

Abstract
Members of the BCL-2 family of proteins either promote or repress programmed cell death. Here we report that neonatal sympathetic neurons undergoing apoptosis after nerve growth factor (NGF) deprivation exhibited a protein synthesis-dependent, caspase-independent subcellular redistribution of BAX from cytosol to mitochondria, followed by a loss of mitochondrial cytochrome c and cell death. Treatment with elevated concentrations of the neuroprotectants KCl or cAMP at the time of deprivation prevented BAX translocation and cytochrome c release. However, administration of KCl or cAMP 12 hr after NGF withdrawal acutely prevented loss of mitochondrial cytochrome c, but not redistribution of BAX; rescue with NGF acutely prevented both events. Overexpression of Bcl-2 neither altered the normal subcellular localization of BAX nor prevented its redistribution with deprivation but did inhibit the subsequent release of cytochromec, caspase activation, and cell death.Bcl-2 overexpression did not prevent cell death induced by cytoplasmic microinjection of cytochrome c into NGF-deprived competent-to-die neurons. These observations suggest that the subcellular redistribution of BAX is a critical event in neuronal apoptosis induced by trophic factor deprivation. BCL-2 acts primarily, if not exclusively, at the level of mitochondria to prevent BAX-mediated cytochrome c release, whereas NGF, KCl, or cAMP may abort the apoptotic program at multiple checkpoints.
Recent evidence implicates the mitochondrion as a critical site at which different apoptotic signals converge. Findings in some non-neuronal models of cell death suggest that a death signal induces translocation of a BH3-containing, proapoptotic BCL-2 family member to the mitochondria, followed by the release of mitochondrial proteins, such as cytochrome c, via an unknown mechanism that may involve the permeability transition pore (PTP) and/or a channel formed by proapoptotic BCL-2 family proteins (Li et al., 1998; Luo et al., 1998; Gross et al., 1999). Once released, cytochrome c forms a complex with Apaf-1 and procaspase-9, which in the presence of ATP or dATP, becomes activated, resulting in further caspase activation, cleavage of cellular substrates, and cell death. However, several questions remain. What is the normal subcellular localization of endogenous BAX in neuronal cells that require Bax expression for cell death? Does BAX translocation require an apoptotic stimulus? Is BAX redistribution necessary for cytochrome c release? Where do BCL-2, caspases, and various neuroprotectants act in this process?
We examined these questions by using the model of trophic factor deprivation in sympathetic neurons. Neonatal sympathetic neurons require nerve growth factor (NGF) for their survival (Levi-Montalcini and Booker, 1960; Gorin and Johnson, 1979; Crowley et al., 1994). In the absence of NGF these neurons undergo an apoptotic cell death (Edwards et al., 1991; Deckwerth and Johnson, 1993), which requiresde novo protein synthesis (Martin et al., 1988) and the development of competence-to-die (Deshmukh and Johnson, 1998). This death is inhibited by caspase inhibitors (Troy et al., 1996; McCarthy et al., 1997), such as boc-aspartyl(OMe)-fluoromethylketone (BAF) (Deshmukh et al., 1996), and neuroprotective agents, such as KCl and cAMP (Rydel and Greene, 1988; Koike et al., 1989; Edwards et al., 1991; Deckwerth and Johnson, 1993; Franklin et al., 1995). Most important, in contrast to other model systems in which BAX translocation has been studied, sympathetic neurons absolutely require the endogenous expression of Bax to undergo apoptosis induced by trophic factor deprivation (Deckwerth et al., 1996). However, because neither the mRNA nor protein level of BAX increases during NGF deprivation-induced apoptosis, BAX must be regulated post-translationally (Greenlund et al., 1995).
Here we report that NGF-deprived sympathetic neurons induced macromolecular synthesis-dependent, caspase-independent subcellular redistribution of BAX, followed by the loss of mitochondrial cytochromec, caspase activation, and apoptosis. Neuroprotective agents, such as KCl and cAMP, blocked the redistribution of both BAX and cytochrome c when added at the time of deprivation but prevented only cytochrome c release when administered 12 hr after NGF withdrawal. Overexpression of Bcl-2 neither altered the normal subcellular localization of BAX nor prevented its redistribution with deprivation but did inhibit the subsequent release of cytochrome c, caspase activation, and cell death. These findings indicate that the subcellular redistribution of BAX is a critical event in neuronal apoptosis induced by trophic factor deprivation. Although BCL-2 acted primarily, if not exclusively, at the level of mitochondria to prevent BAX-mediated cytochrome crelease, neuroprotective agents such as KCl or cAMP aborted the apoptotic program at multiple checkpoints.
MATERIALS AND METHODS
Reagents. All reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated. Collagenase and trypsin were purchased from Worthington Biochemical (Freehold, NJ). The caspase inhibitor, boc-aspartyl(OMe)-fluoromethylketone (BAF), was purchased from Enzyme Systems Products (Livermore, CA). The membrane-permeable cAMP analog, 8-(4-chlorophenylthio)-adenosine 3′:5′-cyclic monophosphate (CPT-cAMP), was used as the source of cAMP. CM1 polyclonal antibody, a gift from Dr. Anu Srinivasan (Idun Pharmaceuticals, La Jolla, CA), was raised against the 13-amino-acid peptide CRGTELDCGIETD, which is found at the C terminus of the p20 subunit of caspase-3 (Armstrong et al., 1997; Namura et al., 1998;Srinivasan et al., 1998b). Medium lacking NGF (AM0) consisted of Eagle’s MEM with Earl’s salts (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal bovine serum, 2 mml-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 μm fluorodeoxyuridine, 20 μm uridine, and 3.3 μg/ml aphidicolin. AM50 medium consisted of AM0 medium plus 50 ng/ml mouse 2.5S NGF (Harlan Bioproducts, Indianapolis, IN). The breeding and genotyping ofBcl-2-overexpressing and Bax-deficient mice have been described previously (Martinou et al., 1994; Knudson et al., 1995;Deckwerth et al., 1996).
