

Abstract
The regulation of glutamate (Glu) release from the excitatory input to dopamine cells in the ventral tegmental area (VTA) during acute withdrawal from morphine was studied in slices from animals treated for 6–7 d with morphine. EPSCs were inhibited by opioid agonists acting at μ-subtype receptors but not by selective δ- or κ-subtype agonists. The opioid inhibition was reduced by 65% with the potassium channel blocker 4-aminopyridine (4-AP; 100 μm) and a 12-lipoxygenase inhibitor, baicalein (5 μm), suggesting that opioids acted via a transduction pathway involving activation of a voltage-dependent potassium conductance by lipoxygenase metabolites as has been shown in the periaqueductal gray (Vaughan et al., 1997). During withdrawal, neither the potency nor the efficacy ofd-Ala-Met-enkephalin-Gly-ol (DAMGO) were changed; however, the blockade of μ-opioid inhibition by both 4-AP and baicalein was reduced. In addition, the potency of baclofen to depress EPSCs by GABA-B receptors and the effects of the GABA-uptake inhibitor NO-711 (10 μm) were increased in withdrawn rats. Finally, group 2 (but not group 4 or 1) metabotropic glutamate receptor-mediated presynaptic inhibition was also enhanced in morphine-withdrawn rats. These results suggest that one of the consequences of withdrawal from chronic morphine is an enhanced presynaptic inhibition of the excitatory inputs to the dopamine cells of the VTA. Inhibition of glutamate release during acute withdrawal would add to the inhibition of dopamine cells that is mediated by an augmented release of GABA (Bonci and Williams, 1997).
The VTA is a component of the endogenous reward circuit that is thought to be activated by many drugs of abuse, including opioids (Bozarth and Wise, 1981). Rats will lever press for microinjection of opioids into the VTA, suggesting that the VTA is one site involved in the reinforcing properties of opioids (Devine and Wise, 1994). Pharmacological evidence suggests that the terminals of afferent fibers from the nucleus accumbens and ventral pallidum mediate GABA-B IPSPs, whereas GABA release from interneurons acts via GABA-A receptors (Johnson and North, 1992a,b;Johnson et al., 1992a; see also Paladini et al., 1998). Opioids cause an acute inhibition of both GABA-A and GABA-B IPSPs (Johnson and North, 1992a), and withdrawal from morphine affects the regulation of synaptic transmission at both synapses by a cAMP-dependent mechanism (Bonci and Williams, 1996, 1997).
Excitatory synaptic input mediated by glutamate is a key component of the regulation of dopamine cell excitability and is known to play an important role in the actions of many drugs of abuse (Kalivas and Stewart, 1991; Kalivas, 1993). The glutamate afferents arise from three primary sources: the medial prefrontal cortex, the pedunculopontine region, and the subthalamic nucleus (Fallon and Loughlin, 1995). One role of the glutamatergic innervation to the VTA is to mediate a switch from pacemaker-like firing in dopamine cells to burst-firing pattern (Gariano and Groves, 1988; Svensson and Tung, 1989; Johnson et al., 1992b; Murase et al., 1993; Zhang et al., 1994). Presynaptic regulation of glutamate release from these excitatory synapses by opioids has not been investigated and could be important in shaping the pattern of activity of dopamine cells.
The goal of the present study was to determine the acute actions of opioids on glutamate-mediated transmission in the VTA and then identify adaptive changes in excitatory synaptic transmission during acute withdrawal from chronic opioid treatment. The results show that opioids acutely inhibit glutamate release and that during withdrawal the transduction pathway mediating this inhibition is not the same as in control. The results also show an increased sensitivity to presynaptic inhibition by both glutamate (autoreceptors) and GABA (heteroreceptors) metabotropic receptors, suggesting that glutamate release would be depressed during acute opioid withdrawal. Because the pattern of dopamine cell activity in the VTA is dependent on excitatory glutamate drive, the present data form a cellular basis for the decreased activity of dopamine cells observed during acute withdrawal from morphine in vivo (Diana et al., 1995).
MATERIALS AND METHODS
Chronic morphine treatment was done using time-released morphine pellets (75 mg of morphine per pellet obtained from National Institute on Drug Abuse). The pellets were implanted subcutaneously in anesthetized rats (ketamine–xylazine), one on the first day and two on days 3 and 5. Experiments were performed on days 6 or 7. This protocol has been shown to produce strong opioid dependence (Chieng and Christie, 1995). Whole-cell recordings of membrane current were made from dopamine neurons in horizontal slices of rat midbrain (holding potential, −70 mV). This method has been described previously (Williams et al., 1984). In brief, rats (Wistar, 160–220 gm) were anesthetized with halothane and killed. The midbrain was sliced (200 μm) in the horizontal plane using a vibratome at 4°C and maintained in physiological saline (morphine-free). Slices (up to three) containing the VTA were stored before being placed in the recording chamber and superfused (2 ml/min) with warmed (35°C) Krebs’ solution–bicarbonate buffer containing the following (in mm): NaCl 126, KCl 2.5, NaH2PO41.2, MgCl2 1.2, CaCl2 2.4, glucose 11, NaHCO3 21.4 saturated with 95% O2 and 5% CO2. All experiments were done after washing slices with morphine-free solution for a minimum of 1 hr and as long as 6 hr. These slices from morphine-treated animals were termed opioid-withdrawn. The superfusion medium contained picrotoxin (100 μm) and strychnine (1 μm) to block GABA-A and glycine-mediated synaptic currents, respectively. There was no effect of this solution on the holding current of the dopamine cells. All drugs were added at the final concentration to the superfusion medium.
