prominent role may be presynaptic modification of neurotransmitter release (for review, see McGehee and Role, 1996; Wonnacott, 1997). This has been clearly documented for nicotinic enhancement of dopaminergic transmission, which may be an important mechanism underlying the addictive properties of nicotine (for review, see Stolerman and Shoaib, 1991; Dani and Heinemann, 1996). More recently, nicotinic enhancement of glutamatergic transmission has been observed in several brain areas (McGehee et al., 1995; Alkondon et al., 1996; Guo et al., 1998), including the hippocampus (Gray et al., 1996) and neocortex (Vidal and Changeux, 1993), in embryonic (McGehee et al., 1995; Guo et al., 1998), cultured (Alkondon et al., 1996), and adult tissue (Gray et al., 1996;Vidal and Changeux, 1993). However, in these studies, nicotine enhanced glutamatergic transmission mediated by AMPA/KA receptors. Hence, our findings demonstrate a novel interaction between nAChR and NMDA receptors that occurs in developing sensory neocortex.
Locus of relevant nicotine and NMDA receptors
The evidence indicates that nicotine acts presynaptically to enhance glutamate release at synapses in which only NMDA receptors are found postsynaptically. Nicotine did not affect postsynaptic depolarizations produced by iontophoretic application of the agonist NMDA (experiments done with synaptic transmission blocked), precluding a postsynaptic interaction between the two receptors and implying that nicotine normally acts presynaptically to enhance glutamate release. Consistent with this interpretation, nicotine administered by itself at higher doses produced postsynaptic depolarizations, but those apparently resulted from activation of postsynaptic NMDA receptors. Thus, nicotine appeared to enhance, or at higher doses elicit, glutamate release.
These results imply that some glutamatergic synapses in developing auditory cortex use only NMDA receptors (see also, Bekkers and Stevens, 1989; Kang, 1995) and that presynaptic nAChRs are restricted to these synapses. The existence of pure NMDA synapses has been proposed for several brain regions during postnatal development in which they may play a crucial role in activity-dependent refinement of synapses (Liao et al., 1995; Durand et al., 1996; Wu et al., 1996; Isaac et al., 1997). Although it is possible that activation of postsynaptic nAChRs could enhance NMDA receptor function (Broide et al., 1996), this does not appear to underlie the present findings.
Involvement of α7 nAChRs
Nicotinic enhancement of the late EPSP involves activation of α7 nAChRs because the effect was blocked by the specific receptor antagonist MLA. Expression of α7 nAChRs in primary sensory cortex is enhanced during postnatal development and declines as the brain matures (Broide et al., 1995, 1996), in parallel to AChE expression (Robertson et al., 1991; this study) and to the actions of nicotine described here. In addition, α7 nAChRs are highly permeable to Ca2+ (Mulle et al., 1992; Séguéla et al., 1993) and could mediate enhanced presynaptic release of neurotransmitter (McGehee et al., 1995; Alkondon et al., 1996; Gray et al., 1996; Coggan et al., 1997; for review, see McGehee and Role, 1996;Wonnacott, 1997). α7 nAChRs desensitize rapidly on exposure to agonist (Castro and Albuquerque, 1993; Zhang et al., 1994), which may explain why bath application of nicotine did not enhance EPSPs and, in fact, prevented enhancement by pulsed nicotine. [Note, however, thatGil et al. (1997) observed MLA-sensitive enhancement of cortical EPSPs with bath application of nicotine. It is possible that our bath flow rate, 3.5–4.0 ml/min, was slower than that used by Gil et al. (1997)(flow rate unreported), and allowed greater time for desensitization]. Finally, neurotrophic roles have been proposed for α7 nAChRs in early developmental processes such as synapse formation and neurite retraction (Pugh and Berg, 1994; Zheng et al., 1994; for review, seeRole and Berg, 1996). Thus, activation of α7 nAChRs may be important for development of glutamate-releasing synapses in sensory neocortex.
Implications for neocortical development
The effectiveness of nicotine in modifying cortical EPSPs parallels heightened expression of AChE and α7 nAChRs in developing sensory cortex. AChE is manufactured and transported to the neocortex by developing thalamocortical neurons, and it delineates their termination zone (Robertson et al., 1991; De Carlos et al., 1995). The most intense AChE and α7 nAChR expression occurs in the first two postnatal weeks, during the ingrowth, branching, and synaptogenesis of thalamocortical axons (Blue and Parnavelas, 1983a,b). Because thalamocortical neurons are glutamatergic (Kaneko and Mizuno, 1988;Kharazia and Weinberg, 1994; Salt et al., 1995) rather than cholinergic, the presence of cholinergic markers on their terminals may indicate their sensitivity to ACh released from ingrowing basal forebrain cholinergic afferents (for review, see Wainer and Mesulam, 1990). A cholinergic function for transiently expressed AChE in thalamocortical development has been questioned, because the arrival of basal forebrain afferents occur at least 1 d after the arrival of thalamic afferents (Robertson and Yu, 1993). However, both afferent systems are in place by the second postnatal week, during which time period we observe the modulatory influence of nAChRs on NMDA receptor-mediated EPSPs. Thus, cholinergic influences may not guide ingrowing thalamocortical axons, but may regulate subsequent synaptic development.
