

Abstract
Human ingestion of “chickling peas” from the plantLathyrus sativus, which contains an excitatory amino acid, l-BOAA (l-β-N-oxalylamino-l-alanine), leads to a progressive corticospinal neurodegenerative disorder, neurolathyrism. Exposure to l-BOAA, but not its optical enantiomer d-BOAA, causes mitochondrial dysfunction as evidenced by loss of complex I activity in vitro in male mouse brain slices and in vivo in selected regions of mouse CNS (lumbosacral cord and motor cortex). Loss of complex I activity in lumbosacral cord after l-BOAA administration to mice was accompanied by concurrent loss of glutathione. The inhibited complex I activity in mitochondria isolated from lumbosacral cord of animals treated with l-BOAA rebounded after incubation with the thiol-reducing agent dithiothreitol, indicating that oxidation of protein thiols to disulfides was responsible for enzyme inhibition. The inhibition of complex I could be abolished by pretreatment with antioxidant thiols such as glutathione ester and α-lipoic acid. Chronic treatment of male mice, but not female mice, withl-BOAA resulted in loss of complex I activity and vacuolation and dendritic swelling of neurons in the motor cortex and lumbar cord, paralleling the regionality of the aforementioned biochemical effects on CNS mitochondria. These results support the view that thiol oxidation and concomitant mitochondrial dysfunction (also implicated in other neurodegenerative disorders), occurring downstream of glutamate receptor activation by l-BOAA, are primary events leading to neurodegeneration. Maintenance of protein thiol homeostasis by thiol delivery agents could potentially offer protection against excitotoxic insults such as those seen withl-BOAA.
- excitatory amino acids
- mitochondrial electron transport
- NADH ubiquinone-1 oxidoreductase (complex I)
- brain
- l-BOAA
- oxidative stress
- glutathione
- protein thiol oxidation
l-β-Oxalylaminoalanine (l-BOAA) is a naturally occurring nonprotein amino acid, first isolated from chickling pea obtained from the plantLathyrus sativus (Rao et al., 1964) grown in the drought-prone areas of Africa and Asia. In humans, ingestion of the chickling pea as a staple diet during famine results in a slowly progressive, neurodegenerative condition known as neurolathyrism (Selye, 1957), a form of motor neuron disease. l-BOAA is generally considered to be the causative agent in neurolathyrism (Spencer et al., 1986). Neurolathyrism in humans is characterized by spastic paraparesis that predominantly targets the Betz cells and the corticospinal tracts (Ludolph and Spencer, 1996). It is a highly prevalent neurotoxic disorder (Haimanot et al., 1990) that predominantly affects males (Roy, 1988). Clinical (Cohn and Streifler, 1981) and neuropathological studies (Streifler et al., 1977) have shown the involvement of upper motor neurons, degeneration of anterior horn cells, and loss of axons in the pyramidal tracts in the lumbar spinal cord in humans affected by neurolathyrism.
l-BOAA exposure was first shown to cause seizures in newborn mice in vivo and degeneration in CNS explant cultures in vitro (Ross and Spencer, 1987), effects that were abrogated in dose-dependent manner by known antagonists of a subclass of glutamate receptor, namely, quisqualic receptors. The unique action of l-BOAA on AMPA receptors was subsequently identified (Ross et al., 1989), and the ability of the specific AMPA receptor antagonist NBQX (2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline) to preventl-BOAA-induced neuronal damage confirmed thatl-BOAA exerted its action through the AMPA receptors (Pearson and Nunn, 1991). In synaptic membrane preparations,l-BOAA prevented the binding of [3H]AMPA, and the ability of l-BOAA to displace [3H]AMPA from membranes prepared from cerebral cortex exceeded that of quisqualic acid or AMPA itself (Bridges et al., 1988; Ross et al., 1989). Although it is generally accepted that l-BOAA causes neurodegeneration through excitotoxic mechanisms, probably involving the AMPA receptors, the actual molecular mechanisms involved in l-BOAA-induced neurotoxicity generally remain uncharacterized.
Mitochondrial dysfunction has been implicated in a variety neurodegenerative disorders. Abnormalities in complex I have been identified in mitochondria from platelet, brain, and muscle of Parkinson’s disease patients (Parker et al., 1989; Mizuno et al., 1995), and cytochrome c oxidase deficiency has been observed in Alzheimer’s disease brain (Parker et al., 1994). The present studies indicate that an inhibition of mitochondrial complex I activity mediated through oxidation of thiol groups in distinctive regions of the CNS is a primary event in thiol-reversible,l-BOAA-induced toxicity.
MATERIALS AND METHODS
Materials.l-BOAA, l-BMAA, AMPA, kainic acid, quisqualic acid, and MK-801 were obtained from Research Biochemicals (Natick, MA). NMDA, N-acetyl aspartyl glutamate (NAAG), and glutamic acid were obtained from Sigma (St. Louis, MO). d-BOAA was procured from Tocris Neuramin and as a gift from Dr. S. L. N. Rao, Osmania University, Hyderabad, India. NBQX and ubiquinone-1 were gifts from Novo Nordisk and Eisai Pharmaceutical Company, respectively. Glutathione isopropyl ester (GSH ester) was obtained as a gift from Yamanouchi Pharmaceutical Company. All other chemicals and reagents were of analytical grade and were obtained from Sigma or Qualigens.
Animals. Male and female Swiss albino mice (3–4 months old; 25–30 gm) or male Wistar rats (3–4 months old, 200–250 gm) were obtained from National Institute of Mental Health and Neurosciences (NIMHANS) Animal Research Facility. Animals had access to pelleted diet (Lipton, Calcutta, India) and water ad libitum.
In vitro studies. Sagittal slices of male mouse or rat brain were prepared using the previously described apparatus developed in our NIMHANS laboratory (Pai et al., 1991). Animals were decapitated, and the brains were quickly removed and kept on a flat surface at 4°C. The slicer was placed above the brain with equal number of blades on either side of the median plane and gently pressed. The slices were carefully transferred using a flat-edged needle to a beaker containing artificial CSF (ACSF). Four slices of uniform thickness (1 mm) were prepared from each mouse brain in a time not exceeding 1–2 min. Slices were incubated in ACSF, pH 7.4, containing sodium chloride (122 mm), potassium chloride (3.1 mm), calcium chloride (1.3 mm), magnesium sulfate (1.2 mm), glucose (10 mm), and glycyl glycine (30 mm) in an oxygen-enriched atmosphere as given by McIlwain (1975) with or without l-BOAA for 1 hr at 37°C.
