

Abstract
The role of neural cell adhesion molecule (NCAM) in the development and maturation of the neuromuscular junction (NMJ) was explored by characterizing structurally and functionally NMJs from postnatal day 11 (P11) to P30 +/+, +/−, and −/− NCAM null mutant mice. Differences in NCAM levels resulted in alterations in the size and shape of NMJs, with −/− NMJs being smaller. Additionally both the withdrawal of polyneuronal innervation and the selective accumulation of synaptic vesicle protein in the presynaptic terminal were delayed. These observations suggest that the bidirectional signaling responsible for these events is impaired at −/− NMJs. Functionally, miniature end plate potential size, end plate potential size, and quantal content did not differ from that of wild type under either normal or low release conditions. However at normal release conditions, −/− NMJs, unlike +/+ NMJs, lacked paired-pulse facilitation. The most striking abnormality was the inability of NCAM null junctions to maintain transmitter output with repetitive stimuli. Combined electrophysiological and FM1-43-labeling studies suggest that NCAM null junctions are unable either to dock or to mobilize a sufficient number of vesicles at high but physiological rates of transmitter release. Taken together our observations show that many aspects of transmission are normal and, thus, that many presynaptic and postsynaptic molecules have assembled properly in the absence of NCAM. However, the fact that NCAM was required for specific aspects of transmission, including paired-pulse facilitation and reliable transmission with repetitive stimuli, suggests that NCAM either is directly involved in these processes or is required for the proper organization and/or function of other molecules underlying these processes.
For proper nervous system function, developing neurons must grow to, and form connections with, their appropriate targets. Subsequent interactions transform these initial connections into highly specialized synapses (for review, seeSanes and Lichtman, 1999). During development of the neuromuscular junction (NMJ), synaptic vesicles that are initially distributed along the length of the motor axon become clustered at presynaptic active zones (Kelly and Zacks, 1969; Lupa and Hall, 1989; Dahm and Landmesser, 1991). Postsynaptically, the density of acetylcholine receptors (AChRs) increases dramatically both by the clustering of diffusely distributed AChRs (Anderson and Cohen, 1977; Frank and Fischbach, 1979; Nitkin et al., 1987) and by transcriptional activation of AChR genes in subsynaptic myonuclei (Usdin and Fischbach, 1986; Fischbach and Rosen, 1997). Postnatally, as the muscle fibers grow, the shape and complexity of the NMJ change, with small uniform plaques of AChRs expanding into pretzel-shaped structures as the nerve terminal grows (Balice-Gordon and Lichtman, 1990). Additionally, there is a withdrawal of all but one motor axon from the initially multiply innervated NMJ (Redfern, 1970;Brown et al., 1976; Balice-Gordon and Lichtman, 1993: Balice-Gordon et al., 1993).
These processes involve the translocation of numerous molecules and presumably alterations in adhesive interactions between prejunctional and postjunctional membranes. Cell adhesion molecules play critical roles in the development and maturation of the DrosophilaNMJ, as well as other vertebrate synapses (for review, see Fields and Itoh, 1996; Landmesser, 1997, 1998). Using the power ofDrosophila genetics, Goodman and colleagues showed that the stability and size of the NMJ were controlled by the relative level of fasciclin II (FasII) on developing muscle and nerve (Schuster et al., 1996a,b). Although synapses initially formed in mutants lacking FasII, they later regressed (Schuster et al., 1996a). Surprisingly, there was enhanced synaptic growth in mutants that expressed only 50% of the FasII protein. This observation becomes understandable by the demonstration that activity-dependent downregulation of FasII was both sufficient and necessary for the presynaptic sprouting that normally accompanies growth of the NMJ (Schuster et al., 1996b).
Does neural cell adhesion molecule (NCAM), the vertebrate homolog of FasII (Lin and Goodman, 1984), play a similar role at vertebrate NMJs? NCAM, specifically its polysialic acid (PSA) moiety, is required for the activity-dependent induction of long-term potentiation (LTP) and long-term depression (LTD) in the hippocampus (Muller et al., 1996). Sanes and colleagues showed recently that NMJs from NCAM null mice were smaller and that postsynaptic junctional fold development was delayed (Moscoso et al., 1998); however no functional studies were performed. To investigate possible roles of NCAM in the formation and maturation of the NMJ, we compared the structure and function of postnatal NMJs in wild-type and NCAM null mice. We found that NCAM-deficient end plates were smaller, synaptic efficacy was compromised, and transmitter release could not be sustained with repetitive stimuli, supporting a role for NCAM in the proper development, function, and plasticity of the synapse.
Parts of this paper have been published previously in abstract form (Rafuse et al., 1998).
MATERIALS AND METHODS
Mice
The NCAM-deficient mice used were originally generated by Cremer et al. (1994) in the C57B/6 strain and were subsequently raised and bred locally. For each experiment wild-type (+/+), heterozygote (+/−), and homozygote (−/−) mice were identified by PCR using primers that distinguish wild-type from mutant alleles (Cremer et al., 1994).
Immunostaining
Mice were killed in a CO2 chamber, and the semitendinosus muscles were quickly dissected and frozen without fixation in ornithine carbamyl transferase (OCT; Miles, Elkhart, IN) in isopentane that was cooled in dry ice. Rhodamine-conjugated α-bungarotoxin (α-BTX; Molecular Probes, Eugene, OR) and SV2 (a gift of K. Buckley, Harvard University) were used to visualize AChRs and synaptic vesicles, respectively. Briefly, 50 μm longitudinal sections were mounted on previously subbed glass slides, fixed in 3.7% formaldehyde for 10 min, rinsed, incubated overnight in rhodamine-conjugated α-BTX and SV2, washed several times in PBS, incubated for 2 hr in fluorescein-conjugated IgG secondary antibody (Sigma, St. Louis, MO), washed in PBS, and, finally, coverslipped with 50% glycerin–PBS containing 0.03 mg/ml p-phenylenediamine to prevent fading. Images were digitally photographed (40×) with an upright Nikon Diaphot 300 microscope equipped with a Javelin Ultrachip CCD camera (Javelin Electronics, Los Angeles, CA). Images were acquired and analyzed using an Argus Hamamatsu Image Processor (Hamamatsu Photonics K.K.) in series with the Metamorph Imaging System (Universal Imaging Corporation, West Chester, PA). The size of the NMJs was quantified by measuring both the area occupied by the AChRs and the circumference of the total end plate region. Acquired images were cropped in Corel Draw to generate the figures. Any modifications to the acquired digital figures were limited to changes in color brightness and contrast.
Muscle fiber cross-sectional area measurements
Mice were killed in a CO2 chamber, and the semitendinosus muscles were quickly removed, pinned at proximal and distal ends to a cork after being extended to their approximatein vivo length, which was measured when the leg was in a position midway between flexion and full extension, mounted in OCT (Miles), and immediately frozen in isopentane that was cooled in dry ice. Fourteen micrometer sections were taken from the midregion of the muscle and dried on a previously subbed glass slide. Sections were incubated overnight (4°C) with a monoclonal antibody (1:100 dilution) that exclusively recognizes slow skeletal muscle myosin (Sigma), fixed in 3.7% formaldehyde for 10 min, rinsed, washed several times in PBS, incubated for 2 hr (room temperature) with a goat anti-mouse fluorescein-conjugated secondary antibody (Sigma), washed in PBS, and, finally, mounted in 50% glycerin–PBS containing 0.03 mg/mlp-phenylenediamine to prevent fading. Images were digitally photographed (20× magnification) with a Nikon Diaphot 300 microscope as described above. Background fluorescence permitted visualization and cross-sectional area measurement of both nonlabeled fast skeletal muscle fibers and labeled slow fibers. The cross-sectional areas of individual muscle fibers in the midbelly region of the semitendinosus muscles were measured using the Metamorph Imaging system (Universal Imaging Corporation).