Although we describe a possible scenario for nAChR modulation of thalamocortical transmission, excitatory corticocortical synapses are also glutamatergic (Salt et al., 1995; for review, see Nieuwenhuys, 1994) and, hence, are potential candidates for regulation by nAChRs. Importantly, Gil et al. (1997) recently reported that nicotine enhances the glutamatergic EPSP elicited by thalamic, but not intracortical, stimulation in somatosensory cortex. Although specific glutamate receptor involvement was not determined, these data indicate that nAChRs may preferentially regulate thalamocortical development.
A potential mechanism for experience-dependent plasticity
During postnatal development, it is thought that behavioral experience shapes neural circuits through activity-dependent synaptic plasticity (for review, see Collingridge and Singer, 1990; Scheetz and Constantine-Paton, 1994). Some forms of developmental plasticity are reduced or prevented by disruption of either cholinergic or glutamatergic function (Bear and Singer, 1986; Kleinschmidt et al., 1987; Fox et al., 1996; Zhu and Waite, 1998). Further, cholinergic denervation of neocortex in young animals by specific immunolesions was shown to decrease cortical size and to retard the development of cortical pyramidal neurons (Hohmann et al., 1991; Robertson et al., 1998). However, pharmacological manipulations that prevent developmental plasticity do not appear to inhibit the ingrowth of thalamocortical axons into correct thalamorecipient layers (Kleinschmidt et al., 1987; Schlaggar et al., 1993; Robertson et al., 1998). Instead, there may be a disruption in the refinement of thalamocortical connectivity (Fox et al., 1996). Thus, cholinergic modulation of glutamatergic function may contribute to synaptic maturation that occurs after the initial establishment of thalamic projections.
For rat auditory cortex, the period of maximal expression of cholinergic markers and susceptibility to modulation by nAChRs corresponds to the time of hearing onset, as measured by scalp and cochlear potentials (Jewett and Romano, 1972; Uziel et al., 1981). The cochlear microphonic can be recorded as early as P8 and is within the adult range by P17 (for review, see Rubsamen and Lippe, 1998). Thus, the initiation and maturation period of audition corresponds to the period of heightened cholinergic function in auditory neocortex.
We propose that synergistic actions between cholinergic and glutamatergic transmission contribute to experience-dependent refinement of cortical circuits during development of the auditory system. During this period, significant behavioral (acoustic) events could evoke ACh release in auditory cortex (for review, see Richardson and DeLong, 1991; Weinberger, 1993), which could activate presynaptic nAChRs to selectively enhance NMDA receptor-mediated synaptic potentials via the mechanisms described here. Enhanced NMDA receptor-mediated EPSPs could then trigger synaptic plasticity (for review, see Collingridge and Singer, 1990; Scheetz and Constantine-Paton, 1994) to establish and/or refine cortical circuits to be used throughout life. After the critical period for experience-dependent plasticity, α7 nAChRs in the middle cortical layers would be downregulated and would no longer modify NMDA synapses. Our findings may extend to the development of sensory cortex in general because cholinergic markers are transiently overexpressed in each primary sensory cortical area (Prusky et al., 1988; Robertson et al., 1991; Broide et al., 1995).
Implications for developmental dysfunction
A potential implication for developmental disorders stems from the finding that nicotine exposure for several minutes precludes enhancement of NMDA receptor-mediated synaptic activity, most likely as a result of nAChR desensitization. If the synergistic action of ACh and NMDA receptors contributes to the refinement of cortical circuits, then exposure to exogenous nicotine would disrupt this process. Because the concentrations of bath-applied nicotine (0.3–0.5 μm) used to induce receptor desensitization in our experiments are similar to levels found in the bloodstream of cigarette smokers (Henningfield et al., 1993), it is possible that exogenous nicotine can impact cortical development. AChE patches are transiently observed in auditory cortex of the human fetus (Krmpotic-Nemanic et al., 1980, 1983), suggesting a similar critical period as in the postnatal rat. It may be relevant that auditory processing is impaired in children exposed prenatally to nicotine via cigarette smoke (McCartney et al., 1994). Although any relationship between the effects of secondhand smoke and brain development remains speculative, future research must determine the developmental function of interactions between nicotinic ACh and NMDA receptors and the degree to which this can be disrupted by exogenous nicotine.
Footnotes
This work was supported by the National Science Foundation (Grant IBN 9510904) and National Institute on Deafness and Other Communication Disorders (Grant DC02967). We thank Ms. N. Patel for her assistance with the AChE histochemistry. Thanks also to Drs. S. Cruikshank, R. Frostig, H. Killackey, R. Robertson, and I. Soltesz and Ms. C. Hsieh for helpful discussions and comments on this manuscript.
Correspondence should be addressed to Dr. Raju Metherate, Department of Psychobiology, University of California, Irvine, 2205 Biological Sciences II, Irvine, CA 92697-4550.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}