

Abstract
During late-embryonic development, retinal neurons lose the ability to attach and extend neurites on the extracellular matrix molecule laminin-1 (LN-1), despite the fact that they retain expression of integrin receptors for LN-1. Here we show that the developmental loss of responsiveness to LN-1 can be reversed by treatments that increase the activation state of integrins. Both extracellular application of Mn2+ (at micromolar concentrations) and viral-mediated neuronal expression of a constitutively active form of the ras-related GTPase R-ras (R-ras38V) potently promoted late-embryonic retinal neurite outgrowth on LN-1 substrata. In both cases, outgrowth was mediated by integrin α6β1 and not α3β1, even though these neurons express α3β1 and use it for outgrowth on other laminin isoforms, as well as on LN-1 that has been proteolytically or conformationally activated (Ivins et al., 1998). Mn2+—and to a much lesser extent R-ras38V—also reversed the developmental loss of retinal neuron responsiveness to type IV collagen, by promoting the function of integrin α1β1. Interestingly, the responses of other late-embryonic CNS neurons to LN-1 were also enhanced by treatments that activate integrin function, but those of peripheral nervous system neurons (dorsal root ganglion neurons) were either not enhanced (embryonic neurons) or only modestly improved (adult neurons). These results suggest that a developmental decline occurs in the activation state of neuronal integrins, particularly among CNS neurons. Such a decline may underlie some of the intrinsic loss of regenerative ability sustained by CNS neurons during development and may be a valid target for therapeutic intervention.
Ligand binding to integrins, the major cell-surface receptors for the extracellular matrix (ECM), initiates signal transduction cascades that can lead to altered gene expression, differentiation, cell motility, or neurite outgrowth (Schwartz et al., 1995). The strength of the integrin–ligand interaction is regulated by a process known as integrin activation. Integrins may be activated from the “outside-in” by changes in extracellular ion concentrations or by integrin-binding monoclonal antibodies (Arroyo et al., 1993; Bazzoni et al., 1995, 1998). Integrins may also be activated from the “inside-out.” For example, treatment of some cells with phorbol esters dramatically increases the adhesive properties of certain integrins (Schwartz et al., 1995). In several cells, introduction of a constitutively active form of the ras-related GTPase R-ras (R-ras38V) enhances integrin-dependent attachment to ECM ligands (Zhang et al., 1996).
The consequences of neuronal interactions with laminins have primarily been studied in vitro. Laminin-1 (LN-1), the most extensively studied isoform, is often less effective than other laminins in stimulating neurite outgrowth. In the retina, responsiveness to LN-1 is under tight developmental control. Retinal neurons are highly LN-1-responsive in early neural development but become virtually nonresponsive at late-embryonic stages (Cohen et al., 1986; Hall et al., 1987), while retaining the ability to respond to other LN isoforms (Cohen and Johnson, 1991; Ivins et al., 1998). Retinal neurons also lose responsiveness to type IV collagen over the same time period (Bradshaw et al., 1995). These changes in neuronal responsiveness to the ECM are paralleled by a loss of regenerative potential in vivo (So et al., 1981; Chen et al., 1995). Thus, developmental changes in neuronal responsiveness to LN-1 may reflect a global change in neuronal physiology that contributes to regenerative failure.
Recently, we showed that the attachment and neurite outgrowth response of late-embryonic retinal neurons to LN-1 could be restored by manipulations of the LN-1 molecule, such as proteolytic cleavages that isolate LN-1's long arm or decoration of the short arms with antibodies (Calof et al., 1994; Ivins et al., 1998). The receptors used by late-embryonic retinal neurons to respond to LN-1 that had been “activated” in these ways were the same β1-containing integrins that early-embryonic retinal neurons use to respond to nonactivated LN-1 and that late-embryonic retinal neurons use to respond to other LN isoforms (Hall et al., 1987; Cohen and Johnson, 1991; Calof et al., 1994; Ivins et al., 1998). These observations suggest that developmental changes in responsiveness to LN-1 are not caused by a loss of appropriate integrins but rather by a relative decrease in integrin function that differentially affects responsiveness to particular ECM ligands. One hypothesis for how such a decrease occurs is via changes in the activation state of integrins.