In vitro isometric tension and electromyogram measurements
Mice were killed in a CO2 chamber, and the semitendinosus muscles, along with the nerve supply and contributing spinal roots (L4–L6), were isolated and immediately placed into well oxygenated (95% O2 and 5% CO2) Tyrode's solution (125 mm NaCl, 24 mm NaHCO3, 5.37 mmKCl, 1 mm MgCl2, 1.8 mmCaCl2, and 5% dextrose). Extra care was taken to ensure that the spinal roots and nerve supply were left intact and not damaged. The proximal end of the muscle was carefully pinned in a Sylgard (Dow Corning, Midland, MI)-coated recording chamber containing well oxygenated Tyrode's solution maintained at 27–29°C. The distal muscle tendons were securely fastened with silk thread (no. 1) and tied to a force transducer (model 373; Harvard Apparatus, Holliston, MA) to measure total muscle force. In all cases a small piece of femur was left in continuity with the tendon to prevent slippage of the thread during maximal contractions. Two fine-tipped polyethylene suction stimulating electrodes [pulled from polyethylene tubing (PE-190; Clay Adams, Parsippany, NJ)] were used to stimulate each of the spinal nerves separately, or together, using a Grass S88 stimulator (Grass, Quincy, MA). Tight electrode seals made it possible to stimulate each spinal nerve separately without the spread of any electric current to the unstimulated spinal nerve (Landmesser and O'Donovan, 1984). Electromyogram (EMGs) were recorded from the midbelly of the semitendinosus muscles using a third polyethylene suction electrode, amplified with a bandwidth between 3 Hz and 10 kHz, and displayed on an oscilloscope (R5030; Tecktronix, Beaverton, OR). Short (0.5–1 msec), monophasic electrical stimuli, isolated from ground with a Grass PISU6P stimulus isolation unit, were used to evoke EMG and force responses that were displayed on a Gould chart recorder (Gould, Cleveland, OH) and stored on analog tape (Vetter, Rebersburg, PA) for later analysis. The signal was digitized in parallel using a Cygnus FLA-01 8 Pole Bessel Filter, connected to a analog-to-digital converter system (DigiData 1200; Axon Instruments, Foster City, CA), on a pentium Dell V-350 computer. Axoscope 8.0 (Axon Instruments) software was used to digitize (50 MHz) and perform measurements. In some cases, 10−7 to 10−5md-tubocurarine (d-TC; Sigma) was added to the Tyrode's solution to reduce the safety margin of transmission (Wernig and Herrera, 1986). At least 30 min was allowed after changing from one perfusion medium to another before EMG and force measurements were taken.
Electrophysiological determination of polyneuronal innervation
The force occlusion technique was used to estimate the presence or absence of polyneuronal innervation in the neonatal mice (Wernig and Herrera, 1986). Briefly, twitch and tetanus force responses were recorded after stimulation of each of the two contributing spinal nerves separately and then together. If each fiber is only innervated by a single motoneuron (i.e., not polyneuronally innervated), then the forces recorded by stimulating each spinal nerve separately should mathematically sum to equal the force recorded after stimulating both spinal nerves simultaneously. If each muscle fiber is innervated by more than one motoneuron (i.e., polyneuronally innervated), then the force evoked by stimulating both nerves simultaneously will be less than the mathematical summation of the forces recorded by stimulating each spinal nerve separately (see Results for further details).
Intracellular recording
Semitendinosus muscles, with the nerve supply intact, were isolated as described above and immediately placed into well oxygenated Tyrode's solution. The muscles were gently extended and pinned flat in a Sylgard (Dow Corning)-coated recording chamber. The nerves were sucked tight into polyethylene stimulating electrodes pulled from polyethylene tubing (PE-190; Clay Adams). Standard electrophysiological techniques were used to record miniature end plate potentials (mepps) and evoked end plate potentials (Epps) in a high-Mg2+, low-Ca2+ Tyrode's solution (125 mm NaCl, 5.37 mm KCl, 24 mmNaHCO3, 12 mmMgCl2, 1 mmCaCl2, and 5% dextrose). Alternatively, evoked Epps alone were recorded in normal Tyrode's solution (1 mmMg2+ and 1.8 mmCa2+) containing between 0.5 and 10 μmd-TC (Sigma) to block muscle contraction. Briefly, sharp glass electrodes were pulled (20–40 MΩ resistance) and filled with 3 m KCl, and single muscle fibers were impaled near the motor end plate. The initial resting potentials were between −70 to −85 mV and usually remained stable throughout the duration of the experiments. Electrophysiological measurements were not recorded if the resting potential decreased by >15% of its original value. Potentials were recorded via an intracellular amplifier (World Precision Instruments) using Axotape or Axoscope software (10 kHz sampling rate; Axon Instruments) and stored simultaneously on a ZIP drive (Iomega, Roy, UT) for later analysis. mepps (100–200) were recorded from each muscle fiber in high-Mg2+, low-Ca2+ Tyrode's solution over a 2–4 min recording period. Single Epps (recorded at 0.5 Hz for 3–4 min), paired-pulse Epps, or trains of Epps (5–200 Hz) were recorded in high-Mg2+, low-Ca2+ Tyrode's solution or in normal Tyrode's solution containing 0.5–10 μmd-TC, using short (0.5–1 msec), monophasic electrical stimuli isolated from ground with a Grass stimulus isolation unit.
FM1-43 optical imaging
FM1-43 dye uptake during a 10 min depolarization with high K+. In an initial series of experiments the dye FM1-43 (Betz and Bewick, 1993; Reid et al., 1999) obtained from Molecular Probes was loaded into presynaptic vesicles by high-K+ depolarization, in a muscle in which end plates have been labeled previously by exposure to 20 μm rhodamine α-bungarotoxin for 1 hr. The extracellular solution bathing a muscle was then switched from normal saline to one containing 60 mm K+ and 12 mm FM1-43 for 7 min. The preparation was then washed with saline containing 12 mm Mg+2and 0.5 mm Ca+2 for 15 min to wash the dye from the bath and to reduce spontaneous release of vesicles during this process. End plates were then visualized with a 40× water immersion objective on a Nikon Microphot fluorescence microscope equipped with the appropriate filter cubes for rhodamine and FM1-43. Different concentrations of FM1-43 and different loading times were tried, with the procedure described above producing the most intense staining of end plates with minimal nonspecific labeling of other structures. Pictures were taken with a Nikon camera at the same exposure and filter settings so that the pattern and intensity of staining could be compared between wild-type and NCAM null junctions.
Dynamic imaging of vesicle release with FM1-43. In a subsequent series of experiments, muscles in which end plates had been made visible by labeling with 20 μm α-bungarotoxin for 30 min had FM1-43 loaded into synaptic vesicles by stimulating the muscle nerve for 10 min at 10 Hz, while the muscle was bathed in normal saline containing 12 μm FM1-43. These parameters produced optimal loading of the dye. After loading, the preparation was washed with high Mg+2 (1.75 mm) and low Ca+2 (0.5 mm) for 10 min to remove the dye from the solution. Images were then captured with a 40× water immersion objective on a Nikon Microphot microscope by means of an SIT camera (Hamamatsu C2400) connected to a frame grabber (Matrox IM-LC) on a computer (Dell pentium V350) in parallel with a videocassette recorder (Toshiba KV-6200A). A neutral density filter was used to reduce light damage and to ensure that the camera was within its linear range. At the same time a second image was captured to show rhodamine α-bungarotoxin labeling to demonstrate overall end plate morphology. The preparation was then returned to normal saline (1.8 mm Ca+2 and 1 mmMg+2) for 5 min, and images were captured every 10 sec. To evaluate changes in the intensity of labeling, the videotape was played back, and the selected frames were captured and digitized using the same computer system. Images were stored digitally and later color-coded for pixel intensity using the Metamorph Image Analysis System. The montage and labeling of the pseudocolored images were created using Corel Draw 7.0. Loss of dye with time after trains of stimuli at different frequencies allowed us to monitor synaptic vesicle exocytosis via the loss of FM1-43 fluorescence.