In experiments performed to evaluate the effects of antioxidant thiols such as glutathione isopropyl ester (1 mm) or α-lipoic acid (100 μm); slices were preincubated with the agents for 30 min at 37°C, before exposure tol-BOAA (1 nm) for 1 hr. To assess the protective effects of glutamate receptor antagonists onl-BOAA-mediated neurotoxicity, sagittal slices of male mouse brain were prepared from animals dosed 4 hr earlier with NBQX (30 mg/kg body weight, i.p.) and incubated without or withl-BOAA (1 nm).
In vivo studies.l-BOAA was dissolved in normal saline (5 or 10 mg/kg body weight, s.c.) and administered to male and female Swiss albino mice. Control animals received vehicle alone. The animals were killed at specified time points, and the brain and spinal cord were removed. Kidney, heart, lung, and the main lobe of the liver were also removed for certain experiments. All dissections and tissue handling were performed at 4°C. The motor cortex (Fig. 1) was dissected out from the brain. The spinal cord was exposed, and the thoracic cord was collected. The lumbar and sacral segments of the cord were dissected out together and used for experiments. In some experiments, the cortex was dissected into three segments, namely, the frontal and parietal cortex, temporal cortex, and occipital cortex, following anatomical landmarks. In experiments performed to assess the effect of the AMPA receptor antagonist NBQX, mice were treated with NBQX (30 mg/kg body weight, i.p., in saline). After 4 hr, l-BOAA (10 mg/kg body weight, s.c., in saline) was administered. Control animals received vehicle alone. In some experiments, the thiol antioxidant α-lipoic acid (20 mg/kg body weight, s.c) was given 0.5 hr beforel-BOAA. All animals were killed 24 hr afterl-BOAA injection, and the lumbosacral cord was dissected.

The region from the mouse brain cortical layer (shaded area) that was dissected out as motor cortex for measurement of enzyme activity in all subsequent experiments
l-BOAA was also administered chronically (5 mg/kg body weight, s.c., daily) to male and female mice, and animals were killed after 5, 10, 20, or 40 d. After 40 d, l-BOAA administration was discontinued and the animals were killed on the 55th day after the first dose of l-BOAA. The motor cortex, thoracic, and lumbosacral segments of the spinal cord were collected for assay of complex I.
Processing of tissue. The tissues (brain slices and cortical regions of mouse brain or spinal cord segments or other tissue) were processed immediately for the assay of activities of complex I, complex II, complex II-III, complex IV, and concentration of glutathione. For the assay of mitochondrial electron transport complexes, tissues were homogenized in sucrose (0.25 m) and centrifuged at 1000 × g for 10 min to obtain a postnuclear supernatant, which was recentrifuged at 17,000 × g for 20 min to obtain a crude mitochondrial pellet. The pellet was resuspended in 0.25 m sucrose and freeze-thawed three times before assay of complex I (Shults et al., 1995), complex II (Hatefi and Stiggall, 1976), complex II-III (King, 1967), or complex IV (Gibson and Hill, 1983).
Assay of NADH: ubiquinone oxidoreductase (complex I) activity. Complex I activity was assayed in brain mitochondrial preparations as rotenone-sensitive NADH:ubiquinone-1 oxidoreductase according to Shults et al. (1995) with minor modifications as described below. The assay was performed in phosphate buffer (35 mm, pH 7.2) containing sodium cyanide (2.65 mm), magnesium chloride (5 mm), EDTA (1 mm), bovine serum albumin (1 mg/ml), and antimycin (2 μg/ml). Brain mitochondria (90–150 μg of protein), coenzyme Q-1 (ubiquinone-1, 0.05 mm final concentration) were added to the assay buffer such that the final assay volume was 0.48 ml. After preincubation of the reaction mixture at room temperature for 2 min, the reaction was initiated by the addition of 0.02 ml of a 5 mm solution of NADH. The decrease in absorbance at 340 nm was monitored over time. The assay was also performed in the presence of rotenone (5 μm final concentration) to determine the rotenone-insensitive complex I activity. The rotenone-sensitive enzyme activity was calculated by subtracting the rotenone-insensitive activity from the total activity and is expressed as nanomoles of NADH oxidized per minute per milligram of protein.
Estimation of glutathione. For estimation of glutathione, the removed tissues were frozen immediately in liquid nitrogen and homogenized in potassium phosphate buffer (100 mm, pH 7.4) containing 1 mm EDTA. Total glutathione levels were estimated according to the method described by Tietze (1969) as follows briefly. To an aliquot of the brain homogenate, an equal volume of 5-sulfosalicylic acid (1%, w/v) was added, mixed, and centrifuged at 10,000 × g for 10 min, and the supernatant was collected and used for assay of GSH by the enzymatic recycling method. Another aliquot of the brain homogenate was used for protein estimation. Protein was estimated by a dye-binding method (Bradford, 1976). Statistical analyses were performed using Student’st test or ANOVA followed by Student–Newman–Keuls, Bonferroni, or Dunnet’s test.
Histology. Brain and total length of the spinal cord from control and mice treated with l-BOAA were fixed by perfusion with Bouin’s fixative, and representative sections were paraffin-embedded, sectioned, and stained with hemotoxylin and eosin or cresyl violet for Nissl.
RESULTS
In vitro studies
Exposure of sagittal slices of male mouse brain to varying concentrations of l-BOAA resulted in a dose-dependent inhibition of complex I. Incubation of slices with 0.1 pml-BOAA inhibited complex I by 28%; at the highest concentration of l-BOAA tested (1 nm), the enzyme activity was inhibited by ∼50% (Fig.2A). Complex I was significantly inhibited (17.8% inhibition) within 5 min of exposure to the toxin (Fig. 2B), and, after 60 min of incubation with l-BOAA, the enzyme activity was inhibited by 51% (Fig. 3). d-BOAA at a similar or even higher concentration had no effect on complex I activity, indicating the stereospecificity of the l-isomer in producing toxicity (data not shown).