Sympathetic neuronal cultures. Primary cultures of sympathetic neurons were established from the superior cervical ganglia of neonatal mice by using a modification of a previously described method (Johnson et al., 1983; Deckwerth et al., 1996; Easton et al., 1997). Briefly, the dissected ganglia were treated with collagenase (1 mg/ml) and then trypsin (2.5 mg/ml) for 30 min each at 37°C. The ganglia were triturated, and the dissociated cells were plated on collagen-coated dishes in AM50.
We observed that neurons from inbred C57BL/6 mice exhibited a loss of mitochondrial cytochrome c and commitment-to-die ∼4–6 hr earlier than neurons from Sprague Dawley rats or outbred ICR mice (see Figs. 1e, 5a; Deckwerth and Johnson, 1993;Deshmukh and Johnson, 1998). We believe that these shifts may be attributable to species and strain differences. Unless indicated otherwise, all control experiments were conducted with C57BL/6 mice, the background strain for both the Bax-deficient andBcl-2 transgenic mice.
Culture conditions. Sympathetic neuronal cultures were grown in AM50 for 4−5 d and then either maintained in AM50 or treated as follows. For NGF deprivation the cultures were rinsed with AM0, followed by the addition of AM0 containing goat anti-mouse 2.5S NGF neutralizing antiserum (anti-NGF) (Ruit et al., 1990). For NGF deprivation in the presence of various reagents, 1 μg/ml cycloheximide, 40 mm KCl, 400 μm CPT-cAMP, or 50 μm BAF was added to AM0 containing anti-NGF. For acute rescue experiments in which NGF, KCl, or CPT-cAMP was readded to cultures after 12 hr of NGF deprivation, the cultures were rinsed with AM0 and then incubated in AM0 medium containing 300 ng/ml NGF, 40 mm KCl, or 400 μm CPT-cAMP, each in the presence of 50 μm BAF.
Immunocytochemistry. Neuronal cultures were immunostained as described previously (Easton et al., 1997; Deshmukh and Johnson, 1998). Briefly, sympathetic neurons (1500−3000 cells) were grown on collagen-coated two-well glass chamber slides (Nalge Nunc, Naperville, IL) in the appropriate medium. Cultures were washed with PBS and fixed with freshly prepared 4% paraformaldehyde in PBS (PFA) for 30 min at 4°C. Cultures then were washed with PBS and Tris-buffered saline (TBS; 100 mm Tris-HCl and 0.9% NaCl, pH 7.6), exposed to blocking solution [TBS containing 5% donkey serum (Chemicon, Temecula, CA) and 0.3% Triton X-100] for 30 min at room temperature (RT) and incubated in the appropriate primary antibody diluted in TBS, 1% donkey serum, and 0.3% Triton X-100 at 4°C overnight. Cultures then were washed with TBS and incubated for 2–4 hr at 4°C in the dark with the appropriate secondary antibody diluted in TBS, 1% donkey serum, and 0.3% Triton X-100. Cultures were washed with TBS and stained with 1 μg/ml bisbenzimide (Hoechst 33258; Molecular Probes, Eugene, OR) for 15−20 min at RT. After being rinsed with TBS, the cultures were coverslipped and examined by fluorescence microscopy. Phospho-c-Jun, Fos, and activated caspases 3 and 7 were detected with the following rabbit polyclonal antibodies at the indicated dilutions: anti-phospho-c-Jun (Ser63) (1:200; New England Biolabs, Beverly, MA), anti-Fos (1:3000; Santa Cruz Biotechnology, Santa Cruz, CA) and CM1 (1:5000; Idun Pharmaceuticals, La Jolla, CA). The secondary antibody was a Cy3-conjugated donkey anti-rabbit IgG (1.5 mg/ml; Jackson ImmunoResearch, West Grove, PA) diluted 1:400. Cytochrome c (1:1000; PharMingen, San Diego, CA), cytochrome oxidase VIc (1:500; Molecular Probes), and human and mouse BCL-2 (1:250; PharMingen) were detected with mouse monoclonal primary antibodies and FITC-conjugated donkey anti-mouse secondary antibody (0.6 mg/ml; Jackson ImmunoResearch) diluted 1:300.
BAX was detected with a rat monoclonal primary antibody (1:250; PharMingen) and a Cy3-conjugated anti-rat secondary antibody (1.5 mg/ml; Jackson ImmunoResearch) diluted 1:400. Several antibodies to BAX, both commercial and proprietary, were tested for specificity in our system. All of the BAX antibodies that we tested stained tissues from Bax-deficient mice. However, the faint, diffuse staining pattern seen in Bax−/−sympathetic neurons with the PharMingen antibody used in these studies did not change during NGF deprivation.
Cell counts: commitment point experiments. Sympathetic neurons from Bcl-2 transgenic mice and their wild-type littermates were deprived of NGF as described above. At various times after deprivation the cultures were rinsed with AM0 and then incubated in AM50 for 5−7 d. Neurons then were counted after being washed with PBS, fixed with freshly prepared 4% PFA for 30 min at 4°C, washed again with PBS, and stained with toluidine blue as described previously (Deckwerth and Johnson, 1993; Deshmukh et al., 1996).
Cell counts: BAX and cytochrome c immunocytochemistry.Sympathetic neurons that had been maintained in NGF for 4–5 d in AM50 were deprived of NGF in the presence of the caspase inhibitor BAF (as described above). At various times after deprivation or acute rescue by readdition of NGF, KCl, or CPT-cAMP the cultures were fixed and immunostained with anti-BAX and/or anti-cytochrome cantibodies. NGF-maintained sympathetic neurons exhibited diffuse BAX and punctate cytochrome c staining patterns. With NGF deprivation the cells showed punctate BAX and diffuse cytochromec staining patterns. For each time point the number of cells that had acquired a punctate staining pattern for BAX or had lost the punctate staining pattern for cytochrome c was determined by a naı̈ve observer from a random sampling of 100–250 cells. For the double immunocytochemistry experiments the staining patterns for both BAX and cytochrome c were assessed in the same neuron. All experiments were conducted in the presence of BAF to prevent any cell loss that otherwise would affect the counts.