Cells were visualized using an upright microscope with infrared illumination, and recordings were made with whole-cell electrodes containing the following (mm): KCl 128, NaCl 20, MgCl21, EGTA 1, CaCl2 0.3, Mg-ATP 2, GTP 0.3, and cAMP 0.2 buffered with HEPES 10, pH 7.3. Electrode resistance was 2–4 MΩ, acceptable access resistance was <15 MΩ, and the holding potential was −70 mV. An Axopatch-200A (Axon Instruments, Foster City, CA) was used to record the data, which were filtered at 1 kHz, digitized at 5 kHz on a DigiData 1200 interface (Axon Instruments) and collected on a personal computer using ACQUIS-1 software (Bio-Logic) or pClamp6. To evoke synaptic currents, stimuli (100–300 μsec duration) were delivered at 0.033 Hz through bipolar tungsten electrodes placed near (30–100 μm) the cell body. Two stimuli were applied at an interval of 50 msec (Manabe et al., 1993), and the paired-pulse ratio was calculated by dividing the amplitude of the EPSC evoked by the second stimulus by the amplitude of the first EPSC evoked by the first stimulus. A change in the paired-pulse ratio is thought to result from the alteration in transmitter release caused by a presynaptic mechanism (Manabe et al., 1993). Evoked EPSC amplitudes were measured by averaging a 2 msec window around the peak and subtracting the average value obtained during a 5 msec window immediately before the stimulus.
Spontaneous miniature EPSCs (mEPSCs) were recorded in the presence of tetrodotoxin (TTX; 300 nm) using pClamp 6.0 (Axon Instruments; 120 sweeps for each condition, 1 sec/sweep). The internal solution contained Cs gluconate instead of KCl. mEPSC amplitude and interinterval time were measured using Axograph 3.6. For this analysis, a template of mEPSCs having the width and time course of a typical synaptic event (a double exponential: f(t) = exp(−t/rise) − exp(−t/decay), where rise = 0.5 msec and decay = 3 msec), was slid along the data trace one point at a time. At each position, this template is optimally scaled and offset to fit the data, and a detection criterion is calculated. The detection criterion is the template scaling factor divided by the goodness-of-fit at each position. An event is detected when this criterion exceeds a threshold and reaches a sharp maximum. The limit of detection was 5 pA.
The fitting of concentration–response curves were calculated according to y = {ymax −ymin/1 + (x/EC50)n} +ymin (where ymax = response in the absence of agonist, ymin = response remaining in presence of maximal agonist concentration,x = concentration, EC50 = concentration of agonist producing 50% of the maximal response andn = slope) with Kaleidagraph software (Abelbeck Software). All values are given as mean ± SEM. Statistical analyses were done with the Mann–Whitney U test, the Kolmogorov–Smirnov, or the Spearman Rank Correlation tests using Statview (Abacus Concepts, Calabasas, CA); p < 0.05 was taken as indicating statistical significance (**p< 0.05; ***p < 0.01). Drugs used were 8-cyclopentyl-1,3-dipropyl-xanthine (DPCPX) from Research Biochemicals (Natick, MA);(2S,1′S,2′S)-2-(2′-carboxycyclopropyl)glycine (l-CCGI), l-2-amino-4phosphono-butyrate (l-AP-4), (S)-4-carboxy-3-hydroxyphenylglycine (4C3HPG), 6-Nitro-7-sulfamoybenzo-[f]quinoxaline-2,3-dione disodium (NBQX), (S)-3,5-dihydroxyphenylglycine [(S)-DHPG] from Tocris Neuramin; TTX, picrotoxin, strychnine, [Met]5enkephalin, forskolin, DAMGO, U69593, [d-Pen]2[d-Pen]5enkephalin, 4-AP from Sigma (St. Louis, MO); baicalein from Biomol (Plymouth Meeting, PA); dendrotoxin from Calbiochem (La Jolla, CA) and Alomone Laboratories (Jerusalem, Israel); and CGP 56999A was generously provided by Dr. A. Sutter at Novartis Pharma.
RESULTS
Acute inhibition by μ-opioids
Whole-cell recordings were made from dopamine cells that were identified by their prominent hyperpolarization-induced inward current (Johnson and North, 1992b). Electrically evoked glutamate EPSCs were completely blocked by NBQX (5 μm). In all experiments other synaptic currents were blocked with receptor antagonists (see Materials and Methods). Slices from both control and morphine-treated animals were prepared and maintained in morphine-free solution.