RESULTS
Alterations in the size and shape of end plates in NCAM null and heterozygous mice
Mammalian neuromuscular junctions are initially formed during embryonic development, and at birth in mice and rats all end plates are polyneuronally innervated (Redfern, 1970; Brown et al., 1976;Balice-Gordon et al., 1993). Fibers become singly innervated during the first 2 postnatal weeks, and in the semitendinosus muscle that was the focus of the present study, we found this process to be complete by postnatal day 11 (P11) in +/+ mice. Thus P11 was chosen as the first time point to assess the effect of the absence of NCAM on the size and shape of end plates.
By P11, AChRs had become clustered at NCAM null end plates at a density comparable with that in +/+ end plates as judged by the intensity of rhodamine α-BTX labeling (Fig.1A,B). Because end plates are complex structures including regions of high AChR density interspersed with regions lacking AChRs, we quantified end plate size in two ways: (1) the total area exhibiting high AChR density (Figs. 1, 2, BTX-binding area histograms) and (2) the total area enclosed by a line encircling the end plate and thus containing areas of both high and low AChR density (Figs. 1, 2, total end plate area histograms). By either measure, end plates from P11 −/− mice were ∼20% smaller than were those from +/+ littermates. Although there was considerable overlap in the end plate size histograms (Fig. 1), those from the NCAM null mice (Fig. 1D) were clearly skewed toward smaller values than were those from +/+ mice (Fig.1C), this reduction being statistically significant (p < 0.05, one-way ANOVA).

Size and shape of end plates in P11 NCAM-deficient and wild-type neuromuscular junctions. A, Three examples of P11 wild-type end plates visualized with rhodamine α-bungarotoxin staining show regions of high AChR density. C, The total area of high AChR density was quantified (BTX-binding area) for 42 end plates, and the values are expressed in the left histogram as the proportion of end plates occurring within each bin size. The right histogram illustrates the total end plate area and includes the area contained within a line encircling the end plate (i.e., including regions of high AChR density as well as the interspersed areas that lack AChRs). B, Three examples of similarly stained P11 NCAM null end plates are shown.D, In general the NCAM null synapses are smaller in size, as is apparent from the histograms showing the distribution of the BTX-binding area (left) and the total end plate area (right) from 46 NCAM null end plates.

Size and shape of end plates from P30 wild-type, heterozygous, and NCAM null mice. A,Left, Two examples of P30 wild-type end plates stained with rhodamine α-bungarotoxin are shown. Right,Histograms show the size distribution for the BTX-binding area (left histogram) and the total end plate area [area enclosed by a line encircling the end plate and containing regions of both high and low ACh receptor density (right histogram)] from 65 wild-type end plates.B, Both the examples of BTX-stained NCAM null end plates (left) and the histograms(right) generated from 64 such end plates show that both the BTX-binding areas (left histogram) and total end plate areas (right histogram) are skewed toward smaller sizes, compared with wild type. C, Left, Two examples of similarly stained end plates from a heterozygous mouse illustrate that such end plates were often less compact.Right, Although the area of membrane containing a high density of ACh receptors was similar to that of wild type (left histogram; 65 junctions analyzed), many junctions had total end plate areas (right histogram) that were larger than that of wild type, because of the less compact shape of the end plates.
As muscle fiber diameter increased during the next several weeks, end plates in both +/+ and −/− mice increased in size. However as shown by the histograms for both BTX-binding area and total end plate area, NCAM null end plates at P30 (Fig. 2B) remained smaller in size than did wild-type end plates (Fig.2A); the histograms were clearly skewed toward smaller sizes, and the mean size was reduced by ∼12%. At P30 we also analyzed +/− mice, which would be expected to have NCAM levels of ∼50% of wild type. The size of these end plates, as measured by the area of the membrane with high AChR density, was not different from control (Fig. 2A,C, left histograms). Thus the apparent reduction in NCAM levels to 50% of wild type did not result in an increase in the junctional area as occurred inDrosophila when FasII was reduced by 50% (Schuster et al., 1996a). We noticed, however, that end plates in +/− mice were much less compact than were those of either +/+ or −/− mice. Although not all end plates were as open in shape as those shown in Figure2C, there was a strong tendency for +/− end plates to be less compact and thus to occupy more total end plate area (Fig. 2, compare A,C, right histograms; the means were statistically different, p < 0.05, one-way ANOVA). These observations suggest that differing levels of NCAM can affect the process by which end plates grow in size during postnatal maturation.
Because end plate size is usually well matched to the diameter of the postsynaptic muscle fiber to maintain effective neuromuscular transmission (Kuno et al., 1971; Harris and Ribchester, 1979), end plates in NCAM null mice might be smaller simply because muscle fibers were smaller. This was in fact suggested in a previous study (Moscoso et al., 1998) that found that end plates in the sternomastoid muscle of NCAM null mice were ∼12% smaller than those of wild type, a reduction similar to what we report in the present study. We therefore measured muscle fiber diameter in P30 +/+ and −/− semitendinosus muscles from frozen cross sections taken through the middle portion of the distal head of the muscle. This head, from which all end plate morphology and most electrophysiology were obtained, is composed primarily of fast-twitch muscle fibers but also contains a small number of slow-twitch fibers that are confined to the internal portion of the muscle (these appear in Fig.3A–C as dark profiles because they were stained with a slow myosin-specific antibody). The process of primary and secondary myogenesis, which produces this pattern of fiber type distribution, appeared to have occurred normally in the NCAM null mice, because both the number and distribution of slow fibers (Fig. 3A,C) were similar to that of wild type (Fig. 3B). Furthermore the mean fiber cross-sectional area did not differ between +/+ and −/−, as indicated by the histograms in Figure 3, D andE, respectively (p > 0.05, one-way ANOVA). The histogram of muscle fiber cross-sectional areas was clearly not skewed toward smaller sizes as was the end plate areahistograms (see Fig. 2) in these mice, as would be expected if the smaller end plate areas were a result of smaller muscle fiber diameters. The slow-twitch fibers (Fig. 3D,E, filled bars) were smaller than the fast-twitch fibers (open bars) in both +/+ and −/−. Thus, overall, the absence of NCAM and PSA does not affect the process of myogenesis or alter the size of the muscle fibers. Although we did not quantify total muscle fiber or motoneuron number, as judged from the size of the muscles and the diameters of the muscle nerves, there did not appear to be a reduction in these parameters in the NCAM null mice.