Microinjections and quantitation of cell death. These experiments were performed as described previously (Deshmukh and Johnson, 1998). Briefly, sympathetic neurons were maintained in NGF for 4 d and treated for 24–48 hr with NGF, anti-NGF, or anti-NGF plus 1 μg/ml cycloheximide. Cells then were microinjected with 5 mg/ml bovine cytochrome c and rhodamine dextran dye. At each time point after the injections the number of microinjected cells that remained viable was determined and expressed as a percentage of the total number of microinjected cells.
Statistics. When indicated, statistical significance was determined by a Student’s t test or by a Mann–Whitney Rank Sum test for parametric and nonparametric data, respectively.
RESULTS
BAX undergoes caspase-independent subcellular redistribution after NGF deprivation in sympathetic neurons
Previous studies in several models of programmed cell death (PCD) show that BAX translocates from the cytosol to mitochondria when overexpressed and/or in response to certain death signals (Hsu et al., 1997; Wolter et al., 1997; Eskes et al., 1998; Gross et al., 1998;Rosse et al., 1998; Saikumar et al., 1998; Zhang et al., 1998; Finucane et al., 1999; McGinnis et al., 1999). We examined whether the translocation of endogenous BAX occurred in NGF-deprived sympathetic neurons. These studies were done in the presence of the caspase inhibitor BAF to prevent any cell death that otherwise might have complicated the determination of cell numbers. BAX redistribution also was observed in the absence of BAF; however, because redistribution was followed very rapidly by cell death in the absence of caspase inhibition, counting cells in the absence of BAF yielded a misleading estimate of the number of cells in which BAX had redistributed (data not shown).
We examined the subcellular localization of BAX by using immunocytochemistry rather than subcellular fractionation because of the impracticality of the latter, given the limited number of primary neurons obtained from each sympathetic ganglion in mice. Recently, two reports (using models of PCD that have not been shown to require endogenous BAX for apoptosis) suggest that BAX in non-neuronal cell lines does not translocate from the cytosol to mitochondria but instead that BAX already at mitochondria undergoes a conformational change that exposes an N-terminal epitope (Goping et al., 1998; Desagher et al., 1999). The immunocytochemistry results reported here cannot distinguish clearly between these two possibilities because the antibody used recognizes an epitope in the N terminus of BAX. However, because the preponderance of evidence in non-neuronal cell lines (see above) suggests that BAX undergoes translocation to mitochondria and not merely a conformational change at mitochondria, we shall, therefore, refer to the apparent redistribution we observed as translocation.
NGF-maintained sympathetic neurons primarily exhibited a diffuse staining pattern, consistent with a predominantly cytosolic localization for BAX (Fig.1a). A small fraction of cells displayed a faint, punctate staining pattern, suggesting that, although BAX normally may reside primarily in the cytosol, a small amount may exist in association with organelle membranes in NGF-maintained cells (data not shown). The pattern of BAX staining was altered in NGF-deprived sympathetic neurons from a diffuse, cytosolic pattern to a punctate pattern, consistent with the redistribution of BAX to intracellular membranes (Fig. 1b). Punctate BAX staining colocalized extensively with mitochondrial markers, such as cytochrome oxidase subunit VIc and Mitotracker Orange CM-H2TMRos (data not shown). As described previously (Deshmukh and Johnson, 1998; Neame et al., 1998), NGF deprivation also induced a change in the pattern of cytochromec staining from a punctate, mitochondrial pattern to a diffuse, cytosolic pattern (Fig.2b, f,j).

NGF deprivation in sympathetic neurons induces subcellular redistribution of BAX, followed by a loss of mitochondrial cytochrome c. Shown are representative photomicrographs of neurons immunostained for BAX (a, c) and the corresponding bisbenzimide-stained nuclei (b, d). Neurons were maintained for 5 d in vitro (DIV) in the presence of NGF and then switched to medium containing NGF (a, b) or deprived of NGF in the presence of 50 μm BAF for an additional 48 hr (c, d). The time course of development of punctate BAX staining (●) and loss of punctate cytochromec staining (▪) are shown (e). At various times after NGF deprivation, sympathetic neurons were fixed and immunostained for BAX or cytochrome c. Then the number of cells exhibiting punctate BAX or diffuse cytochrome cstaining was determined, with 200–250 cells counted for each time point. Mean ± SEM; n ≥ 3 independent experiments. Points indicated by asterisks are statistically different (*p = 0.002; **p = 0.035). Scale bar, 5 μm.

BAX and cytochrome c undergo subcellular redistribution during NGF deprivation in sympathetic neurons. Shown are representative photomicrographs of neurons immunostained for BAX (a, e, i), cytochrome c (b, f, j), or both (c, g, k) and the corresponding bisbenzimide-stained nuclei (d, h, l). Neurons either were maintained in NGF (a–d) or were deprived of NGF in the presence of 50 μm BAF for 12 hr (e–h) or 24 hr (i–l). The cells were fixed and immunostained for BAX and cytochrome c. Then the number of neurons (m) exhibiting diffuse BAX and punctate cytochrome c staining (State 1), punctate BAX and punctate cytochrome c staining (State 2), or punctate BAX and diffuse cytochrome cstaining (State 3) was determined, with 100–150 cells counted for each time point. Mean ± SEM; n ≥ 3. Scale bar, 10 μm.
The time courses for BAX and cytochrome c redistribution after NGF deprivation in sympathetic neurons were determined (see Fig.1e). Fifty percent of the neurons exhibited a punctate, mitochondrial BAX staining pattern by 11−13 hr, with nearly all of the neurons displaying redistribution of BAX by 24 hr. BAX redistribution was followed rapidly, but not instantaneously, by a loss of mitochondrial cytochrome c staining. Fifty percent of the neurons exhibited a diffuse, cytosolic pattern of cytochromec staining by 14−16 hr, with essentially all of the cells showing a loss of mitochondrial cytochrome c by 24 hr. The time course with which sympathetic neurons became committed to die (i.e., could not be rescued by NGF readdition) paralleled the time course of cytochrome c release, suggesting that the loss of mitochondrial cytochrome c commits these cells to die (see Figs. 1e, 5; data not shown).