In control slices, the opioid agonists [Met]5enkephalin (ME; 10 μm) and DAMGO (10 μm) had no effect on the holding current but depressed the amplitude of the evoked EPSCs by ∼50% (Fig. 1). The inhibition caused by ME (10 μm) was completely blocked by naloxone (1 μm; n = 3) and the selective μ-receptor antagonist CTAP (1 μm; n = 9; Fig. 1). Thus μ-opioid receptors were responsible for the inhibition. This suggestion was further supported by the observation that neither the selective δ agonist [D-Pen]2[D-Pen]5enkephalin (DPDPE; 1 μm) nor the selective κ agonist U69593 (1 μm) affected EPSCs (Fig.1C).

Inhibition of glutamate EPSCs by opioids is mediated by activation of μ-opioid receptors. A,Top, Superimposed current traces showing the effect of ME (10 μm) in control (left) and in the presence of CTAP (1 μm; right). The plot below shows the entire experiment, plotting the amplitude of the EPSC as a function of time. In this and other plots, the barsindicate the period during which the superfusion solution contained the indicated drug. B, Summarized results demonstrating the inhibition of evoked EPSCs by ME (10 μm;n = 6). In this and other plots, the amplitude of the evoked EPSC was normalized for each cell using the mean amplitude recorded during the first 10 min and plotted as a function of time.C, Summary of the experiments showing the inhibition of the EPSC by ME (10 μm; 49 ± 9%;n = 6), DAMGO (10 μm; 51 ± 9%;n = 4), DPDPE (1 μm; 7 ± 6%;n = 7), U69593 (1 μm; 4 ± 14%;n = 7), and ME (10 μm) plus CTAP (1 μm) (6 ± 5%; n = 9).
This inhibition was thought to result from presynaptic inhibition of glutamate release based on two observations. First ME caused a shift in the paired-pulse ratio toward facilitation (mean inhibition, 55 ± 3%; mean increase in paired-pulse ratio, 146 ± 8%;n = 32) and, although there was considerable variation from cell to cell, the shift in the paired-pulse ratio was related to the amount of inhibition (Fig. 2). The second method used to determine the site of opioid action was by measuring the effect of opioids on the frequency of spontaneous mEPSCs. The μ-opioid agonist, DAMGO (10 μm) decreased the frequency of spontaneous mEPSCs, from 2.8 ± 0.2 Hz in control to 1.6 ± 0.2 Hz (Mann–Whitney U test, p= 0.0031; n = 9; Fig.3C) and caused a significant shift of the cumulative frequency distribution to the right (Fig.3B). The decrease in frequency caused by DAMGO was completely reversed by naloxone (1 μm, Fig.3B). During the inhibition caused by DAMGO neither the mean amplitude of the mEPSCs (12.3 ± 2.7 pA and 12.5 ± 3; Mann–Whitney U test, p = 0.69; Fig. 3), nor the cumulative amplitude distribution were modified (Kolmogorov–Smirnov test, p < 0.9999). Taken together there results indicate that μ-subtype opioid receptors located on presynaptic glutamate terminals inhibit EPSCs to the dopamine cells of the VTA.

Inhibition of the EPSC by ME (10 μm) is accompanied by a change in the paired-pulse ratio toward facilitation. Top, Three superimposed current traces illustrating the effect of ME on EPSCs induced using a paired-pulse protocol. The inhibition of the first EPSC (S1) by ME is larger than the second (S2). The inset shows the second EPSC (S2) after normalizing the amplitude of the EPSCs evoked by the first stimulus. This illustration indicates a shift toward facilitation. Bottom is a plot of the inhibition induced by ME against the paired-pulse ratio for 32 cells. Theline is constructed from a linear regression analysis and indicates that the variables correlated significantly (ρ = 0.62; p = 0.0004, Spearman rank correlation). Theopen circle indicates the average inhibition and shift in paired-pulse ratio for all these cells (change in paired-pulse ratio = 146 ± 8%; ME-induced inhibition = 55 ± 3%).

DAMGO (10 μm) inhibits the frequency but not the amplitude of mEPSCs. A, Raw traces illustrating mEPSCs and the effect of DAMGO and DAMGO plus naloxone.B, The interevent interval distribution curve was significantly shifted to the right by DAMGO and reversed in presence of naloxone (Kolmogorov–Smirnov test, p < 0.0001).C, The effect of DAMGO on the average frequency (left, Mann–Whitney U test,p = 0.0031) and amplitude (right, Mann–Whitney U test, p = 0.69).