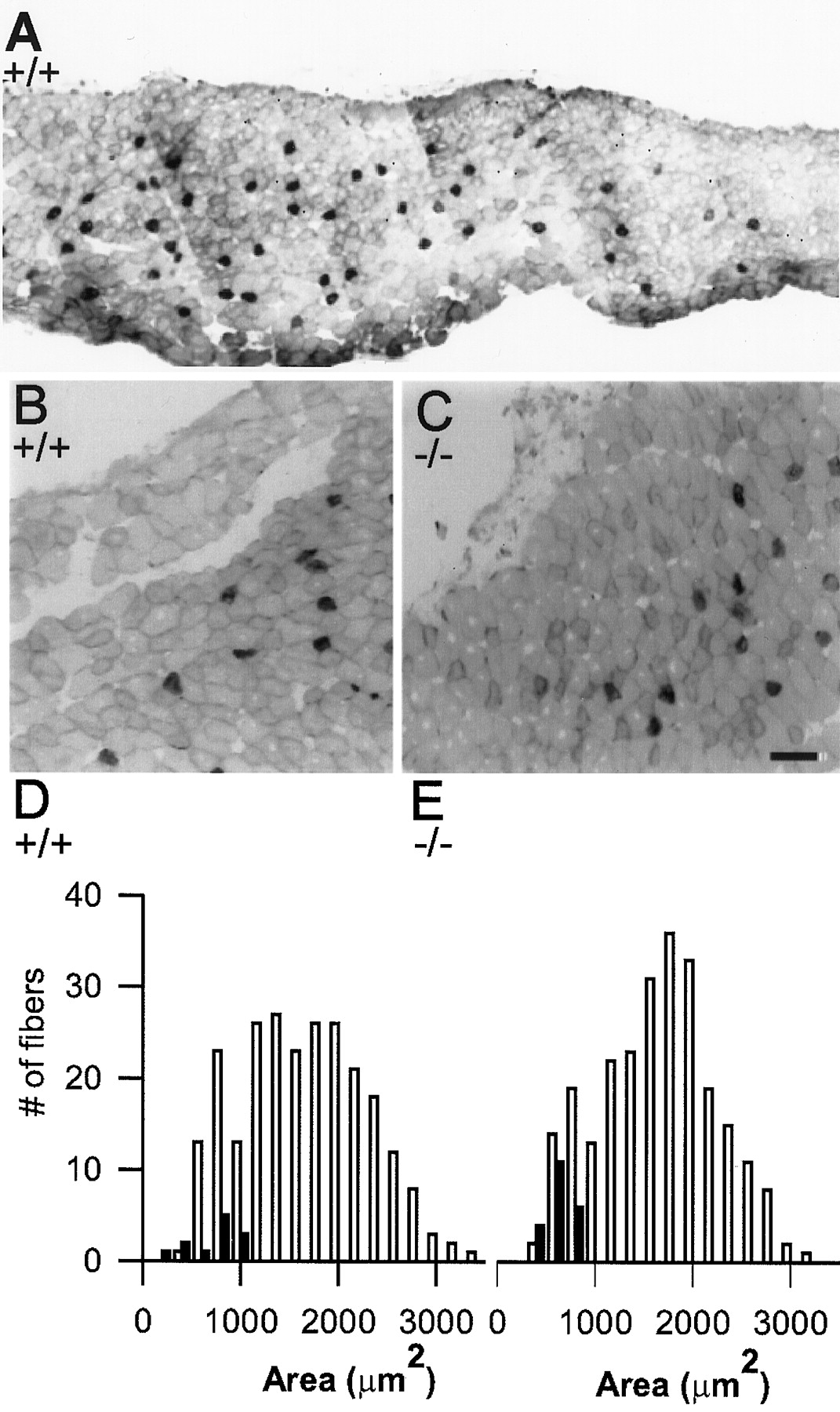
Size and spatial distribution of fast- and slow-twitch muscle fibers in the semitendinosus muscle from wild-type and NCAM null P30 mice. A, Low-magnification view of a frozen cross section from a wild-type muscle stained with an antibody specific for slow myosin. The slow-twitch muscle fibers (stained darkly) are sparsely distributed throughout the interior of the muscle, interspersed with larger fast-twitch muscle fibers that comprise the majority of the muscle. B, C, Higher magnification views of portions of wild-type (B) and NCAM null (C) muscles showing that the distribution and size of both slow- and fast-twitch muscle fibers are similar. D, E, Histograms showing the cross-sectional area distributions of all fibers within a square region taken from the center of a wild-type (D) and NCAM null (E) muscle and extending from the top to the bottom surface. The slow-twitch fibers are illustrated as filled bars; fast-twitch fibers are open bars in thehistograms. It is clear that thehistogram of muscle fiber cross-sectional areas from the NCAM-deficient muscle (E) is not highly skewed toward smaller sizes as were the end plate areahistograms from the (−/−) mice shown in Figure 2. Scale bar: A, 82 μm; B, C, 50 μm.
NCAM null mice exhibit a delay in the withdrawal of polyneuronal innervation
We estimated the extent of polyneuronal innervation from P0 to P30 functionally by the method of tension occlusion. If all muscle fibers were singly innervated, then the tension obtained by stimulating those fibers innervated by spinal nerve L4 alone and the tension obtained by stimulating those innervated by L5 alone should sum linearly when L4 and L5 are stimulated simultaneously. We found that by this criteria both +/+ and +/− semitendinosus muscles were no longer polyneuronally innervated by P11 (see, for example, Fig.4, top). In contrast, P11 −/− muscles did not exhibit any summation of tension and appeared to be completely polyneuronally innervated (Fig. 4, middle). It is also apparent that the P11 −/− muscle exhibited extensive fatigue during the 1 sec, 50 Hz train. The reason for this fatigue, which was not observed under similar conditions in +/+ or +/− muscles, will be considered below. In this figure, muscles were activated by a 1 sec train at 50 Hz. Comparable data were also obtained when twitch tension was analyzed (data not shown). By P17 (Fig. 4, bottom), withdrawal of polyneuronal innervation also had taken place in the −/− mice. Thus the absence of NCAM delays, but does not prevent, the process of neonatal synapse elimination. Similar observations have been made morphologically for NCAM null sternomastoid muscles (Moscoso et al., 1998).

Tension produced by semitendinosus muscles in response to a 50 Hz, 1 sec train of stimuli applied to spinal nerves L4, L5, or L4 + L5. Top, Traces from a P11 heterozygote show that the tension produced by stimulating both nerves simultaneously is equivalent to the sum of the tensions produced by stimulating L4 and L5 separately, indicating that the majority of the muscle fibers are no longer polyneuronally innervated.Middle, In contrast, a P11 homozygous muscle does not exhibit any summation of tension, indicating that most muscle fibers are still polyneuronally innervated. In addition the NCAM null muscle is unable to maintain tension for 1 sec when stimulated at the 50 Hz frequency. Bottom, By P17, the NCAM null muscle exhibits summation of tension, indicating that synapse elimination is primarily complete. By P17 the NCAM null muscle is also able to maintain tension when stimulated at 50 Hz for 1 sec.
NCAM null mice exhibit a delay in the selective localization of the synaptic vesicle antigen SV2 to the end plate
After the growing tip of an axon stops and forms a synapse, there is a rapid redistribution of synaptic vesicle proteins from an initial diffuse localization along the nerve to the presynaptic terminal where they become highly concentrated (Lupa and Hall, 1989; Dahm and Landmesser, 1991). Thus by P11 in +/+ end plates, SV2 immunolabeling (Fig. 5A) was confined to the region immediately overlaying the end plate as visualized with BTX staining (Fig. 5B). In contrast, in NCAM null end plates, SV2 immunolabeling remained high within the nerve, as well as at the end plate. This allowed us to confirm by morphological means that these NCAM null junctions were polyneuronally innervated (Fig.5C,E, arrows indicate separate axons). Synaptic vesicle proteins gradually became cleared from the intramuscular axons in NCAM null mice, but even at P30, pale labeling of the preterminal axon was often visible in NCAM null but not wild-type synapses. These observations suggest that the process by which synaptic vesicle proteins (and presumably synaptic vesicles) become selectively targeted to the presynaptic terminal occurs less effectively in NCAM-deficient mice.

The distribution of the synaptic vesicle protein SV2 at P11 wild-type and NCAM null end plates. A, In the wild-type muscle, the synaptic vesicle antigen SV2 selectively localized to the presynaptic nerve terminals. B, The same wild-type terminals stained with rhodamine α-bungarotoxin, showing good colocalization with the SV2 staining shown inA. C, E, Two examples of SV2 staining from a P11 NCAM null muscle, showing extensive staining for SV2 all along the intramuscular axons, in addition to the presynaptic terminal overlying the end plate. This extensive axonal staining also allows morphological confirmation that NCAM null junctions are polyneuronally innervated at this stage of development. Arrows indicate separate axons that innervate the same end plate. D, F, α-Bungarotoxin labeling of the same NCAM null end plates shown inC and E, respectively. Scale bar, 50 μm.