As a step toward testing this hypothesis, the effects of both outside-in and inside-out regulators of integrin function on the responses of neurons to laminins and other ECM molecules were studied. The data show that changes in integrin activation differentially affect neuronal responsiveness to different ECM ligands and that the developmental loss of CNS neurite outgrowth in response to ECM ligands can be reversed by increasing integrin activation. Interestingly, among the different types of neurons tested [CNS vs peripheral nervous system (PNS); embryonic vs adult], the ability of integrin activators to improve neurite outgrowth on ECM substrata tended to vary inversely with the regenerative potential of the neurons, suggesting that a low state of integrin activation may be characteristic of poorly regenerating neurons.
MATERIALS AND METHODS
Cell culture. Neuronal cell cultures were established and maintained from embryonic day 18 rat retinas, hippocampi, and cortices and embryonic day 16 mouse retinas under serum-free conditions as described (Ivins et al., 1998). Sensory neurons from embryonic day 7 chick embryo dorsal root ganglia (DRG) were enzymatically dissociated and cultured under serum-free conditions as described (Kindt and Lander, 1995). Adult mouse DRG were prepared and cultured on LN-1 as described (Birgbauer et al., 1991) in F-12 media supplemented with 10% fetal bovine serum and 100 ng/ml 2.5 S nerve growth factor (NGF). Embryonic mouse DRG were cultured in DMEM/F-12 (1:1) supplemented with 0.5% BSA, N2 components, and 100 ng/ml NGF. ECM components were prepared and used as described previously (Ivins et al., 1998). Collagen IV (Collaborative Research, Bedford, MA) was used at a coating concentration of 24 μg/ml and was applied overnight at 4°C.
Antibodies. Function-blocking monoclonal hamster anti-rat and -mouse α1 [clone Ha31/8 (Mendrick et al., 1995)], α2 [clone Ha1/29 (Mendrick and Kelly, 1993)], and β1 [clone Ha2/5 (Mendrick et al., 1995)] integrins were obtained from PharMingen (San Diego, CA) and were used in cell culture experiments at a final concentration of 10 μg/ml. Ralph 3.1 (DeFreitas et al., 1995), a function-blocking anti-rat α3 integrin mouse monoclonal antibody was prepared and used as described (Ivins et al., 1998). The function-blocking rat anti-mouse α6 integrin monoclonal antibody GoH3 was obtained from AMAC (Westbrook, ME) and was used at a concentration of 2 μg/ml. The preparation and use of all anti-LN-1 antibodies have also been described (Calof et al., 1994; Ivins et al., 1998). All antibodies or other drugs were added to cultures at the time of plating.
Immunocytochemistry and morphometry. For β-galactosidase histochemistry, cells were fixed by underlaying the culture medium for 10 min at room temperature with 1% formaldehyde and 0.2% glutaraldehyde in PBS containing 5% sucrose. The fixative was removed, and cells were reacted with a solution containing 5-bromo-4-chloro-3-indolyl-β-d-galactoside (0.4 mg/ml), K3Fe(CN)6 (5 mm), K4Fe(CN)6(5 mm), MgCl2 (1 mm), deoxycholate (0.01%), and NP-40 (0.02%) in PBS for times ranging from 1 hr to overnight at 37°C.
Immunocytochemistry was performed essentially as described (Ivins et al., 1997) by the use of spent culture supernatants from the 9E10 hybridoma (American Type Culture Collection, Bethesda, MD) as the primary antibody and either Cy3- or HRP-conjugated anti-mouse antibodies (Jackson ImmunoResearch, West Chester, PA) for visualization.
Morphometric analysis of neurite lengths was performed on fixed cultures from images captured with a CCD camera (ORCA; Hamamatsu Photonics) and Openlab 2.0.7 software (Improvision) running on a Macintosh G3 computer. For routine analysis, cultures were scored for the number of cells or clumps of cells bearing neurites greater than two cell body diameters. Under the culture conditions used here, neurites from retinal neurons were only rarely observed on LN-1 or collagen IV, unless integrin function was activated. Treatment with integrin activators had a minimal effect on the size of the neuronal clumps that form in these cultures and was not taken into account when the percent of neurite-bearing cells was calculated.