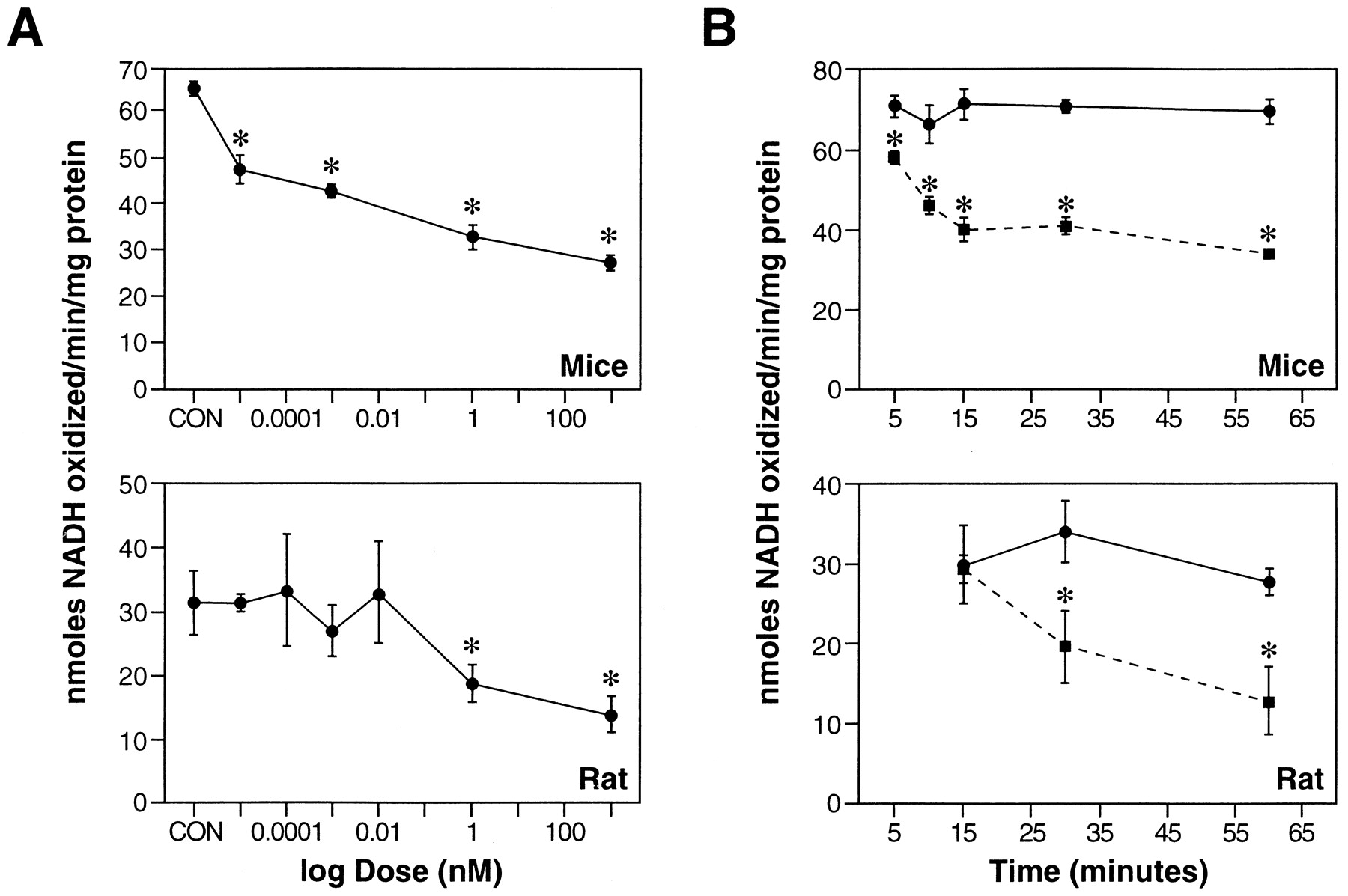
Effect of l-BOAA on complex I activity in mouse and rat brain slices. A, Sagittal slices of mouse or rat brain were incubated in ACSF with varying concentrations (0.01 pm to 1 μm) ofl-BOAA for 1 hr, and the activity of complex I was assayed in the crude mitochondrial preparation. B, Time-dependent inhibition of complex I activity by l-BOAA in mouse and rat brain slices. Sagittal slices of mouse brain were incubated in ACSF without (•–•) or with l-BOAA (1 nm, ▪–▪) for varying intervals (5–60 min), and the activity of complex I was assayed in the crude mitochondrial preparation. The enzyme activity is expressed as nanomoles of NADH oxidized per minute per milligram of protein. Values are mean ± SEM (n = 4–6 slices).Asterisks indicate values significantly different from respective control (p < 0.01).

Effect of l-BOAA on GSH levels in mouse brain slices (A) and effect of GSH on complex I activity inhibited by l-BOAA (B). A, Sagittal slices of mouse brain were incubated in ACSF without (•–•) and withl-BOAA (1 nm, ○–○) for varying intervals (5–30 min). GSH levels are expressed as nanomoles of GSH per milligram of protein. Values are mean ± SEM (n = 13–17 slices). B, Mitochondria were prepared from untreated and l-BOAA-treated mouse brain slices, and complex I activity was assayed. Two aliquots of this preparation were incubated with and without GSH (2 mm) for 30 min at 37°C. Thereafter, the activity of complex I was assayed and expressed as nanomoles of NADH oxidized per minute per milligram of protein. Values are mean ± SEM (n = 5, wherein each sample was prepared by pooling mitochondria from 3 slices).Asterisks in A and Bdenote values significantly different from respective controls (p < 0.05).
A marked difference in the response to toxicity was observed when brain slices prepared from male rats and mice were exposed tol-BOAA. The minimum toxic dose required to inhibit complex I significantly in rat brain slices was 1 nm (Fig.2A), as compared with the minimum toxic dose of 0.01 pm required to inhibit the enzyme in mouse brain slices.