As seen in Figure 1e, BAX redistribution to the mitochondria preceded loss of mitochondrial cytochrome c by 1−3 hr, suggesting that a transient “state” exists in a sympathetic neuron undergoing NGF deprivation-induced death in which both BAX and cytochrome c show mitochondrial localization. Immunocytochemistry for BAX and cytochrome c confirmed this prediction (Fig. 2). Sympathetic neurons maintained in NGF existed in “state 1,” in which BAX was distributed primarily throughout the cytoplasm and cytochrome c was localized mitochondrially (Fig. 2a–d, m). After 24 hr of NGF deprivation nearly all of the cells resided in “state 3,” with BAX and cytochromec predominantly localized to the mitochondria and cytosol, respectively (Fig. 2i–m). However, at an intermediate time point such as 12 hr after deprivation, we found evidence that cells transited through a short-lived “state 2,” in which both BAX and cytochrome c were localized mitochondrially (Fig. 2e–h, m). Thus, NGF deprivation in sympathetic neurons induced a caspase-independent subcellular redistribution of BAX, followed by a loss of cytochrome cfrom the mitochondria.
Redistribution of BAX and Cytochrome c is inhibited by the neuroprotective agents cycloheximide, cAMP, and KCl
Inhibitors of macromolecular synthesis such as cycloheximide prevent NGF deprivation-induced cell death in sympathetic neurons (Martin et al., 1988), aborting the PCD pathway upstream of cytochromec release and caspase activation (Deshmukh and Johnson, 1998; Neame et al., 1998) (data not shown). To determine whetherde novo protein synthesis was required for the redistribution of BAX during neuronal death, we examined the ability of cycloheximide to block this event. NGF-deprived, cycloheximide-saved neurons maintained a diffuse pattern of BAX staining even after 48 hr of NGF deprivation (Fig. 3a). Accordingly, cycloheximide also prevents the loss of mitochondrial cytochrome c during NGF deprivation (Fig. 3b) (Deshmukh and Johnson, 1998; Neame et al., 1998).

Effects of neuroprotectants on the redistribution of BAX and cytochrome c after NGF deprivation in sympathetic neurons. Neurons (5 DIV) were maintained in NGF or deprived of NGF in the presence of 50 μm BAF, 1 μg/ml cycloheximide (CHX), 40 mm KCl, or 400 μm CPT-cAMP. After 48 hr the neurons were fixed and immunostained for BAX (a) or cytochromec (b). Then the number of cells exhibiting punctate BAX staining or diffuse cytochrome cstaining was determined, with 200–250 cells counted for each condition. Mean ± SEM; n ≥ 3.
Although NGF is the physiological survival factor for sympathetic neurons in vivo, several other agents promote their survivalin vitro. Of these, two robust neuroprotectants are the cell-permeable cAMP analog, CPT-cAMP, and depolarizing concentrations of KCl (Rydel and Greene, 1988; Koike et al., 1989; Edwards et al., 1991; Deckwerth and Johnson, 1993; Franklin et al., 1995). Therefore, we examined the effects of these agents on the redistribution of BAX and cytochrome c during sympathetic neuronal death. Even after 48 hr of NGF deprivation in the presence of either KCl (40 mm) or CPT-cAMP (400 μm), neurons exhibited minimal redistribution of BAX and cytochromec (Fig. 3a,b).
Thus, NGF deprivation in sympathetic neurons induced a macromolecular synthesis-dependent, caspase-independent redistribution of BAX and cytochrome c. Potent neuroprotectants, such as KCl and cAMP, prevented both events.
Rescue with NGF, KCl, or cAMP prevents further release of cytochrome c, but only NGF acutely prevents further redistribution of BAX during neuronal death
NGF readdition within the first 12 hr after deprivation can fully rescue sympathetic neurons from neonatal rats (Edwards et al., 1991;Deckwerth and Johnson, 1993). Thereafter, sympathetic neurons start becoming irreversibly committed to die. Only 50% can be rescued by NGF readdition after 22 hr of deprivation; essentially none can be rescued after 48 hr of deprivation. (In neurons from C57BL/6 mice, these time points are shifted ∼4–6 hr earlier than in neurons from Sprague Dawley rats; see Materials and Methods.) Like NGF, either KCl, cAMP, or BAF can arrest sympathetic neuronal death post-translationally. Indeed, the time courses with which sympathetic neurons are rescued by NGF, KCl, cAMP, or BAF are indistinguishable (Edwards et al., 1991;Deckwerth and Johnson, 1993; Franklin et al., 1995; Deshmukh et al., 1996).
As described above, treatment with elevated concentrations of KCl or cAMP at the time of NGF withdrawal prevented the redistribution of BAX and cytochrome c (Fig. 3). To study the mechanisms by which NGF, KCl, or cAMP arrests sympathetic neuronal death post-translationally, we examined whether treatment with each of these agents 12 hr after NGF withdrawal acutely prevented further redistribution of BAX and cytochrome c. Cells were deprived of NGF in the presence of BAF for 12 hr, and the pattern of BAX or cytochrome c staining was assessed. In parallel cultures NGF, KCl, or cAMP was added back, and the pattern of BAX or cytochromec staining was determined 12 hr later. As shown in Figure4b, rescue with NGF, KCl, or cAMP acutely prevented any further loss of mitochondrial cytochromec in NGF-deprived sympathetic neurons, indicating that each of these agents rapidly can suppress cytochrome c release. However, only NGF, but not KCl or cAMP, prevented the further redistribution of BAX to mitochondria. These results suggested that cAMP and KCl acted after BAX redistributed to mitochondria to prevent cytochrome c release and cell death in sympathetic neurons.