Synaptic efficacy is reduced in NCAM-deficient muscles during repetitive stimulation
While making tension recordings to assess the degree of polyneuronal innervation, we observed that NCAM null muscles often exhibited a falloff in tension during the 1 sec stimulation trains especially at higher stimulus frequencies (100 Hz or higher), whereas wild-type muscles were able to maintain tension effectively (Fig. 4,middle). Because NCAM null muscles could maintain tension when directly stimulated (data not shown), this suggested that the decline in tension was synaptic in origin.
To address this possibility more directly, we recorded compound action potentials (EMGs) with suction electrodes from +/+ and −/− muscles in Tyrode's solution containing normal Ca2+and Mg2+. As shown in the example in Figure 6A, the wild-type junction (top) maintained the original level of transmission for the first five stimuli during a 200 Hz train. Even by the end of the 1 sec train, the compound action potentials were ∼50% of the original amplitude. In contrast, the NCAM null junction (Fig.6A, bottom) showed a decrement in transmission even during the first five stimuli, and transmission had failed completely by the end of the train. Although there was some variation in the degree of transmission failure from muscle to muscle, in general all of the NCAM null muscles showed a severe decrement in transmission at higher stimulus frequencies (i.e., 100 and 200 Hz; Fig.6B), indicating that in most fibers transmission had fallen below threshold for action potential generation. This appears to be a result of defective synaptic transmission, because direct recording of compound action potentials from the muscle nerve as it enters the muscle indicated that NCAM-deficient axons were able to conduct action potentials without failure even at repetition rates in excess of 200 Hz (data not shown). This decline in effective transmission was accentuated in preparations in whichd-TC had partially blocked the NMJ (data not shown). This inability of NCAM null junctions to maintain transmission at high repetition rates does not appear to reflect simply a delay in synaptic maturation, because P65 junctions exhibited the same defect (Fig. 6B).

EMG recordings from P30 wild-type and NCAM null muscles reveal differences in the efficacy of NCAM null end plates when activated repetitively. All data were obtained from muscles with physiological levels of Ca2+ (1.8 mm) and Mg2+ (1 mm). A, Examples of EMGs from a wild-type (top) and NCAM null (bottom) muscle stimulated at 200 Hz. B,Bar graphs showing the ratios of the amplitude of the fifth over the first response (left) and the last over the first response (right) at P30 (open bars, wild type; black filled bars, NCAM null) and at P65 (grayfilled bars, NCAM null). The bars represent the mean ± SE from 28 recordings from six muscles from three different wild-type mice, 22 recordings from four muscles from three different NCAM null mice at P30, and 14 recordings from two different muscles from one NCAM null mouse at P65. Each recording was performed twice with 1 sec trains of 10, 20, 50, 100, and 200 Hz with 8–10 sec between each train.
In summary these observations show that NCAM null junctions are more prone to failure especially with repetitive stimulation. The retardation of junctional fold development that has been described (Moscoso et al., 1998) could reduce the safety factor for transmission (Wood and Slater, 1997). Additionally a reduction in the amount of transmitter released could contribute to the observed fatigue. To define better which presynaptic or postsynaptic changes might contribute to this reduction in synaptic efficacy, we characterized different parameters of transmission at P30 junctions using intracellular recordings, limiting our studies to +/+ and −/− mice.
Properties of synaptic transmission at P30 junctions under conditions of low transmitter release
To prevent contraction and to allow for quantification of both spontaneous mepps and evoked Epps, we reduced the probability of release by raising Mg2+ to 12 mm and lowering Ca2+ to 1 mm, resulting in the release of only several quanta per stimulus. Under these conditions, mepp amplitudes were unimodally distributed, and for all the cells that were analyzed, the entire distribution was clearly above noise level. The mean mepp amplitude (Fig. 7A) did not differ between +/+ and −/− junctions, indicating that a single quantum of transmitter produced the same postsynaptic depolarization in both cases. Because muscle fiber diameters did not differ on average between +/+ and −/− (see Fig. 3), the input resistance of the fibers would be expected to be similar. This suggests that both the amount of transmitter contained per vesicle and its effect, which would be dependent on the density of ACh receptors, are essentially normal at junctions lacking NCAM. There was however a slight decrease in the frequency of mepps compared with that at wild-type junctions (Fig.7B).

Parameters of synaptic transmission at P30 wild-type and NCAM null junctions under conditions of low transmitter release (extracellular Ca+2, 1 mm; Mg+2, 12 mm). A,Bar graph showing mean mepp amplitude (Amp.) ± SD. There was no statistical difference between +/+ and −/− end plates (p > 0.05, one-way ANOVA). B, Bar graph showing mepp frequency, which was slightly reduced at the −/− junctions (p < 0.05, one-way ANOVA).C, Bar graph showing Epp amplitude in response to a single suprathreshold stimulus to the nerve. The −/− and +/+ junctions were not statistically different (p > 0.05, one-way ANOVA).D, Bar graph showing mean quantal content ± SD for Epps evoked by a single stimulus and calculated from the ratio of mean Epp amplitude/mean mepp amplitude. The quantal contents were not statistically different between +/+ and −/− junctions. E, Responses to pairs of pulses to determine paired-pulse facilitation. Top, Tracefrom +/+ junctions showing facilitation at 10, 6, and 4 msec intervals (left to right).Bottom, Trace from a −/− junction showing similar facilitation at the three intervals. F,Bar graph showing the mean amplitude ± SD of the second Epp of the pair as a percent of the amplitude of the first Epp in the pair at 10, 6, and 4 msec intervals. Both +/+ and −/− junctions showed a similar amount of facilitation at each interval. Data were obtained from 10 +/+ and 10 −/− junctions.
When release was evoked by single suprathreshold stimuli to the nerve, the Epp amplitude also did not differ from that of wild type (Fig.7C). Furthermore quantal content calculated by the ratio of the Epp/mepp amplitude was ∼2 for both +/+ and −/− junctions (Fig.7D). Finally, NCAM-deficient junctions exhibited paired-pulse facilitation to the same level as wild-type junctions at 10, 6, and 4 msec intervals (Fig. 7E,F). Taken together these observations suggest that most of the presynaptic and postsynaptic machinery required for normal synaptic transmission has become properly assembled at NMJs in the absence of NCAM.
Properties of synaptic transmission at P30 junctions under normal levels of transmitter release
To study synaptic transmission at levels of transmitter release that would be expected in vivo, we recorded from junctions in normal Ca2+ and Mg2+, by using d-TC (5 μm) to reduce transmission to below threshold for muscle action potential generation, thereby preventing contraction. Under these conditions Epp amplitude did not differ between +/+ and −/− junctions (Fig.8A). Because this concentration of d-TC reduced mepp amplitude to within the noise level, we estimated quantal content from the coefficient of variation of Epp amplitude (Miyamoto, 1975; Clements and Silver, 2000). The number of quanta released by single stimuli did not differ significantly between wild-type and NCAM null junctions and was ∼60 at this stage of development (Fig. 8B). However, unlike wild-type synapses that continued to show paired-pulse facilitation at normal levels of transmitter release, the NCAM-deficient synapses failed to show paired-pulse facilitation at intervals between 6 and 50 msec. Examples of traces at the 6 and 10 msec intervals are shown in Figure 8, C andD. At the 4 msec interval, the NCAM-deficient synapses in fact exhibited a moderate degree of depression (Fig.8C,D).