To evaluate if l-BOAA had any effect on other mitochondrial respiratory chain enzymes, thereby contributing to mitochondrial dysfunction, we assessed the activities of some of these enzymes in male mouse brain slices incubated with l-BOAA for 1 hr.l-BOAA (1 nm) significantly inhibited the activities of succinate dehydrogenase (complex II) and succinate-cytochrome c oxidase (complex II-III) by 28 and 30%, respectively. The concentration of l-BOAA at which these enzymes were inhibited was profoundly higher (1 nm) than that required to inhibit complex I (0.01 pm).l-BOAA had no effect on cytochrome c oxidase (complex IV) activity even at the highest tested concentration (data not shown).
The neurotoxic effect of l-BOAA (0.1 pm) was compared with other glutamate agonists: l-glutamic acid (10 μm and 1 mm), NAAG (0.1 pm and 1 μm), NMDA (1 nm and 1 μm), quisqualic acid (1 nm and 1 μm), AMPA (100 μm and 1 mm), kainic acid (1 μmand 1 mm), or L-BMAA (1 mm). The lower concentrations of the agonists selected represent the minimum toxic dose that was required to induce significant leakage of lactate dehydrogenase from the slices into the medium (Pai and Ravindranath, 1993). In addition, higher concentrations (10- to 100-fold) of these agonists were also tested to determine their effect on mitochondrial function. Only glutamic acid and AMPA significantly inhibited complex I, albeit at millimolar concentrations; the other agonists tested had no effect on complex I activity at any of the concentrations tested (Table 1).
Comparative toxicities of l-BOAA with other glutamate agonists
Exposure of male mouse brain slices to l-BOAA (1 nm) resulted in a small, transient decrease (8.7%) of the endogenous thiol antioxidant GSH 10 min after incubation with the toxin (Fig. 3A). The GSH levels recovered to control values within 15 min and were not significantly different from controls at all other time points. To determine whether the loss in complex I activity was caused by oxidation of protein thiols, mitochondria prepared froml-BOAA-treated slices were incubated without and with GSH (2 mm). In mitochondrial incubations containing GSH, complex I activity was restored, whereas in incubations with buffer alone, the enzyme activity remained inhibited (Fig. 3B), suggesting the involvement of thiol oxidation as a cause of complex I inhibition by l-BOAA.
To determine the mechanism of complex I inhibition byl-BOAA, experiments were designed to assess if previous exposure to thiol antioxidants could protect against l-BOAA neurotoxicity. Pretreatment of male mouse brain slices with GSH isopropyl ester (1 mm) 0.5 hr before exposure to the toxin (1 nm) protected against l-BOAA-mediated inhibition of complex I (Table 2). Similarly, pretreatment with α-lipoic acid (100 μm) also afforded protection, indicating that inhibition of complex I may be caused by the loss of thiol homeostasis (Table 2). The AMPA receptor antagonist NBQX also afforded protection againstl-BOAA-mediated inhibition of complex I in mouse brain slices (Table 2).
Effect of glutamate receptor antagonist and thiol antioxidants on l-BOAA-induced neurotoxicity
In vivo studies
In male mice given a single dose of l-BOAA (10 mg/kg body weight, s.c.), selective loss of mitochondrial complex I activity was observed in the frontoparietal cortex (Fig.4A, wherein the motor cortex is located, Fig. 1) and the lumbosacral segment of the spinal cord (Fig. 4B) within 0.5 hr after administration. Complex I activity was not significantly decreased (Fig.4A–D) in other brain regions (temporal and occipital cortex, striatum, hippocampus, brainstem, thalamus, and cerebellum), other segments of spinal cord (cervical and thoracic cord) and in non-CNS tissue (heart, lung, kidney, and liver). In all further experiments, the area depicted in Figure 1 was designated as motor cortex and was dissected out for biochemical estimations.

Effect of l-BOAA administration on complex I activity in different regions of mouse brain cortex (A), different segments of spinal cord (B), various regions of brain (C), and in other organs (D) of mice. Mice were administered a single dose of l-BOAA (10 mg/kg body weight, s.c.) and killed 0.5 hr after the injection (■). Controls received vehicle alone (saline, ■). Mitochondria prepared from the tissues were used to measure complex I activity. F/P CT, Frontoparietal cortex; T CT, temporal cortex; O CT, occipital cortex; C CD, cervical cord;T CD, thoracic cord; L/S CD, lumbosacral cord; ST, striatum;HP, hippocampus; BS, brainstem;TH, thalamus; CE, cerebellum;HT, heart; LG, lung; KD, kidney; LV, liver. Values are mean ± SEM (n = 4 animals). Asterisks indicate values significantly different from respective controls (p < 0.01).
Administration of the optical antipode of the glutamate agonist, namely, d-BOAA (5 and 10 mg/kg body weight, s.c.) to male mice resulted in no change in complex I activity in either lumbosacral region of the spinal cord or motor cortex (Fig.5A,B, respectively). In contrast, administration of the same dose ofl-BOAA resulted in significant inhibition of complex I activity in both regions of the CNS within 0.5 hr. Although the enzyme activity in the motor cortex was inhibited (Fig. 5B) after administration of the lower dose of l-BOAA (5 mg/kg body weight), the activity in the lumbosacral cord was affected only after administration of higher dose of the glutamate agonist (Fig.5A).

Effect of administration of varying doses of d-BOAA and l-BOAA on complex I activity in mouse brain lumbosacral segment of spinal cord (A) and motor cortex (B). Mice were administered varying doses of d-BOAA orl-BOAA (5 or 10 mg/kg body weight, s.c). Control animals received vehicle alone. The animals were killed 0.5 hr after administration of amino acids. The motor cortex (B) and lumbosacral cord (A) were dissected out and complex I activity was measured in the mitochondrial preparation. Values are mean ± SEM (n = 3–5 animals).Asterisks indicate values significantly different from control (p < 0.05).
When we followed the effect of l-BOAA on complex I activity in motor cortex and lumbosacral cord over a time period, a striking difference was noticed. A sustained loss of enzyme activity (Fig.6B) was observed in the lumbosacral cord after a single dose of l-BOAA (10 mg/kg body weight), whereas in the motor cortex, the enzyme activity was inhibited at 0.5 hr after the dose, but rebounded at 1 hr and returned to near normal levels by 4 hr. However, at the end of 24 hr, complex I activity remained inhibited (Fig. 6A). Under the same conditions, the enzyme activity in the thoracic segment of the cord was unaffected. Complex I activity was also unaffected in the hippocampus and striatum after l-BOAA administration (5 or 10 mg/kg body weight).