NGF, KCl, and cAMP differ in their ability to rescue the redistribution of BAX and cytochrome c during sympathetic neuronal death. At 0, 6, 12, 18, or 24 hr after NGF deprivation in the presence of 50 μm BAF (●), sympathetic neurons were fixed and immunostained for BAX (a) or cytochrome c(b). In parallel cultures the neurons that were deprived of NGF in the presence of 50 μm BAF for 12 hr were washed and rescued with 300 ng/ml NGF (▴), 40 mm KCl (▪), or 400 μm CPT-cAMP (♦). After an additional 12 hr the number of cells exhibiting punctate BAX or diffuse cytochromec staining was determined, with 200–250 cells counted for each time point. In a, the neurons rescued with NGF were statistically different from those rescued with BAF, KCl, or CPT-cAMP (p < 0.001). In b, the neurons rescued with BAF were statistically different from those rescued with NGF, KCl, or CPT-cAMP (p < 0.001). Mean ± SEM; n ≥ 3.
If cells were rescued acutely with NGF and observed 5 d (not 12 hr) later, all cells returned to “state 1” (see Fig. 2) in which BAX was diffuse and cytochrome c was punctate (data not shown). We currently are investigating whether this phenomenon reflects reversibility in BAX redistribution or degradation of mitochondrial BAX and de novo synthesis of cytosolic BAX.
Overexpression of Bcl-2 inhibits cytochromec release, caspase activation, and cell death, but not BAX redistribution, during sympathetic neuronal death
Sympathetic neurons from Bax-deficient mice do not undergo apoptosis after NGF deprivation (Deckwerth et al., 1996). Similarly, overexpression of Bcl-2 or inhibition of caspases—by either genetic deletion or peptide inhibitors—protects numerous neuronal subpopulations, including sympathetic neurons, from developmental and induced cell death (Martinou et al., 1994; Greenlund et al., 1995; Deshmukh et al., 1996; Kuida et al., 1996, 1998; McCarthy et al., 1997; Hakem et al., 1998; Woo et al., 1998). To compare mechanistically the sites of action for BCL-2, BAX, and caspases, we examined sympathetic neuronal death in transgenic mice overexpressingBcl-2. These mice, which have been described extensively (Martinou et al., 1994), overexpress human Bcl-2 behind the neuron-specific enolase promoter, which is expressed from embryonic day 12.5 into adulthood. Western blot analysis confirmed expression of the transgene in sympathetic neurons (data not shown).
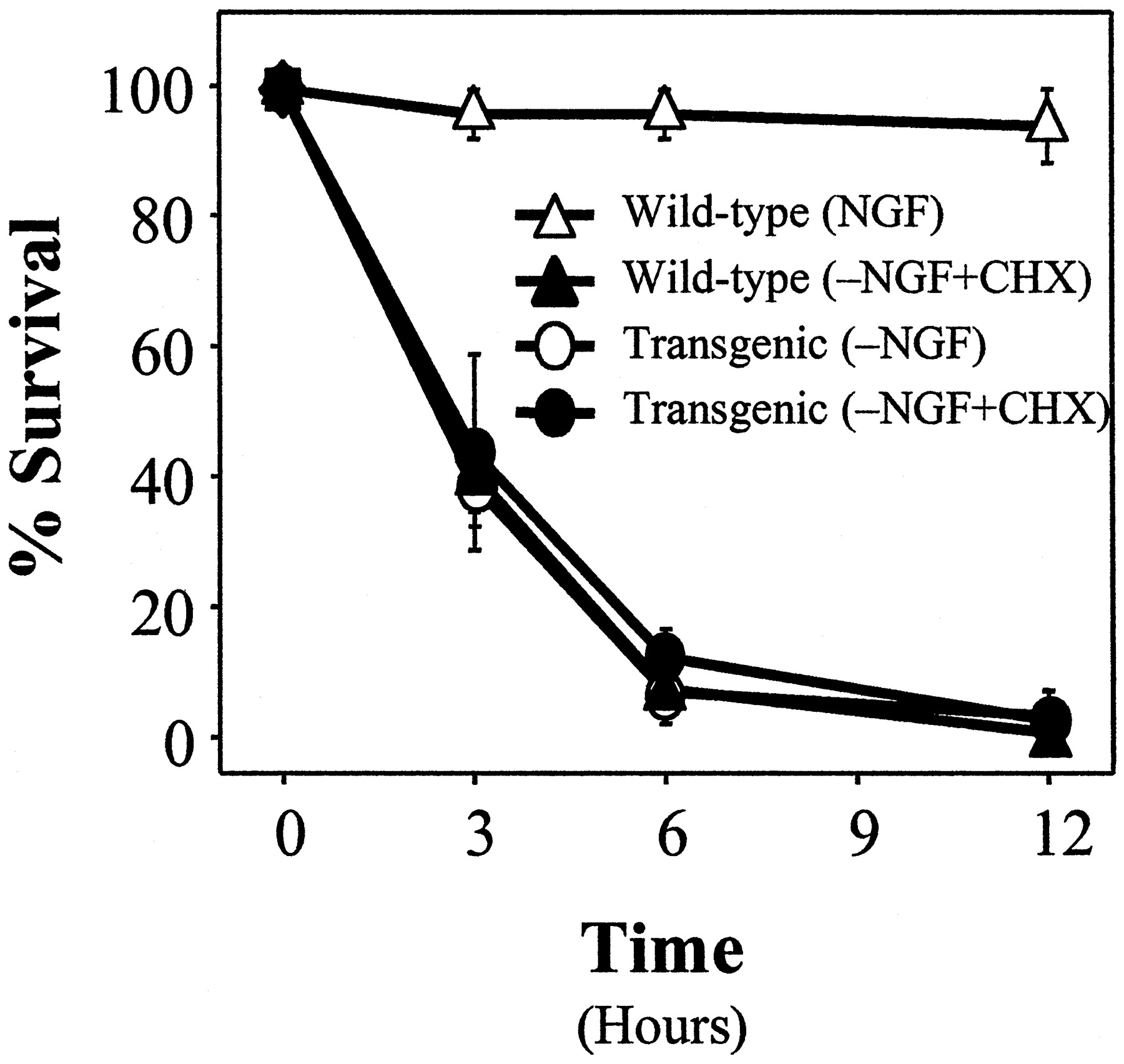
To assess the extent of protection afforded by overexpression ofBcl-2, we determined the time course of commitment-to-die in neurons from Bcl-2 transgenic mice. Sympathetic neurons from both transgenic and wild-type mice were deprived of NGF. At various times after deprivation, NGF was readded and cell survival was assessed 5–7 d later. As shown in Figure 5,Bcl-2 overexpression significantly delayed commitment-to-die from ∼12 hr to ∼3 d; however, unlike Bax deletion, overexpression of Bcl-2 did not prevent NGF deprivation-induced cell death completely in sympathetic neurons. Other studies (e.g., c-Jun phosphorylation and c-fos induction) indicated that Bcl-2 overexpression aborted NGF deprivation-induced cell death at a point that is mechanistically indistinguishable fromBax deletion (Deckwerth et al., 1998) (data not shown).