Opioid inhibition during withdrawal from chronic morphine
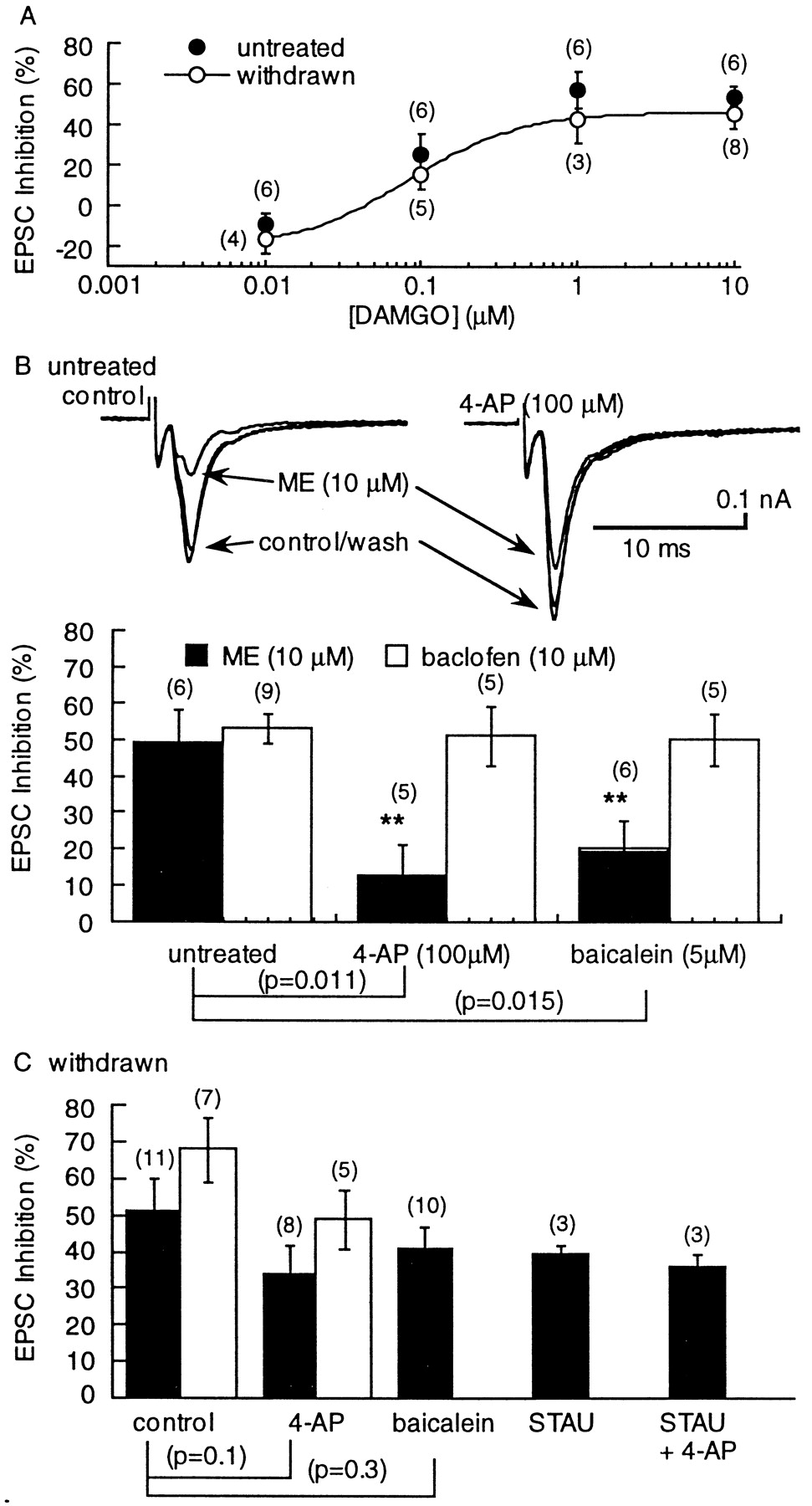
Neither the sensitivity (EC50 = 0.08 μm) nor the maximal effect of DAMGO were changed during withdrawal from morphine (Fig.4A). Based on recent reports indicating that the transduction mechanism for opioid inhibition of transmitter release was dramatically altered during opioid withdrawal (nucleus accumbens, Chieng and Williams, 1998; periaqueductal gray (PAG), Ingram et al., 1998) the mechanism of opioid inhibition of glutamate release was investigated in the VTA.

The sensitivity of the EPSC to inhibition by DAMGO was not changed, however, the blockade of the opioid inhibition by 4-AP and baicalein was reduced during opioid withdrawal. A, Concentration– response curves showing the inhibition by DAMGO in untreated (solid circles) and withdrawn (open circles) slices. B, An illustration of the effect of 4-AP on the EPSC and the resulting reduction in the inhibition caused by ME. Left, Three superimposed traces showing the inhibition caused by ME. Right, The same cells after superfusion with 4-AP (100 μm). The amplitude of the EPSC is larger, and the inhibition caused by ME is reduced. The bar graph below summarizes the results obtained with 4-AP and baicalein on the inhibition caused by ME and baclofen. The inhibition caused by ME was significantly reduced by both 4-AP and baicalein, whereas the inhibition caused by baclofen was not changed. Csummarizes results using withdrawn slices showing that the blockade of the ME-induced inhibition caused by 4-AP and baicalein was significantly reduced. In addition, the ME-induced inhibition was not significantly decreased in the presence of the kinase inhibitor staurosporine (STAU; 1 μm; 20 min) or staurosporine plus 4-AP.