Parameters of synaptic transmission at P30 wild-type and NCAM null junctions at normal levels of transmitter release. All data were collected in a physiological saline containingin vivo levels of Ca+2 (1.8 mm) and Mg+2 (1 mm) and 5 μmd-TC to partially block end plates to avoid muscle contraction. A, Mean Epp amplitudes ± SD for P30 +/+ and −/− junctions were not statistically different.B, Quantal content, calculated from the coefficient of variation of Epp amplitude (see Materials and Methods for additional details), also did not differ between +/+ and −/− junctions (p > 0.05, one-way ANOVA for both A, B). C, Epps to pairs of pulses separated by 10, 6, or 4 msec (left to right) to assay paired-pulse facilitation are shown. Top,Trace from a +/+ junction shows that the response to the second stimulus is facilitated at all intervals. Bottom,Trace from an NCAM null junction that did not exhibit facilitation at any of the intervals is shown. D,Bar graphs show the mean amplitude ± SD of the second Epp as a percent of the first. Although the wild-type junctions (open bars) exhibited a modest degree of facilitation at 10, 6, and 4 msec intervals, the NCAM null junctions (filled bars) did not exhibit facilitation at the 10 and 6 msec intervals and were in fact depressed at the 4 msec interval.
NCAM-deficient synapses are unable to sustain transmitter output with repetitive stimuli
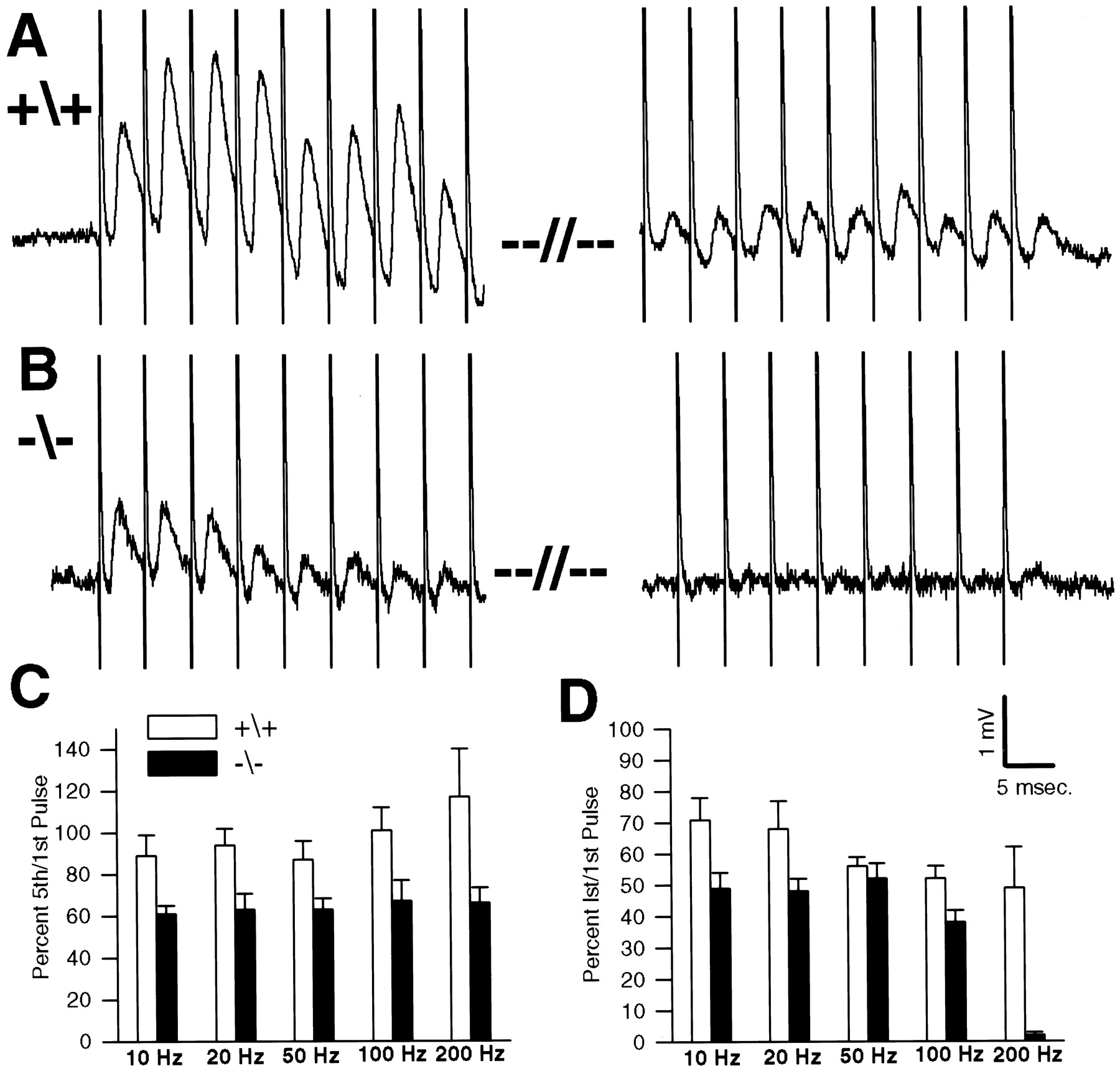
P30 wild-type and NCAM-deficient junctions were next stimulated in 5 μmd-TC and normal Ca2+ and Mg2+with 1 sec trains of stimuli at 10, 20, 50, 100, and 200 Hz. In agreement with the extracellular recordings of compound action potentials, we observed that NCAM-deficient junctions were unable to maintain effective transmission when challenged with multiple stimuli. Such a deficiency was most pronounced at high repetition rates as shown by the 200 Hz, 1 sec train illustrated in Figure9A (the traceon the left shows Epps occurring at the beginning of the train; the trace on the right occurs at the end of the train). The wild-type junction shows some facilitation of Epp amplitude during the first few stimuli and even at this high stimulation rate was able to maintain Epp amplitude at ∼50% of the initial value at the end of the 1 sec train (see also Fig.9C,D, bar graphs). In contrast, the NCAM null synapse did not show facilitation, and even by the ninth stimulus, Epp amplitude was already reduced to 25% of the initial value (Fig.9B). Transmission at this junction had essentially failed by the end of the 1 sec train. Although most extreme at high stimulus rates, as illustrated by the bar graph in Figure9D, the NCAM-deficient synapses showed some falloff in efficacy of transmission even at moderate repetition rates (i.e., 10 and 20 Hz; Fig. 9C).

Inability of NCAM null junctions to maintain transmitter output when stimulated repetitively. All data were obtained from intracellular recordings from muscles in saline with physiological levels of Ca+2 (1.8 mm) and Mg+2 (1 mm) and 5 μmd-TC to block contraction. A, Epps from a wild-type junction during a 1 sec, 200 Hz train are shown. The Epps exhibited facilitation during the beginning of the train (left) and even by the end of the train (right) were ∼50% of the initial Epp amplitude.B, A similar trace from an NCAM null junction is shown. There is no facilitation at the beginning of the train (left), and even by the ninth stimulus, Epp amplitude was reduced to less than one-half of the initial Epp. By the end of the train (right), transmission had almost completely failed. C, D, Bar graphs show the mean amplitude ± SD of the fifth and the last (lst) Epp in the train, respectively, as a percent of the first Epp for a number of junctions at a variety of different stimulus repetition rates. C, Compared with wild type, the NCAM null junctions show a rapid depression in transmission to ∼60% by the fifth pulse at all frequencies tested. D, By the end of the 1 sec train, the NCAM null junctions are more depressed than wild type at all intervals except for 50 Hz. The depression with respect to wild type is most extreme at 200 Hz where the wild-type junctions maintain transmission at ∼50% of the initial value, whereas the NCAM null junctions are almost completely blocked.