Overexpression of Bcl-2 inhibits NGF deprivation-induced cell death in sympathetic neurons. Neurons from wild-type (▴) and Bcl-2 transgenic (●) mice were maintained in NGF for 5 d and then deprived of NGF for various periods. At 1, 2, 3, or 7 d after deprivation the cells were washed and rescued with 50 ng/ml NGF. Then 5 d later the neurons were fixed, stained with toluidine blue, and counted. Mean ± SEM;n ≥ 3.
Bax deletion prevents the loss of cytochrome cfrom mitochondria (Deshmukh and Johnson, 1998). Similarly, overexpression of Bcl-2 orBcl-XL inhibits cytochrome crelease induced by different apoptotic stimuli in several non-neuronal cell lines (Kharbanda et al., 1997; Kluck et al., 1997; Yang et al., 1997; Duckett et al., 1998; Srinivasan et al., 1998a; Finucane et al., 1999). As shown in Figure6b, Bcl-2overexpression inhibited the loss of mitochondrial cytochromec during NGF deprivation in sympathetic neurons. Accordingly, we found that overexpression of Bcl-2 inhibited caspase activation, as determined by staining with the polyclonal antibody CM1, which recognizes activated caspases 3 and 7 (data not shown).

Overexpression of Bcl-2 inhibits the redistribution of cytochrome c, but not BAX, in NGF-deprived sympathetic neurons. Neurons from wild-type (▴) orBcl-2 transgenic (●) mice were maintained in NGF for 5 d and then deprived of NGF in the presence of 50 μm BAF for various periods. At 0, 12, 24, or 48 hr after deprivation the cells were fixed and immunostained for BAX (a) or cytochrome c(b); then the number of cells exhibiting punctate BAX or diffuse cytochrome c staining was determined, with 200–250 cells counted for each time point. Mean ± SEM;n ≥ 3.
To determine whether overexpression of Bcl-2 affects the subcellular localization of endogenous BAX during cell death, we examined the time course of BAX redistribution in sympathetic neurons from Bcl-2 transgenic mice. As shown in Figure6a, Bcl-2 overexpression neither prevented nor altered the rate of subcellular redistribution of endogenous BAX after NGF deprivation. We also found that overexpression of Bcl-2had no obvious effect on the distribution of endogenous BAX in NGF-maintained sympathetic neurons (Fig. 6a, 0 hr; data not shown). Thus, the results shown in Figure 6 indicate that the overexpression of Bcl-2 in sympathetic neurons inhibited cell death induced by NGF deprivation by preventing BAX-mediated cytochrome c release.
Overexpression of Bcl-2 does not inhibit cell death induced by microinjection of cytochrome c
As described above, the overexpression of Bcl-2 did not prevent BAX redistribution, but it did inhibit the loss of mitochondrial cytochrome c and caspase activation (Fig. 6). These findings indicate that BCL-2 acted at the level of mitochondria to inhibit NGF deprivation-induced cell death in sympathetic neurons. Recent reports suggest that overexpression of anti-apoptotic BCL-2 family members, such as Bcl-2 andBcl-XL, can prevent caspase activation and cell death downstream of or independent from cytochrome crelease (Li et al., 1997; Rosse et al., 1998). To determine whether BCL-2 can prevent neuronal death even after cytochrome ctranslocates to the cytosol, we examined whether overexpression ofBcl-2 inhibited the cell death induced by microinjection of cytochrome c into NGF-deprived sympathetic neurons. Previously, we have shown that microinjection of cytochromec into NGF-maintained neurons does not induce cell death. However, microinjection of cytochrome c into NGF-deprived, cycloheximide-treated or NGF-deprived, Bax-deficient neurons induces rapid cell death that is inhibitable by BAF, suggesting that NGF withdrawal induces two parallel processes, both of which are required for cell death: loss of mitochondrial cytochrome cand the development of competence-to-die (Deshmukh and Johnson, 1998).
As shown in Figure 7, microinjection of cytochrome c into NGF-deprived,Bcl-2-overexpressing sympathetic neurons in the presence or absence of cycloheximide induced cell death. Furthermore, the extent and time course of death after the injection of cytochrome cwas not affected by overexpression of Bcl-2. These findings also indicate that Bcl-2 overexpression—like Baxdeletion and cycloheximide treatment—did not prevent the development of competence-to-die. Taken together, the results shown in Figures 6and 7 indicate that BCL-2 appears to act solely at the level of mitochondria to prevent BAX-mediated cytochrome c release and cell death in NGF-deprived sympathetic neurons.