Given the well known effect of chronic morphine treatment to upregulate adenylyl cyclase activity (Sharma et al., 1975; Terwilliger et al., 1991), the role of the cAMP cascade at this excitatory synapse in the VTA was first examined. The amplitude of EPSCs was increased by the adenylyl cyclase activator forskolin. However, the augmentation was not different in morphine-withdrawn and control slices (control, 143 ± 13%, n = 15; withdrawn, 140 ± 10%,n = 11). The transduction mechanism was further investigated with the use of the nonselective kinase inhibitor staurosporine (STAU). The opioid inhibition was not affected by STAU (1 μm; 20 min; Fig. 4C) in both control and withdrawn slices. Another consequence of the activation of adenylyl cyclase is an increase in extracellular adenosine that has been shown to inhibit GABA IPSCs in the VTA (Bonci and Williams, 1996) and nucleus accumbens (Chieng and Williams, 1998) and glutamatergic EPSCs in the hippocampus (Manzoni et al., 1995; Brundege et al., 1997). At this glutamate synapse in the VTA, however, blockade of A1 adenosine receptors with DPCPX (200 nm, 20 min) neither augmented basal synaptic transmission (109 ± 7% change in untreated,n = 13; 100 ± 6% change in withdrawn,n = 9), nor increased the effect of forskolin in slices from untreated or withdrawn animals (untreated 137 ± 11%,n = 7; withdrawn 135 ± 20%, n = 5). Thus, there was no detectable upregulation of the adenylyl cyclase–protein kinase A cascade caused by morphine withdrawal at this glutamate synapse in the VTA. This result differs from the increase in GABA-A IPSPs in withdrawn slices mediated by an upregulation of the cAMP-dependent cascade in the VTA (Bonci and Williams, 1997), nucleus accumbens (Chieng and Williams, 1998), PAG (Ingram et al., 1998), and dorsal raphe (Jolas et al., 1998).
It was recently reported that opioids inhibit GABA release via a transduction pathway involving the activation of a voltage-dependent potassium conductance by lipoxygenase metabolites in the brainstem PAG (Vaughan et al., 1997). This mechanism of opioid inhibition was tested in the VTA. In untreated rats, the ME-induced inhibition of evoked EPSCs was significantly reduced by the voltage-dependent potassium channel blocker 4-AP (100 μm; Fig. 4B). In withdrawn slices, however, the inhibition caused by ME was not significantly changed by 4-AP (Fig. 4C). Preincubation of the slices with the 12-lipoxygenase inhibitor baicalein reduced the ME-mediated EPSC depression in control but not in morphine-withdrawn slices (Fig. 4). Interestingly, the effects of 4-AP and baicalein were selective for the μ-opioid receptor inhibition because GABA-B receptor-mediated inhibition by baclofen was insensitive to both compounds (Fig. 4). These results suggest that during morphine withdrawal there is a change in the transduction pathway that normally mediates presynaptic inhibition. Because the degree of inhibition mediated by the 4-AP–baicalein-sensitive pathway was reduced, and the inhibition caused by opioids was not changed, it appears that in withdrawn slices there has been an upregulation of another mechanism that mediates presynaptic inhibition.
Augmented presynaptic inhibition by GABA-B receptors
In slices from untreated rats, baclofen decreased the amplitude of evoked EPSCs (see Fig. 6) and reduced the mean frequency of spontaneous mEPSCs (from 2.8 ± 0.2 to 1.3 ± 0.2 Hz;n = 9; Mann–Whitney U test,p = 0.0012; Fig. 5) and shifted the interevent interval distribution curve significantly to the right (Kolmogorov–Smirnov test, p < 0.001). Despite having a powerful postsynaptic action, baclofen did not have a significant effect on the average amplitude mEPSCs (untreated, 12.3 ± 2.7 pA, n = 13; withdrawn, 8.9 ± 2.2 pA, n = 9, Mann–Whitney U test,p = 0.39) or the cumulative amplitude distribution of mEPSCs (Kolmogorov–Smirnov test, p = 0.35). Thus, in addition to the activation of potassium currents in dopamine cells, GABA-B receptors also inhibit glutamate EPSCs by a presynaptic mechanism. The effects of baclofen were always reversed by the GABA-B antagonist CGP 56999A (1 μm).

Presynaptic inhibition of spontaneous mEPSCs by baclofen. A, Raw current traces illustrating the inhibition of mEPSCs by baclofen and the reversal of that inhibition by CGP 56999A. B, Plots of the average amplitude (left) and frequency (right) of mEPSCs in control (con, solid bars), in baclofen (B; 10 μm), and in CGP56999A (CGP; open bars). Baclofen (10 μm) caused inhibition of the frequency of mEPSCs (from 2.8 ± 0.2 to 1.3 ± 0.3 Hz; Mann–WhitneyU test, p = 0.0012) without significantly reducing the average amplitude (control, 12.3 ± 2.7 pA; withdrawn 8.9 ± 2.2; Mann–Whitney U test,p = 0.39).