Thus one of the most striking defects in the NCAM-deficient NMJ is an inability to sustain transmission at an optimal level over time. This would most likely represent a presynaptic defect in the ability either to release synaptic vesicles or to mobilize vesicles from the reserve pool, after those in the readily releasable pool (Kuromi and Kidikoro, 1998; Stevens and Sullivan, 1998; Wang and Kaczmarek, 1998) have been exhausted. To confirm that the reduction in Epp amplitude does in fact represent a reduction in the number of quanta (vesicles) released per stimulus, we calculated quantal content from the coefficient of variation of Epp amplitude (Miyamoto, 1975) at several wild-type and NCAM-deficient junctions after a series of 1 sec trains separated by sufficient time for full recovery. We chose the 10th stimulus of the 200 Hz train to compare because in NCAM null synapses, Epp amplitude was considerably depressed but not completely blocked as at the end of the train. As an example, the quantal content in one junction was reduced from 94 to 18 by the 10th stimulus. We quantified this reduction for eight NCAM null junctions and found a mean reduction of 68.8% (range, 41–82%).
The data showing a difference in synaptic depression between wild-type and NCAM null junctions were obtained in the presence of 5 μmd-TC to prevent muscle contraction. Although this is an accepted procedure, it is known thatd-TC can enhance synaptic depression at NMJs by acting on presynaptic ACh receptors (Magelby et al., 1981; Tian et al., 1994). However we observed a similar difference between +/+ and −/− junctions in synaptic depression in the absence of d-TC, while recording EMGs during repetitive stimulation as described previously (Fig. 6). Thus it does not seem that differences in the sensitivity of wild-type and NCAM null junctions to this action ofd-TC can account for our observations.
One explanation for the excessive depression at NCAM null junctions would be that the pool of releasable vesicles is so small that all have been exhausted by the end of the 200 Hz, 1 sec train (Schneggenburger et al., 1999). Alternatively, the NCAM-deficient synapses may have difficulty in mobilizing or docking vesicles at these high rates (Pozzo-Miller et al., 1999; Wu and Borst, 1999). To distinguish better between these possibilities, we have made preliminary observations of transmitter release properties using the dye FM1-43 (Betz and Bewick, 1993; Reid et al., 1999).
Aspects of synaptic vesicle pool size and release dynamics as revealed by FM1-43 labeling
The fluorescent dye FM1-43 is taken up into synaptic vesicles during the process of exocytosis/endocytosis and can be used in conjunction with electrophysiology to estimate synaptic vesicle pool size and to monitor both temporally and spatially synaptic vesicle dynamics (Betz and Bewick, 1993; Reid et al., 1999). After determining optimum conditions for loading with FM1-43 (see Materials and Methods for details), we found that with low-frequency stimulation both wild-type and NCAM null synapses could be loaded with dye to approximately the same level. Although there was some variation from junction to junction, as shown by the photos in Figure10, junctions from both types of animals exhibited similar intensities of staining, which was unevenly distributed, with intense hot spots of staining interspersed with lower intensity, somewhat more diffuse staining. These patterns are similar to those described by others at both mouse and rat NMJs (Ribchester et al., 1994; Reid et al., 1999). When comparing FM1-43 labeling with rhodamine BTX staining of the same junctions to delineate the end plate area, all regions of the NCAM-deficient end plates appeared capable of being loaded.

Wild-type and NCAM null junctions labeled by FM1-43 dye uptake to indicate synaptic vesicles participating in the exocytotic/endocytotic cycle. A, B, Two wild-type junctions visualized with rhodamine α-bungarotoxin to label postsynaptic ACh receptors. C, D, The same junctions inA and B, respectively, visualized by FM1-43 taken up during a 7 min depolarization in 60 mm KCl containing 12 mm FM1-43. E, F, Two NCAM null junctions visualized with rhodamine α-bungarotoxin. G, H, The same junctions in E and F, respectively, visualized by FM1-43 after depolarization as described above. Scale bar, 30 μm.
To visualize FM1-43 labeling dynamically and to compare the loss of dye with the amount of transmitter release detected electrophysiologically, we acquired FM1-43 images with a digital camera after complete loading and at various intervals during stimulation at 200 Hz. In Figure11 the first pair of images (left) shows a wild-type (top) and NCAM-deficient (bottom) junction at the end of FM1-43 loading, when the intensity of staining has been color-coded. At the end of a 1 sec, 200 Hz train, dye has been lost from both junctions, but a large amount of labeled vesicles remains in both. After an additional 30 sec of stimulation, much of the remaining dye can be released from the NCAM-deficient junction. Thus the failure in transmission cannot be caused simply by a very small pool of releasable vesicles, which is completely exhausted by the end of the 1 sec train. The observations rather suggest some defect in the NCAM-deficient junctions either in docking vesicles at the presynaptic active zones at these high stimulus rates or in mobilizing vesicles from the reserve to the readily releasable pool (Stevens and Sullivan, 1998; Wang and Kaczmarek, 1998). Nevertheless, as Figure 11 shows, a consistent finding, which at least superficially seems at odds with this suggestion, was that NCAM-deficient junctions lost FM1-43 labeling much more rapidly than did wild-type junctions (L. Polo-Parada and L. T. Landmesser, unpublished observations). Furthermore this rate of destaining was inconsistent with the much smaller number of quanta detected electrophysiologically. One possibility is that many of the vesicles loaded with FM1-43 in the −/− junctions do not contain transmitter. Another possibility is that at the NCAM-deficient junctions, vesicles are also released at sites that are not directly apposed to the postsynaptic muscle membrane and therefore are not detected by intracellular recordings. Ongoing experiments are being performed to distinguish among these possibilities and to determine the cellular mechanism underlying the depression we have observed.

Loss of FM1-43 label from a wild-type (top row) and NCAM null (bottom row) junction by electrical stimulation of the nerve. The two junctions were loaded by stimulating the nerve for 10 min at 10 Hz in normal saline.Left, After washout of FM1-43 from the bath in high-Mg+2, low-Ca+2 saline for 10 min, the muscle was perfused with physiological saline, and an image was captured. Middle, An image was captured at the end of a 1 sec, 200 Hz train. Right, Dye remaining after another 30 sec of stimulation at 200 Hz is shown. All images were captured with a digital camera, and pixel intensity was later color-coded by means of the Metamorph imaging program, withred indicating the highest intensity.
DISCUSSION
Although many aspects of neuromuscular maturation occurred normally in NCAM null mice (see also Moscoso et al., 1998), we detected a number of differences including the size and shape of end plates and delays in the withdrawal of polyneuronal innervation and the selective targeting of synaptic vesicle proteins to the presynaptic terminal. Most striking were specific functional defects in synaptic transmission: an absence of paired-pulse facilitation and an inability to maintain effective transmission with repetitive stimuli. Each of these differences will be discussed in turn.
NCAM can act as a homophilic adhesion molecule (Rutishauser and Jessell, 1988) and could contribute to initial NMJ formation by promoting interactions between the presynaptic and postsynaptic membrane. However the large width of the synaptic cleft and the interposition of a basal lamina would seem to preclude such homophilic interactions across postnatal NMJs. It seems more likely that NCAM, which postnatally becomes concentrated both prejunctionally and postjunctionally (Covault and Sanes, 1986), would interact heterophilically with other molecules in the synaptic basal lamina, such as neural agrin (Ferns et al., 1993), to which it can bind (Burg et al., 1995). NCAM might also interact with other molecules enriched in synaptic regions including synaptic laminins (Sanes et al., 1990;Patton et al., 1997; Sanes and Lichtman, 1999). Such heterophilic interactions, acting via adhesion or signaling in either acis or trans manner, could contribute to the structural and functional alterations that we documented. PSA can also modulate NCAM function (Rutishauser and Landmesser, 1996), and a role for it cannot be excluded until effects of its enzymatic removal on junctional properties of +/+ mice are determined. However we found that junctional PSA levels were quite low during the postnatal period we investigated.