Overexpression of Bcl-2 does not prevent cell death induced by cytoplasmic microinjection of cytochromec into NGF-deprived sympathetic neurons. Cells from wild-type (▵ or ▴) or Bcl-2 transgenic (○ or ●) mice were maintained in NGF for 5 d and treated for 24–48 hr with NGF (▵), anti-NGF (○), or anti-NGF plus 1 μg/ml cycloheximide (CHX; ▴ and ●). Then the cells were microinjected with 5 mg/ml bovine cytochrome c and rhodamine dextran dye. At each time point after the injections the number of microinjected cells that remained viable was determined and expressed as a percentage of the total number of microinjected cells. Mean ± SEM; n ≥ 3.
DISCUSSION
BAX and cytochrome c undergo subcellular redistribution during sympathetic neuronal death
Despite the widespread expression of Bax in vivo,Bax-deficient mice exhibit relatively modest defects in non-neuronal lineages, perhaps because of functional redundancy with other proapoptotic BCL-2 family members (Knudson et al., 1995). In contrast, their phenotype in neuronal lineages, whether central or peripheral, is dramatic (Deckwerth et al., 1996; Miller et al., 1997;Shindler et al., 1997; White et al., 1998). For example, despite the coexpression of multiple proapoptotic BCL-2 family members, cell death induced by trophic factor deprivation in sympathetic and motor neurons is remarkably dependent on BAX alone. However, because neither the mRNA nor protein level of BAX increases during NGF deprivation-induced apoptosis in sympathetic neurons (Greenlund et al., 1995), BAX must be regulated post-translationally.
Recent evidence suggests that this regulation occurs at the level of subcellular localization of BAX. In several models of cell death the overexpression of Bax and/or certain death signals induce translocation of BAX from the cytosol to mitochondria (Hsu et al., 1997; Wolter et al., 1997), followed by cytochrome crelease, mitochondrial dysfunction, caspase activation, and cell death (Gross et al., 1998; Rosse et al., 1998; Saikumar et al., 1998; Zhang et al., 1998; Finucane et al., 1999; McGinnis et al., 1999). However, to our knowledge, none of these non-neuronal models of PCD requires endogenous Bax expression for cell death; in fact, a few have been shown specifically not to require BAX (Knudson et al., 1995). Moreover, Bax overexpression per se drives mitochondrial translocation (Rosse et al., 1998; our unpublished observations) and induces a cell death that may not require caspases and is not purely apoptotic (Xiang et al., 1996).
Neonatal sympathetic neurons deprived of NGF undergo an apoptotic cell death that requires de novo protein synthesis (Martin et al., 1988), Bax expression (Deckwerth et al., 1996), and the development of competence-to-die (Deshmukh and Johnson, 1998). In this study we found that NGF deprivation in sympathetic neurons induced a protein synthesis-dependent, caspase-independent redistribution of BAX from cytosol to mitochondria (see Figs. 1-3). This was followed rapidly, but not instantaneously, by cytochrome c release and commitment-to-die (see Figs. 2, 5). The identity of the specific gene product(s) induced during NGF deprivation and required for BAX redistribution remains unknown.
Recently, Goping et al. (1998) and Desagher et al. (1999) suggested that BAX does not translocate from the cytosol to mitochondria but instead that BAX already at mitochondria undergoes a conformational change that exposes an N-terminal epitope. The immunocytochemical findings reported here cannot distinguish clearly between the translocation of BAX to mitochondria and a conformational change in BAX at mitochondria. We currently are examining the feasibility of subcellular fractionation experiments to resolve this issue.
The precise mechanism by which BAX mediates loss of mitochondrial cytochrome c is unclear. Recent evidence suggests that two potentially interrelated mechanisms may mediate this release: the calcium-inducible, cyclosporin A (CsA)-sensitive permeability transition pore (PTP) and a Mg2+-sensitive, CsA-insensitive pathway that depends on BAX (or perhaps more generally on BH3 domain-containing, proapoptotic BCL-2 family members) (Eskes et al., 1998; Marzo et al., 1998; Narita et al., 1998). If BAX and the PTP mediate two biochemically and molecularly distinct pathways for cytochrome c release, four observations argue against a significant role for the PTP in sympathetic neuronal death. First, primary sympathetic neurons absolutely require BAX alone for cytochromec release (Deshmukh and Johnson, 1998), caspase activation, and apoptosis (Deckwerth et al., 1996). Second, mitochondria in NGF-deprived neurons do not exhibit the ultrastructural changes such as swelling and outer membrane rupture that should accompany permeability transition (Martin et al., 1988; Martinou et al., 1999). Third, nontoxic concentrations of CsA neither prevented nor delayed cytochromec release or cell death induced by NGF deprivation in murine sympathetic neurons (our unpublished observations). Fourth, we find that a significant delay in time occurred between cytochromec release and the loss of mitochondrial membrane potential (our unpublished observations). Thus, our data suggest that permeability transition does not mediate cytochrome crelease during trophic factor deprivation-induced cell death in neurons.
Acute rescue with NGF, but not KCl or cAMP, prevents redistribution of BAX
Cell death induced by NGF deprivation in sympathetic neurons can be arrested late in the apoptotic process by the readdition of NGF (Deckwerth and Johnson, 1993), KCl (Franklin et al., 1995), cAMP (Edwards et al., 1991), or BAF (Deshmukh et al., 1996). In fact, the time courses with which sympathetic neurons are rescued by NGF, KCl, cAMP, or BAF are indistinguishable. However, BAF, but not NGF, can inhibit sympathetic neuronal death after cytochrome crelease and caspase activation (Deshmukh and Johnson, 1998).
In this study we have identified other important differences in the post-translational mechanisms by which NGF, KCl, cAMP, or BAF aborts NGF deprivation-induced cell death in sympathetic neurons. Specifically, we found that NGF, KCl, or cAMP, but not BAF, prevented redistribution of BAX and cytochrome c when added at the time of deprivation (see Fig. 3). Similarly, acute rescue with NGF, KCl, or cAMP, but not BAF, after 12 hr of deprivation prevented further loss of cytochrome c from mitochondria (see Fig.4b). However, only NGF acutely prevented further redistribution of BAX (see Fig. 4a). These observations suggest that KCl or cAMP can act after BAX has redistributed to mitochondria to prevent cytochrome c release and cell death in sympathetic neurons. Our results cannot exclude the possibility that NGF also may act after BAX translocation to inhibit cytochromec release. These findings also imply that, although NGF, KCl, or cAMP each can abort the cell death cascade in sympathetic neurons at multiple checkpoints, only NGF, the physiological survival factor, may inhibit apoptosis at all known points before cytochromec release and caspase activation.