As a control experiment, concentration–response curves to baclofen were constructed measuring the inhibition of electrically evoked EPSCs in withdrawn and untreated slices. In withdrawn slices, the sensitivity but not the maximal effect of baclofen was increased (Fig.6A). The EC50 for baclofen was 2.1 ± 0.8 μm in control and 0.5 ± 0.3 μm in withdrawn slices (9–13 experiments). This unexpected increase in the sensitivity to baclofen may suggest that withdrawal from morphine has altered inhibition by receptors unrelated to opioids.

Presynaptic inhibition by GABA-B receptors was increased in withdrawn slices. A, Concentration–response curves to baclofen in untreated and morphine-withdrawn slices showing that the sensitivity but not the maximal effect of baclofen was increased. The EC50 for the GABA-B agonist was 2.1 ± 0.8 μm in untreated slices and 0.5 ± 0.3 μm in morphine-withdrawn slices.B, The GABA-uptake blocker NO-711 (10 μm) slightly reduced the amplitude of evoked EPSCs in slices from untreated animals (16.8 ± 5%) but significantly inhibited EPSCs in morphine-withdrawn slices (48 ± 7%; Mann–WhitneyU test, p = 0.005). The NO-711-induced inhibition was reversed by CGP 56999A (1 μm). C, Plot of the amplitude of the EPSC amplitude (left axis) and paired-pulse ratio (right axis) showing that the depression of evoked EPSCs in the presence of NO-711 was accompanied by a shift in the paired-pulse ratio toward facilitation.
The increased sensitivity to GABA-B-mediated inhibition was further examined using endogenous GABA. It has been shown in slices of hippocampus that under conditions where there was an increase in GABA release, the spatial range of over which GABA acts was increased (Isaacson et al., 1993). Because it has been shown that GABA-A IPSPs in the VTA were augmented during morphine withdrawal (Bonci and Williams, 1997), it was possible that the increased GABA-A IPSCs could overwhelm normal uptake mechanisms to increase the extracellular concentration of GABA and result in the activation of GABA-B receptors on glutamate-releasing terminals. The GABA-B antagonist CGP 56999A had no effect by itself in control or withdrawn slices (untreated, 108 ± 6%, n = 11; withdrawn, 109 ± 3%,n = 8). The role of endogenous GABA was examined further using the GABA-uptake blocker NO-711 (Suzdak et al., 1992). Inhibition of GABA reuptake was expected to increase the likelihood to detect GABA at glutamate-releasing terminals. Although NO-711 (10 μm) had little effect on the amplitude of EPSCs in control slices, it caused an inhibition of EPSCs in withdrawn slices (Fig. 6), the inhibition was accompanied by an increase in the paired-pulse ratio (Fig. 6) and was completely blocked by CGP 56999A (1 μm). Thus, in withdrawn slices after blockade of GABA reuptake, endogenous GABA activated GABA-B receptors to presynaptically inhibit glutamate release. This result supports previous work indicating an increase in GABA release during opioid withdrawal, however also indicates that normal GABA reuptake processes are capable of limiting the diffusion of GABA even in withdrawn slices.
Augmented presynaptic inhibition by metabotropic glutamate receptors
To further examine the changes in presynaptic regulation of glutamate release induced by chronic morphine treatment, the inhibition of glutamate release by metabotropic glutamate receptors (mGluRs) was examined. Several mGluRs have been cloned and classified into three families: group 1 consists of mGluR1 and mGluR5 both positively coupled to phospholipase C, whereas group 2 receptors (mGluR2/3) and group 3 mGluRs (mGluR4/6/7/8) are negatively coupled to adenylyl cyclase (Nakanishi, 1992). Activation of all mGluRs have been shown to decrease transmitter release at many sites in the brain (Nakanishi, 1992), including the VTA (Bonci et al., 1997; Wigmore and Lacey, 1998).
In agreement with previous reports (Bonci et al., 1997; Wigmore and Lacey, 1998), application of selective agonists (Conn and Pin, 1997) of group 2 [(S)-4C3HPG, 50 μm] or group 3 mGluRs (l-AP-4, 10 μm) caused an inhibition of the evoked EPSC that was accompanied by an increase in the paired-pulse ratio. The group 2 agonist (S)-4C3HPG (50 μm) caused a 17 ± 3.5% inhibition of the EPSC (n = 9) and a 122 ± 4% change in the paired-pulse ratio. The group 3 agonist l-AP-4 (10 μm) caused a 46 ± 11% inhibition of the evoked EPSC (n = 5) and a 197 ± 20% change in the paired-pulse ratio (see also Fig. 7, legend).

Increased mGluR inhibition in morphine-withdrawn slices. A, Evoked EPSCs were inhibited by the selective group 2 mGluR agonists (S)-4C3HPG (50 μm) and LCCG1 (1 μm). The inhibition was significantly larger in withdrawn slices compared with untreated controls. The (S)-4C3HPG inhibition was 17 ± 4% in untreated slices and 50 ± 4% in withdrawn slices (Mann–Whitney U test, p = 0.018). A change in the paired-pulse ratio from 122 ± 4% in control to 146 ± 13% in withdrawn slices suggests that the increase in sensitivity resulted from a presynaptic action. The LCCG1 inhibition was 28 ± 4.6% in naive slices and 40 ± 3.5% in morphine-withdrawn slices (Mann–Whitney U test,p = 0.048). B, There was no effect of morphine withdrawal on group 1 (specifically activated by (S)-DHPG, 100 μm) or group 3 (selectively activated by l-AP-4) mGluRs.