Complex I activity and GSH concentration in the motor cortex (A) and lumbosacral cord (B) at various time periods after a single dose of l-BOAA. Mice were administered l-BOAA (10 mg/kg body weight, s.c.), whereas control animals received vehicle alone. The animals were killed at indicated time periods. The motor cortex (A) and the lumbosacral segment of the spinal cord (B) were dissected out and used for the assay of complex I (in mitochondrial preparation) or GSH (in tissue homogenate). The control values were similar at all time points examined. Values are mean ± SEM (n = 4 animals). Asterisks indicate values significantly different from corresponding control (p < 0.05).
Complex II activity in both motor cortex and spinal cord was inhibited only 4 and 1 hr after the l-BOAA dose, respectively, while remaining unaffected in the thoracic cord. Thus, inhibition of complex II was observed several hours after the inhibition of complex I (data not shown).
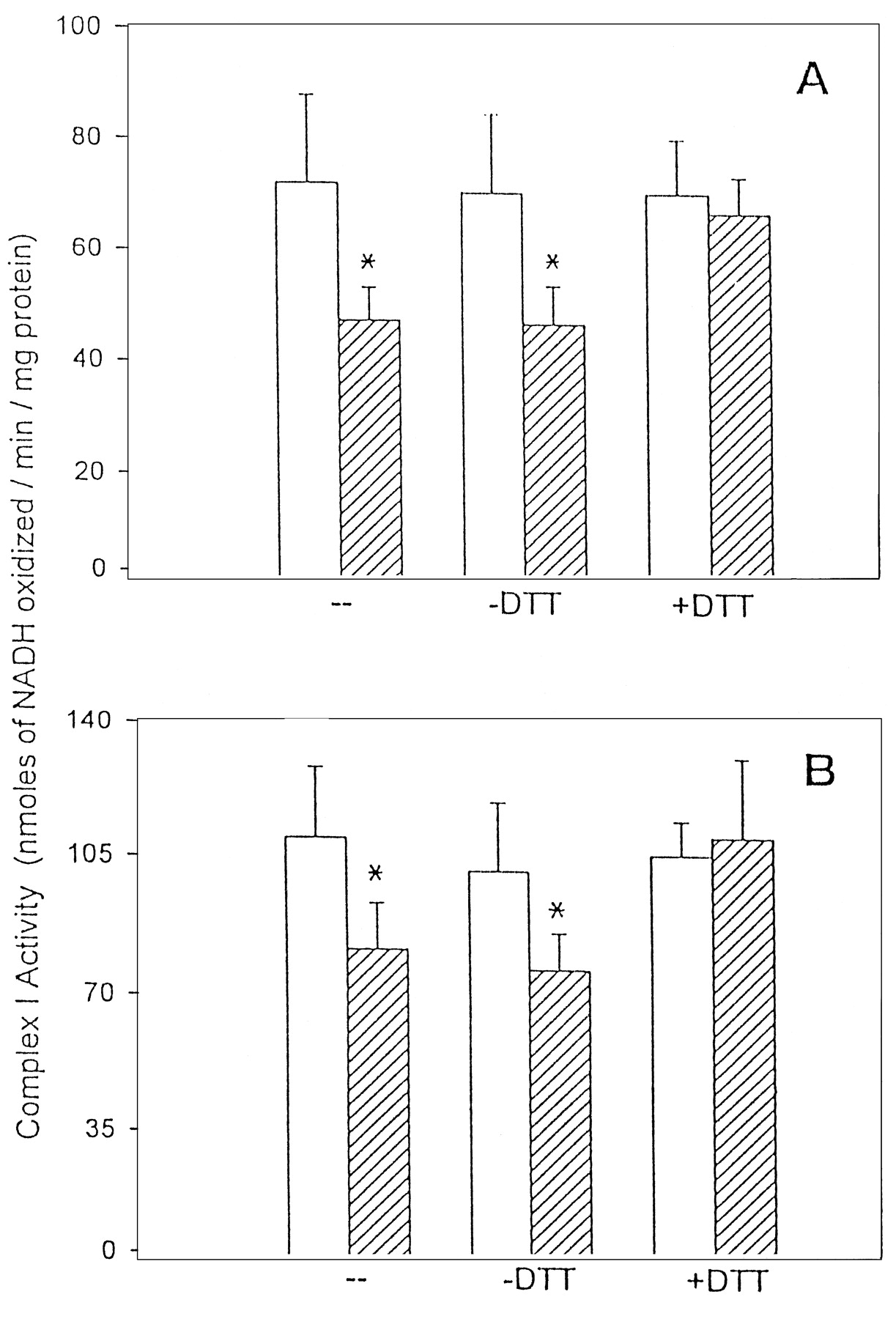
We measured the total GSH content and complex I activity in motor cortex (Fig. 6A) and lumbosacral cord (Fig.6B) over a 24 hr period after a single dose ofl-BOAA (10 mg/kg body weight). A small but significant decrease in GSH levels was seen in the motor cortex at 0.5 and 1 hr after the administration of l-BOAA. The GSH levels, thereafter, were similar to control levels. Interestingly, in the motor cortex, the complex I activity showed a transient decrease 0.5 hr afterl-BOAA dose, and thereafter rebounded to enzyme activity higher than controls, however, after 24 hr, complex I activity in the motor cortex was significantly lower than controls, indicating that a single dose of l-BOAA resulted in a long-term inhibition (Fig. 6A). In the lumbosacral region of the spinal cord, sustained loss of both complex I and GSH was observed from 0.5 to 24 hr after the administration of a single dose of l-BOAA (Fig. 6B). It was only 48 hr after administration of a single dose of l-BOAA that both GSH and complex I returned to near control levels in both motor cortex and lumbosacral cord (data not shown). Mitochondria from the motor cortex (Fig.7A) and lumbosacral region of spinal cord (Fig. 7B) of mice treated withl-BOAA (10 mg/kg body weight) were incubated in vitro with the thiol reductant dithiothreitol, and complex I activity was assayed. Dithiothreitol completely reversed the action ofl-BOAA and restored the complex I activity to levels comparable with those in corresponding controls (Fig.7A,B), indicating that the inhibition of enzyme activity was caused by oxidation of thiols to protein disulfides.