Alterations in size and shape of NMJs that lack NCAM
The usual tight relationship between end plate size and muscle fiber diameter (Kuno et al., 1971; Harris and Ribchester, 1979) ensures effective transmission. Such size matching was impaired in NCAM null mutants because end plates were smaller although fiber diameter was normal. We also observed differences in the shape and compactness of end plates in both −/− and +/− compared with +/+ mice. Furthermore, the +/− and −/− mice differed from each other. The diverse end plate shapes result from the loss of AChRs under portions of the initially plaque-shaped endings during the first few postnatal weeks (Balice-Gordon and Lichtman, 1993), a process that appears to involve bidirectional signaling. Our results indicate that this process is somehow affected by differing NCAM levels. Subsequent growth without shape change has been proposed to be mediated by adhesion between terminal and muscle fibers, resulting in intercalary terminal growth as the muscle fiber increases in size (Balice-Gordon and Lichtman, 1990). This could also be affected by alterations in NCAM-mediated adhesion or signaling. Ectopic expression of muscle NCAM in mice was shown recently to result in terminal sprouting and increased NMJ area (Walsh et al., 2000). In Aplysia (Mayford et al., 1992; Zhu et al., 1995) and Drosophila (Schuster et al., 1996a,b) alterations in levels of NCAM-like molecules also affect synaptic growth and plasticity. Additional insight into how differing levels of NCAM affect the size and shape of mouse NMJs, and how this might relate to the observations in invertebrates, will probably require dynamic observations of junctions over time (Balice-Gordon and Lichtman, 1990).
Presynaptic structural maturation is delayed in the absence of NCAM
Immature motor axons have synaptic vesicle proteins diffusely distributed (Lupa and Hall, 1989; Dahm and Landmesser, 1991) and are able to release ACh (Chow and Poo, 1985) along their length. Contact with a muscle fiber results in specialization of the presynaptic terminal, with synaptic vesicle proteins becoming concentrated there and cleared from the rest of the axon (Lupa and Hall, 1989; Dahm and Landmesser, 1991). Although the synaptic vesicle protein SV2 became concentrated at NCAM null junctions, this was delayed and incomplete even by P30. This observation also suggests some impairment in the process of bidirectional signaling at the −/− NMJ.
Neonatal synapse elimination is delayed in NCAM null junctions
Neonatal synapse elimination is a competitive (Betz et al., 1989;Balice-Gordon and Lichtman, 1994) and activity-dependent (O'Brien et al., 1978; Thompson, 1985; Busetto et al., 2000) process by which all but one of the multiple nerve terminals are eliminated from the NMJ (for review, see Sanes and Lichtman, 1999). We found that synapse elimination was significantly delayed in the NCAM null semitendinosus muscle, as also observed for the sternomastoid muscle (Moscoso et al., 1998). Why this should result from lack of NCAM is not immediately apparent, although a general retardation in development seems unlikely. Nerve ingrowth and initial synapse formation occurred between embryonic day 12 (E12) and E14 as in +/+ mice (S. Banerjee and L. T. Landmesser, unpublished observations), and other aspects of neuromuscular development occurred on schedule including secondary myogenesis and growth of muscle fibers.
Because synapse elimination is activity dependent, the fact that NCAM null junctions were less effective, especially with repetitive stimulation, might have contributed to this delay. Alternatively, the competitive process by which one terminal becomes functionally stronger at the expense of the other (Balice-Gordon et al., 1993; Colman et al., 1997) might be affected more directly by the absence of NCAM.Kopp and Balice-Gordon (1999) found recently that differences in the probability of transmitter release between competing terminals precede subsequent structural alterations and synapse elimination. The absence of paired-pulse facilitation under normal release conditions at NCAM null junctions suggests that the release probability may already be maximal. This in turn could impede early events in the divergence of synaptic strength that contribute to synapse elimination.
Alterations in the function of NCAM null junctions
At P30, many properties of transmission appeared normal in NCAM null homozygotes. Mepp amplitude was normal, as were Epp amplitude and quantal content at both low and normal levels of transmitter release. That both Epp amplitude and quantal content were normal, although the junctional areas in NCAM −/− mice were skewed toward smaller sizes, suggests that some functional compensation (see Sandrock et al., 1997) may have occurred. Paired-pulse facilitation was also normal under low transmitter release conditions. Taken together these observations indicate that the large number of molecules required for both presynaptic and postsynaptic function (Sanes and Lichtman, 1999) become appropriately organized without NCAM.
Nevertheless we observed two striking differences in transmission. First, at normal levels of extracellular Ca+2 and Mg+2, NCAM null junctions did not exhibit paired-pulse facilitation, suggesting that some aspect of the release process had become saturated. This might occur if intracellular Ca+2 levels were elevated or if the release machinery was more sensitive to Ca+2 in −/− terminals, resulting in a maximal probability of release even to the first stimulus. Alternatively, the entire readily releasable pool of vesicles (Kuromi and Kidikoro, 1998; Stevens and Sullivan, 1998; Wang and Kaczmarek, 1998; Schneggenburger et al., 1999) might be released by each stimulus, leaving no additional vesicles or docking sites for enhanced release to the second stimulus (for review, see Zucker, 1999). Ongoing studies to measure presynaptic Ca+2, to estimate the binomial release parameters n and p, and to measure vesicle pool sizes with FM1-43 imaging and EM should help distinguish among these possibilities.
The second major defect was a profound inability of NCAM null junctions to maintain transmitter output with repetitive stimuli. Because quantal content to single stimuli did not differ from +/+, this synaptic fatigue cannot result from an increase in the number of released vesicles to compensate for a postsynaptic reduction in efficacy. This process occurred in neuregulin heterozygotes (Sandrock et al., 1997) and resulted in synaptic fatigue. Total vesicle pool size appeared approximately normal on the basis of the amount of FM1-43 that could be loaded, and all of this dye was releasable by stimuli to the nerve. Thus NCAM null junctions appear to have some defect in docking and releasing the appropriate number of vesicles at high stimulus repetition rates. Whether this is caused by some defect in the machinery involved in docking and release or in mobilizing vesicles to replenish those lost (see Pozzo-Miller et al., 1999) remains to be determined. As noted previously there may be additional alterations in vesicle cycling and release at the NCAM-deficient junctions that may contribute to the observed depression.
Although NCAM null mice lack hippocampal LTP and LTD, this appears to be caused by a lack of PSA (Muller et al., 1996). It will be interesting to see whether the alterations we have found at the NMJ also occur in central synapses. A more complete characterization of the dynamics of synaptic vesicle cycling should help to define the mechanisms underlying the transmission defects and thus the potential role of NCAM. It is possible that presynaptic NCAM, acting in acis manner, helps to organize other molecules involved in transmitter release (see Thomas et al., 1997). If true, this would be a novel role for NCAM, and we are currently determining whether there are any alterations in the level or distribution of such molecules in NCAM-deficient NMJs.
Footnotes
This work was supported by National Institutes of Health Grants NS 19640 and NS 23678 from the National Institute of Neurological Diseases and Stroke and by a McKnight Senior Investigator Award. We thank Harold Cremer for making available the NCAM null mice and Urs Rutishauser and Huaiyu Hu for supplying us with mice and for initial help in genotyping, this latter effort being supported by National Institutes of Health Grant HD 18369 to Urs Rutishauser. We also thank Roman Fatkiyev for much of the anatomical analysis and Christian Bose for improved genotyping. We thank Guillermo Pilar, Ben Strowbridge, Christian Bose, M. Usiak, and M. Gartz Hanson for helpful advice on the experiments and this manuscript.
V.F.R. and L.P.-P. contributed equally to this work.
Correspondence should be addressed to Dr. Lynn Landmesser, Department of Neurosciences, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106-4975. E-mail: ltl{at}po.cwru.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}