During morphine withdrawal, the inhibition of EPSCs induced by the selective activation of group 2 mGluRs by either (S)-4C3HPG (50 μm) or LCCG1 (1 μm) was enhanced (Fig. 7A). In contrast, the inhibition caused by the group 1 and 3 mGluR-specific agonists (S)-DHPG (100 μm) andl-AP-4 (10 μm) was identical in both groups (Fig. 7B). Thus, withdrawal from chronic morphine caused a selective increase of presynaptic inhibition induced by group 2 mGluRs.
DISCUSSION
The results show that opioids acting on μ-opioid receptors inhibit glutamate EPSCs in dopamine cells in the VTA. In control animals, this inhibition is mediated by a mechanism that was sensitive to blockade of a voltage-dependent potassium conductance by 4-AP and to baicalein, an agent that blocks the metabolism of arachidonic acid by the lipoxygenase pathway. During acute withdrawal from chronic morphine treatment there was a fundamental change in the presynaptic regulation of the glutamate release at this synapse. First, the transduction pathway responsible for the opioid receptor-mediated inhibition was less sensitive to agents that disrupt the normal inhibition (4-AP and baicalein). Second, there was an enhancement of the presynaptic inhibition mediated by GABA and glutamate metabotropic receptors. This increased sensitivity would serve to limit glutamate release onto dopamine cells and decrease their excitability.
Mechanism of μ-opioid inhibition
The mechanism of presynaptic inhibition caused by opioids has been the subject of considerable effort that, until recently, has revolved around two effectors, activation of an inwardly rectifying potassium conductance and inhibition of voltage-dependent calcium conductance (Di Chiara and North, 1992). A third mechanism involving the activation of a voltage-dependent potassium conductance has been shown to inhibit glutamate EPSCs at hippocampal mossy fibers (Simmons and Chavkin, 1996). In the PAG, arachidonic acid metabolites have been shown to be responsible for the activation of this voltage-dependent potassium conductance and the resulting inhibition of GABA-A IPSCs (Vaughan et al., 1997). Although μ-opioid receptors on glutamate terminals in the PAG were not coupled to this pathway (Vaughan et al., 1997), the present results suggest that this mechanism may be extended to excitatory synapses in the VTA.
In the PAG, the opioid-induced inhibition was blocked by dendrotoxin, a more selective potassium channel blocker than 4-AP (Vaughan et al., 1997). In the present study, inconsistent results were obtained with dendrotoxin, using a similar protocol and dendrotoxin purchased from two commercial sources. In eight cells, the ME-induced inhibition was tested first in dendrotoxin (100 nm) and then in 4-AP (100 μm). In three cells, both dendrotoxin and 4-AP caused a similar blockade of the ME-induced inhibition, however when all the results were averaged there was no significant effect of dendrotoxin in the same group of cells in which the ME inhibition was significantly decreased by 4-AP [control, 45 ± 6%; dendrotoxin (100 nm), 41 ± 8%; 4-AP (100 μm), 18 ± 3.5%). In all experiments, dendrotoxin caused an obvious increase in the rate of spontaneous EPSCs, suggesting that it was somewhat effective. These results may suggest that dendrotoxin blocked a subset of channels that were insensitive to inhibition by ME. The effects of 4-AP and baicalein were not, however, totally nonselective because the GABA-B-mediated inhibition was not blocked by either 4-AP or baicalein. Selectivity of these compounds may also be suggested by the observation that the blockade of opioid action was significantly reduced during withdrawal. A similar reduction in sensitivity to these compounds was observed in the PAG in withdrawn slices (Ingram et al., 1998).
Transduction mechanisms change during withdrawal
Chronic use of opioids results in tolerance to and dependence on the drug (Johnson and Fleming, 1989). Withdrawal results from adaptive changes in response to chronic drug abuse, such as an upregulation of the cAMP-dependent cascade (Sharma et al., 1975; Terwilliger et al., 1991). Withdrawal from chronic morphine treatment has been shown to augment μ-opioid receptor-mediated inhibition of GABA IPSCs in the nucleus accumbens (Chieng and Williams, 1998) and in the PAG (Ingram et al., 1998). In withdrawn slices of nucleus accumbens, the maximal inhibition caused by μ-opioid receptor activation was increased (Chieng and Williams, 1998). Similarly, in the PAG the sensitivity to μ-opioid receptor agonists was increased (Ingram et al., 1998). In both those studies the increased opioid action was dependent on the activity of adenylyl cyclase. In each study, after inhibition of cAMP-dependent kinase activity (with staurosporine or H89), the increased effect of opioids was reduced to control values. Those results indicated that the upregulated cAMP cascade during withdrawal not only increased transmitter release but was itself sensitive to inhibition by opioids. In the present study, there was no indication that the cAMP-dependent pathway had an effect on glutamate release during withdrawal.