Reversal by dithiothreitol ofl-BOAA-mediated inhibition of complex I in the motor cortex (A) and lumbosacral cord (B). Mice were administered l-BOAA (10 mg/kg body weight, s.c.) and killed 0.5 hr after dosing. Mitochondria, prepared by pooling the tissue from three animals, were freeze-thawed three times, and complex I activity was assayed. Thereafter, mitochondria from each sample were divided into two aliquots and incubated with and without dithiothreitol (3.5 mm) for 30 min at 37°C. After the incubation, the samples were dialyzed, and the activity of complex I was assayed before and after incubation with dithiothreitol. Values are mean ± SEM (n = 5 samples wherein the mitochondria prepared from three animals were pooled to make one sample).Asterisks indicate values significantly different from control (p < 0.05).
Pretreatment of mice with the AMPA receptor antagonist NBQX (30 mg/kg body weight, i.p.) 4 hr before l-BOAA administration completely abolished l-BOAA-induced (10 mg/kg body weight, s.c.) complex I inhibition in the lumbosacral cord, indicating that the mitochondrial enzyme loss was mediated through downstream events of glutamate receptor activation (Fig. 8,top). Interestingly, pretreatment of mice with the thiol antioxidant α-lipoic acid also abolished l-BOAA-mediated inhibition of complex I activity (Fig. 8, bottom), indicating that thiol antioxidant could potentially reverse the neurotoxic effects of l-BOAA.

Effect of pretreatment of mice with NBQX (top) and α-lipoic acid (bottom) on complex I activity in lumbosacral cord after l-BOAA administration. Top, Two sets of mice were administered NBQX (30 mg/kg body weight, i.p.) and, after 4 hr, one set was dosed with l-BOAA (10 mg/kg body weight, s.c.), whereas the other set received vehicle (saline) alone. Bottom, Two sets of mice were pretreated with α-lipoic acid (20 mg/kg body weight, s.c.) and, after 1 hr, one set was given l-BOAA as described in the top panel. Another group of mice received onlyl-BOAA, and the control animals received vehicle alone. The animals were killed 24 hr after l-BOAA administration, the lumbosacral segment of spinal cord was dissected out, and complex I activity was estimated in the isolated mitochondria. Values are mean ± SD (n = 4 animals).Asterisks denote values significantly different from control (p < 0.01).
Neurolathyrism is known to occur in humans after consumption of at least 300 gm/d of the chickling peas from the plant Lathyrus sativus for a period of at least 3 months (Streifler et al., 1977). The concentration of l-BOAA in the pea varies from 0.1 to 2.5% (Roy, 1988) Assuming the average body weight of humans as 60 kg and the least l-BOAA concentration in the pea as 0.1% (w/w), the daily intake of l-BOAA would be 5 mg/kg body weight. Male mice were administered 5 mg/kg body weight ofl-BOAA chronically for up to 40 d, after which one group of animals was maintained untreated for an additional 15 d before killing. Complex I activity in the lumbosacral cord (Fig.9) showed marked reduction after 5, 10, 20, and 40 d of l-BOAA exposure (19, 29, 20, and 29% decrease), whereas in the motor cortex (Fig. 9), the activity was significantly decreased (48%) only after 40 d. After withdrawal of l-BOAA, the complex I activity in the lumbosacral cord recovered to control levels while remaining significantly depressed in the motor cortex (Fig. 9).
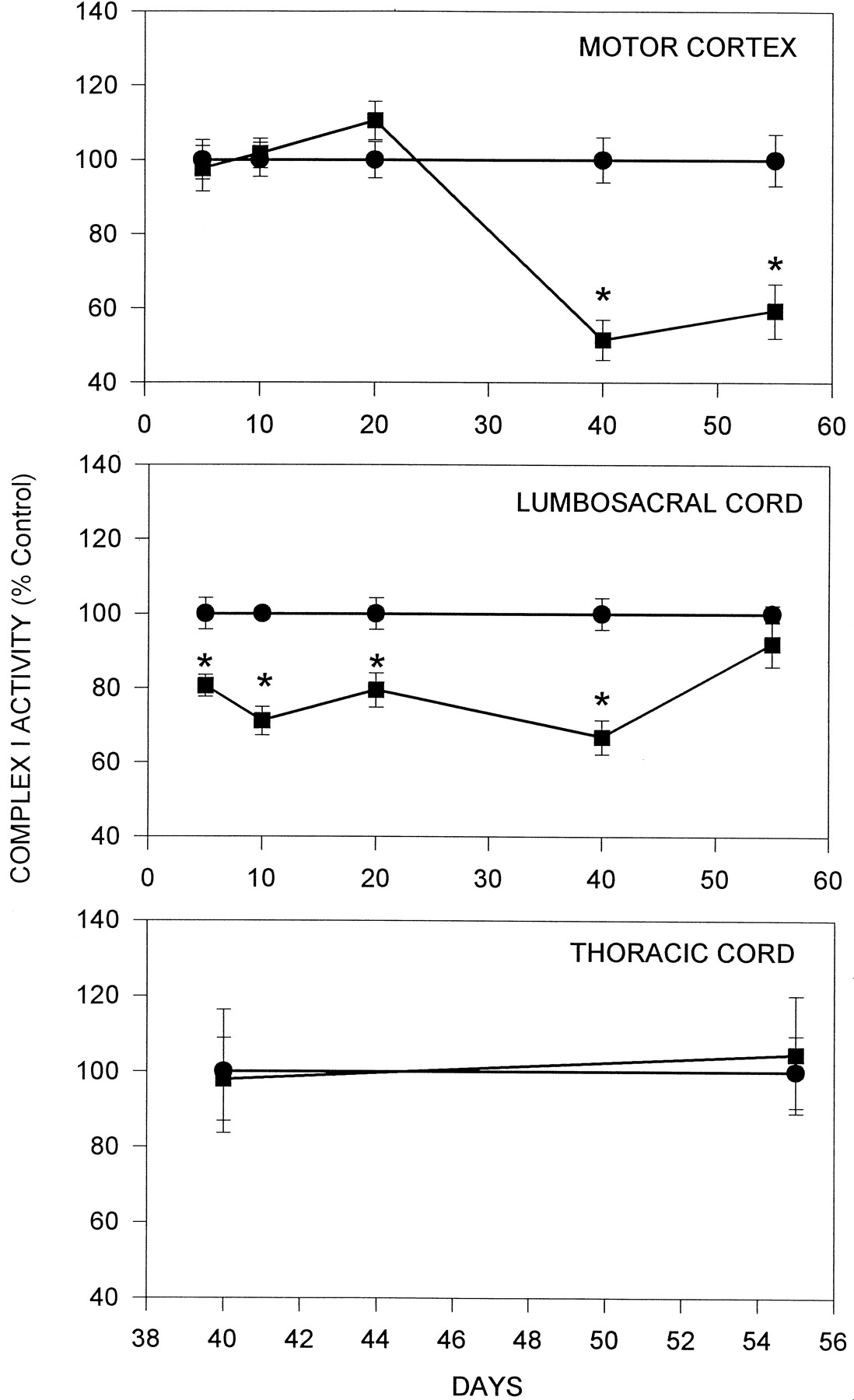
Effect of chronic administration ofl-BOAA on complex I activity in thoracic (top panel), lumbosacral segment of mouse spinal cord (middle panel) and motor cortex (top panel). Mice were administered l-BOAA (5 mg/kg body weight. s.c., daily) and killed on days 5, 10, 20, and 40 (▪–▪). l-BOAA administration was stopped after 40 d, and the animals were kept on normal diet for 15 d before killing. Control animals received vehicle alone (•–•). After killing, the motor cortex (top), thoracic, and lumbosacral segments of the spinal cord (middle) were removed and assayed for complex I activity. Values are mean ± SD (n = 6 animals). Asterisks denote values significantly different from control (p < 0.01).