Bcl-2 overexpression prevents redistribution of cytochrome c, but not BAX, during PCD
Bax deletion and, to a lesser extent, caspase inhibition protect sympathetic neurons against NGF deprivation-induced apoptosis (Deckwerth et al., 1996; Deshmukh et al., 1996; Troy et al., 1996; McCarthy et al., 1997). In this study we found thatBcl-2 overexpression significantly delayed, but did not prevent, sympathetic neuronal death (see Fig. 5). Overexpression ofBcl-2 aborted the apoptotic cascade induced by NGF deprivation at a point that is mechanistically indistinguishable fromBax deletion.
In several non-neuronal cell lines the overexpression ofBcl-2 or Bcl-XL prevents—andBax promotes—the translocation of cytochrome cand cell death (Kharbanda et al., 1997; Kluck et al., 1997; Yang et al., 1997; Gross et al., 1998; Rosse et al., 1998; Finucane et al., 1999). To examine further the actions of BAX and BCL-2 in regulating the loss of cytochrome c from mitochondria in primary neurons, we examined the subcellular localization of BAX and cytochromec during NGF deprivation in Bcl-2-overexpressing sympathetic neurons. We observed that Bcl-2 overexpression, like Bax deletion, prevented the loss of mitochondrial cytochrome c to approximately the same extent that it inhibited cell death induced by NGF deprivation (compare Figs. 5,6b). We also found that overexpression of Bcl-2did not prevent—nor did it alter the kinetics of—the subcellular redistribution of endogenous BAX (see Fig. 6a).
Last, whether BCL-2 or BCL-XL can prevent caspase activation and cell death downstream of or independent from cytochromec release is unclear (Li et al., 1997; Rosse et al., 1998;Zhivotovsky et al., 1998). Presumably, this anti-apoptotic activity would be related to the interaction of BCL-2 or BCL-XL with Apaf-1 at the level of the apoptosome (Pan et al., 1998). However, in NGF-deprived sympathetic neurons in the presence or absence of cycloheximide, we observed that Bcl-2overexpression neither prevented nor altered the rate of cell death induced by microinjection of cytochrome c (see Fig.6c). Taken together, these observations indicate that BCL-2 functions primarily, if not exclusively, at the level of mitochondria to inhibit BAX-mediated cytochrome c release.
The role of macromolecular synthesis in apoptosis
Unlike apoptosis induced by NGF deprivation in sympathetic neurons, which requires de novo protein synthesis, Fas- or TNF-mediated cell death is facilitated by the inhibition of macromolecular synthesis. Recently, several groups reported that activation of the Fas and TNF-R1 receptors causes receptor oligomerization, recruitment and activation of procaspase-8, and N-terminal cleavage of BID; this is followed by translocation of the C-terminal fragment to mitochondria, cytochrome crelease, and cell death (Li et al., 1998; Luo et al., 1998; Gross et al., 1999). In light of our findings, we propose that a critical difference between macromolecular synthesis-dependent (e.g., NGF deprivation in sympathetic neurons) and independent (e.g., Fas or TNF treatment in non-neuronal cells) paradigms of cell death may be that the former require the expression of new gene products to induce the translocation of a BH3-domain-containing, proapoptotic BCL-2 family member to mitochondria, whereas the latter do not. In NGF-deprived sympathetic neurons, BAX alone appears to serve the role of this proapoptotic, BH3-containing protein that mediates the loss of cytochrome c, caspase activation, and cell death (Fig.8). In other models of cell death the identity of the proapoptotic, BH3-containing protein responsible for the loss of mitochondrial cytochrome c may vary according to cell type and apoptotic stimulus.

Schematic diagram of the sequence of critical events that occurs during sympathetic neuronal death induced by NGF deprivation. ROS, Reactive oxygen species;SOD, superoxide dismutase; CHX, cycloheximide; cyt c, cytochrome c. CHX appears to inhibit the induction of a gene required for the translocation of BAX to mitochondria. The identity of the specific gene product, the expression of which may be regulated by c-Jun, is unknown. Although NGF, KCl, or cAMP can block the cell death pathway in sympathetic neurons at multiple checkpoints, only NGF, the physiological survival factor, inhibits apoptosis at all known points before cytochrome c release and caspase activation.
Footnotes
This work was supported by National Institutes of Health Grant AG-12947 (E.M.J.) and a Paralyzed Veterans of America Spinal Cord Research Foundation grant (M.D.). We thank Drs. C. Michael Knudson (University of Iowa, Iowa City, IA) and Stanley Korsmeyer (Dana Farber Cancer Institute, Boston, MA) for the Bax-deficient mice; Drs. Jean-Claude Martinou (Serono Pharmaceutical Research Institute, Geneva, Switzerland) and Susumu Tonegawa (Massachusetts Institute of Technology, Boston, MA) for the Bcl-2 transgenic mice; and Dr. Anu Srinivasan (Idun Pharmaceuticals, La Jolla, CA) for the CM1 antibody. We also thank Krista Moulder, Dr. Brian Tsui-Pierchala, Charles Harris, Melanie Leitner, and Patricia A. Osborne for critical review of this manuscript; Dr. Malú Tansey for assistance with immunoblots; and Mary Bloomgren for secretarial assistance.
Correspondence should be addressed to Dr. Eugene M. Johnson, Jr., Department of Molecular Biology and Pharmacology, Washington University School of Medicine, 4566 Scott Avenue, Box 8103, St. Louis, MO 63110.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}