There was however a clear change in the transduction mechanism responsible for the opioid inhibition of transmitter release. In withdrawn slices, opioids mediated an inhibition of transmitter release by a mechanism that was not sensitive to 4-AP and baicalein, indicating that there was an upregulation of an alternate, and as yet unidentified, mechanism. The increased sensitivity to mGluR- and GABA-B-receptor-mediated inhibition at this synapse may suggest that a common transduction pathway mediates inhibition for all three receptors during acute morphine withdrawal. The present work further indicates that the mechanisms responsible for presynaptic inhibition are not static and can vary with the experimental conditions and after chronic drug treatment.
Increased sensitivity of presynaptic GABA and glutamate metabotropic receptors
Withdrawal from chronic morphine enhanced the potency of GABA-B and mGluR agonists to inhibit glutamate release. Similarly, an augmented inhibition of NMDA-mediated EPSCs by mGluR agonists after chronic morphine treatment has been reported in the nucleus accumbens (Martin et al., 1999). It is interesting to note that mGluRs have sequence similarities with GABA-B receptors (Kaupmann et al., 1997) that may result in common coupling mechanisms, thus equivalent changes induced by withdrawal. Drugs acting at GABA-B receptor have been proposed as possible pharmacotherapeutic agents in drug addiction (Roberts et al., 1996; Roberts and Andrews, 1997). Likewise, inhibitory mGluRs have been described at the excitatory terminals to midbrain dopamine cells (Bonci et al., 1997; Wigmore and Lacey, 1998), and recent evidence indicates a role for mGluRs in morphine and nicotine withdrawal syndromes (Fundytus and Coderre, 1997a,b; Helton et al., 1997) as well as the sensitization to amphetamine (Kim and Vezina, 1998). We show here that the presynaptic inhibition induced by group 2 (but not group 3) mGluRs was enhanced in VTA slices prepared from morphine-withdrawn rats. This observation is in agreement with a report describing that a selective group 2 mGluR agonist attenuates the severity of precipitated morphine withdrawal syndromes in rats (Fundytus and Coderre, 1997a,b). The present report supports the hypothesis that presynaptic GABA-B and mGluR receptors or the signal transduction pathway mediated by these receptors may be affected in similar ways by drugs of abuse.
Increased extracellular GABA during opiate withdrawal
GABA-B-mediated heterosynaptic depression regulates excitatory transmission in the hippocampus, and transmitter uptake has been shown to control the spatial range of synaptically released GABA (Isaacson et al., 1993). The hypothesis that withdrawal from chronic morphine augments GABA tone at glutamate synapses in the VTA was explored. There was no GABA tone as determined by the effect of the GABA-B antagonist CGP 56999A in either control or withdrawn slices, suggesting that reuptake of GABA was sufficient to limit the extent of GABA diffusion. After blockade of GABA reuptake, with NO-711, the EPSC was significantly inhibited in withdrawn slices, and this inhibition was reversed by the GABA-B antagonist CGP 56999A. Thus, in withdrawn slices, NO711 mediated an inhibition resulting from the activation of GABA-B receptors. Two mechanisms could account for this observation. First, enhanced GABA release during withdrawal (Bonci and Williams, 1997) could mediate this heterosynaptic effect. Second, the increased sensitivity of GABA-B receptors to reduce glutamate transmission in withdrawn slices found in the present study could account for this result. The physiological significance of either mechanism is the same, an increase in GABA-mediated inhibition of glutamate release.
In summary, withdrawal from chronic morphine was found to modify excitatory terminals to dopamine cells in two ways that may be linked. First, the mechanism of μ-opioid inhibition was altered and second, the actions of agonists of metabotropic receptors to both glutamate and GABA were enhanced. The augmented presynaptic inhibition by GABA and glutamate metabotropic receptors will ultimately reduce glutamate release and therefore decrease dopamine cell firing. The decrease in glutamate release, along with the increased probability of GABA release from interneurons would not only reduce the excitability of dopamine neurons but also limit the occurrence of burst firing. These results provide a cellular basis for the profound decrease of mesolimbic dopaminergic neuronal activity observed in vivo in morphine-withdrawn rats (Diana et al., 1995).
Footnotes
This research was supported by Institut National de la Santé et de la Recherche Médicale, Fondation Simone et Cino Del Ducca, the National Institute on Drug Abuse (NIDA)/INVEST program, and NIDA Grant DA 08163. We thank Drs. J. Brundege, J. Bockaert, P. Chavis, and S. Ingram for critical reading of this manuscript and Dr. A. Sutter at Novartis Pharma (Switzerland) for generously providing CGP 56999A.
Correspondence should be addressed to J. T. Williams, Vollum Institute, Oregon Health Sciences University L-474, 3181 Southwest Sam Jackson Park Road, Portland, OR 97201.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}