Morphological evaluation was performed on the brain and spinal cord of animals that received l-BOAA for 40 d (Fig.10A–D). Pyramidal neurons of layers 3 and 5 in the motor cortex revealed cytoplasmic vacuolation and swelling of the apical dendrites. Similar pathology was also noted in the neurons of the motor cranial nerve nuclei. In the spinal cord, the brunt of the pathology was seen in the lumbar and sacral segments, with relative sparing of the cervical and thoracic segments. The medial and intermediate group of neurons corresponding to laminae 7 and 8 of the gray horn in the lumbar level of the spinal cord were abnormal, whereas the lateral group of neurons (lamina 9 of the gray horn) were relatively preserved. The spinal neurons showed cytoplasmic vacuolation, dendritic swelling, and depletion of Nissl substance. The neuronal contour was distorted significantly in some by multiple vacuolation. However, the nucleus and nucleolus were intact. Some of the vacuoles had a central, dark granule. An increase in astrocytes was noted, many of them having a pale watery nucleus and multiple obvious nucleoli, unlike in controls. These astrocytes were seen as small clusters in the neuropil. In mice, wherein l-BOAA was administered for 40 d and then discontinued for 15 d, the pathological changes of neuronal vacuolation receded.

Lumbar cord (A,B) and motor cortex (C, D) sections from control animals and those treated with l-BOAA (5 mg/kg body weight for 40 d). A, Anterior horn neurons of the lumbar spinal cord from control animal.B, Similar section as A from animals chronically administered l-BOAA; anterior horn neurons show cytoplasmic vacuolation (arrowheads), whereas the nucleus and nucleolus are relatively well preserved. The vacuolation of the apical dendrites resulting in distortion of the neuronal contour is seen at higher magnification in inset I; inset II depicts the pale area in cytoplasm of the large neuron that extends along the dendrite and indents the adjacent cell.C, Cortical pyramidal neurons in layer V of motor cortex from control mouse. D, A similar section asC from l-BOAA-treated mouse showing vacuolation and clearing along the apical dendrites (arrowheads) and depletion of Nissl substance in the pyramidal neurons. Scale bar, 25 μm.
The effect of l-BOAA on mitochondrial complex I was gender-specific. Exposure of brain slices from female mice tol-BOAA had no effect on the complex I activity even at concentrations up to 1 μm (data not shown). Administration of a single dose of l-BOAA (10 mg/kg body weight, s.c.; Fig.11A) or chronic exposure to l-BOAA (5 mg/day, s.c; Fig.11B) for 40 d did not have any effect on the complex I activity in the motor cortex or lumbosacral cord, whereas the activity was significantly inhibited in similarly treated groups of male mice.

Effect of l-BOAA on mitochondrial complex I in male and female mice. A, Male and female mice were administered a single dose of l-BOAA (10 mg/kg body weight) and killed 24 hr later. B, Male and female mice were treated daily with l-BOAA (5 mg/kg body weight, s.c.) for 40 d. Complex I activity was estimated in the motor cortex (MC) and lumbosacral cord (LSC) in vehicle-treated (■) and l-BOAA-treated (▪) animals. Values are mean ± SEM (n = 5–6 animals).Asterisks indicate values significantly different from corresponding controls (p < 0.05).
DISCUSSION
Animal models for neurolathyrism that mimic the progressive degeneration of Betz cells in the motor cortex and the corticospinal tract have not been reported. Monkeys fed 10- to 20-fold higher doses than that necessary to produce irreversible neurolathyrism in humans develop the initial signs of the disease as evidenced by central motor deficits involving the corticospinal tracts (Ludolph and Spencer, 1996). Administration of large doses of the toxic principlel-BOAA to rodents, however, results in seizures (ShashiVardan et al., 1997). Pathological effects in the animal model of l-BOAA toxicity defined in the present study share remarkable resemblance to the regiospecific effects seen in the human disease. Selective inhibition of mitochondrial complex I (Fig. 4) and pathological changes (Fig. 10) were seen in the lumbosacral cord and the motor cortex after acute or chronic exposure to l-BOAA with no apparent changes in all the other regions of the CNS that were examined. The similarity with the human disease was also seen in the gender-specific effects of l-BOAA; the female mice were relatively resistant to the toxic effects (Fig. 11). In humans, the incidence of neurolathyrism is more common in men, whereas women are less prone to the disease, although the intake of the chickling pea from Lathyrus sativus is not significantly different (Roy, 1988). It has been reported that pretreatment of cortical neuronal cultures with estrogen protects them from excitotoxic insults when the cells were challenged with glutamate (Behl et al., 1997). Although the mechanism by which estrogens protect the neuronal cell is not known, it is hypothesized that estrogen-like compounds, in general, are able to protect the neurons from damage by oxidative stress-mediated events.
The inhibition of complex I is a sensitive and selective index ofl-BOAA toxicity. In vitro, in brain slices, complex I is inhibited by very low concentrations of l-BOAA in a dose- and time-dependent manner, an effect not shared by other glutamate agonists tested, with the exception of AMPA and glutamate, which inhibit complex I at very much higher doses. We had earlier reported that l-BOAA inhibits NADH-dehydrogenase activityin vitro in mouse brain slices (Pai and Ravindranath, 1993), however, measurement of rotenone-sensitive complex I activity is a more sensitive and selective index of l-BOAA toxicity.
The mechanism of inactivation of complex I seems to be a downstream event of l-BOAA action on excitatory amino acid receptors, because the loss in complex I activity was blocked both in vitro and in vivo by specific antagonists of AMPA receptor. The neurotoxic effects of l-BOAA seem to be predominant in the motor cortex and lumbosacral region of the spinal cord, and this specificity does not seem to reflect the distribution of AMPA receptors in the CNS. The observations made herein do seem to suggest that hitherto undetected high-affinity binding sites forl-BOAA may exist in the CNS. Earlier studies have shown that administration of 14C-l-BOAA to rats resulted in maximal accumulation of the radiolabel in the spinal cord (Rao, 1978). Nevertheless, the regions of the CNS affected byl-BOAA bear a remarkable similarity to the regions affected in human forms of the disease.
The results described herein provide the first evidence thatl-BOAA, an excitatory amino acid, can cause mitochondrial dysfunction through inhibition of complex I in vivo in specific regions of the CNS. Furthermore, the inhibition of complex I activity was associated with oxidation of protein thiol groups in complex I (Dupius et al., 1991), because the enzyme activity rebounded after incubating the mitochondria with the thiol-reducing agent dithiothreitol. From the experimental evidence presented in this study, wherein the oxidative stress generated (measured as loss of cellular antioxidant thiol GSH) followed the inhibition of complex I activity (Fig. 6); loss of mitochondrial complex I seems to be a primary event after exposure to l-BOAA. But because selective cell populations are affected within specific regions of the brain byl-BOAA, the measurement of GSH in sagittal slices of whole brain as performed in the in vitro experiments may not reflect subtle changes in the antioxidant status in the targeted cells. However, the reversal of the loss in complex I by thiol reductants seems to indicate that oxidative stress and protein thiol oxidation may be primary events in l-BOAA induced mitochondrial dysfunction. One of the downstream events of excessive stimulation of glutamate receptors is the generation of reactive oxygen species leading to oxidative stress-mediated cellular damage (Murphy et al., 1989).
Mitochondrial dysfunction has been recognized as a primary event in glutamate neurotoxicity (Schinder et al., 1996), particularly that mediated through NMDA receptors. The activation of NMDA receptors leads to massive increase in intracellular calcium, which results in the alteration in the mitochondrial membrane potential and opening of the mitochondrial permeability transition pore (White and Reynolds, 1996). However, pore-opening through voltage-dependent channels is directly associated with an oxidation–reduction-sensitive dithiol, and the cross-linking of the sulfhydryl groups increases the probability of the mitochondrial permeability transition pore opening, an event that can be blocked by the thiol reductant dithiothreitol (Petronilli et al., 1994). Thus, the mitochondrial dysfunction caused through mitochondrial permeability transition pore opening is also sensitive to the oxidation of protein thiols in a manner similar to that seen in the present study with reference to the inhibitory effect of l-BOAA on complex I. We have earlier shown that brain mitochondria (Ravindranath and Reed, 1990) are severalfold more vulnerable to oxidative stress-mediated damage as compared with liver mitochondria (Olafsdottir and Reed, 1988), and the major consequence of oxidative stress on brain mitochondria is the formation of protein–glutathione-mixed disulfide (rather than increase in levels of oxidized GSH, GSSG as seen in liver mitochondria). Taken together these studies demonstrate the vulnerability of brain mitochondrial proteins to oxidative modification through formation of mixed disulfides.
The involvement of excitatory amino acids in the pathogenesis of motor neuron disease has been demonstrated both in sporadic cases (Rothstein et al., 1990) and in chemically induced forms of the disease (Spencer et al., 1986, 1987). More recent developments include the discovery of mitochondrial dysfunction in a variety of neurodegenerative disorders, including motor neuron disease (Chang et al., 1995; Mizuno et al., 1995). This study provides evidence indicating a mechanism for neurodegeneration through mitochondrial dysfunction chemically induced through excitatory amino acids. Increased glutaminergic activity has been shown to be associated with Parkinson’s disease (Verma and Kulkarni, 1995), and glutamate antagonists (Stauch Slusher et al., 1995) have been shown to alleviate Parkinsonism in experimental animals. Recent hypotheses on role of excitatory amino acids in pathogenesis of sporadic motor neuron disease have also resulted in new putative therapies that include anti-excitotoxicity agents (Rothstein, 1996). Although the loss of complex I activity has been demonstrated in Parkinson’s disease patients (Parker et al., 1989); the present study indicates a similar dysfunction in chemically induced motor neuron disease, in experimental animals. It remains to be seen if similar dysfunction occurs in humans in sporadic forms of the disease.
In conclusion, the present investigation demonstrated that excitatory amino acids like l-BOAA cause mitochondrial dysfunction in selected regions of the CNS that can be reversed in vitro by thiol reductants. The results indicate an initiating role for oxidative stress and the potential importance of maintaining protein thiol homeostasis in brain through administration of thiol antioxidants.
Footnotes
This work was supported by a grant from the United States–India fund for cultural, educational, and scientific cooperation. We thank Dr. S. L. N. Rao, Osmania University, Hyderabad, India for the generous gift of d-BOAA.
Correspondence should be addressed to Dr. V. Ravindranath, Department of Neurochemistry, Hosur Road, National Institute of Mental Health and Neurosciences, Bangalore 560 029, India.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}