Viral constructs. The herpes simplex virus (HSV) amplicons pHSVpuc and pHSVlac (Lim et al., 1996) were the generous gift of Dr. Filip Lim (Universidad Autonoma de Madrid, Madrid, Spain). These vectors use the HSV IE4/5 promoter to drive high levels of transgene expression rapidly in a wide range of cells including postmitotic neurons. pHSVpuc was further modified to encode an internal ribosome entry site (IRES) driving expression of either enhanced green fluorescent protein (eGFP) (Clontech, Palo Alto, CA) or bacterial β-galactosidase (lacZ). pHSV–IRES–GFP was created by subcloning the IRES fragment of pIRES–GFP (Clontech) into the shuttle vector pLitmus-29 (New England Biolabs, Beverly, MA) as anNco1–Pst1 fragment to make pLitmus–IRES. eGFP was excised from pIRES–GFP as anNco1–Sac1 fragment and subcloned into pLitmus–IRES linearized with the same enzymes to make pLitmus–IRES–GFP. The IRES–GFP fragment was then excised withBglII and Sac1 and subcloned into pHSV linearized with BamHI and Sac1 to create pHSV–IRES–GFP. The generation of pHSV—IRES–lacZ has been described previously (Ivins et al., 1999). cDNAs encoding wild-type R-ras and R-ras38V (both the generous gift of Dr. Alan Hall, University College, London, UK) were subcloned into pBluescript as ClaI–EcoRI fragments and subsequently subcloned as Sal1–Spe1 fragments into pHSV–IRES–GFP linearized with Sal1 andXba1 to generate amplicons directing expression of bicistronic mRNAs, facilitating the identification of transgene-expressing cells. The wild-type and R-ras38V constructs also contain an N-terminal myc-epitope tag to allow immunocytochemical verification of transgene expression.
Viral constructs were packaged in 2-2 cells (Smith et al., 1992) by the use of the replication-incompetent 5dl1.2 strain of HSV-1 (McCarthy et al., 1989) as a helper virus, according to methods described previously (Lim et al., 1996) and titers determined on cultures of pheochromocytoma-12 cells. Neuronal cultures were infected in serum-free medium at a multiplicity of infection (moi) of 0.5–1 within 2 hr of plating and fixed by sucrose underlay 16–18 hr later. Toxic effects were sometimes seen when higher rates of infection were attempted.
RESULTS
Outside-in integrin activation restores retinal neurite outgrowth on LN-1
When assayed early in nervous system development, retinal neurons from rat, mouse, and chick respond to LN-1 substrata with vigorous neurite outgrowth, in a manner dependent on the actions of integrin α6β1, a known LN-1 receptor (Aumailley et al., 1990). This response is lost relatively abruptly at approximately the stage in late embryogenesis that retinal axons reach their CNS targets yet is not correlated with a loss of expression of α6β1 (de Curtis and Reichardt, 1993). Recently, we showed that these neurons not only continue to express α6β1 but can still use it—together with another integrin, α3β1—to support vigorous neurite outgrowth on LN-1, provided that the LN-1 has first been modified via proteolytic cleavage or the binding of domain-specific antibodies (Ivins et al., 1998). Thus, late-embryonic retinal neurons retain not only LN-1 receptors but all of the signaling downstream of those receptors that is necessary for triggering neurite growth.
To test whether the failure of these receptors on late-embryonic retinal neurons to promote outgrowth on unmodified LN-1 might be caused by a low level of integrin activation, we exposed cultures of retinal neurons to micromolar amounts of Mn2+, a broadly acting outside-in activator of integrins (Bazzoni et al., 1995;Schwartz et al., 1995). As can be seen in Figure1, retinal neurons cultured from embryonic day 18 rats failed to extend neurites on an LN-1 substratum (Fig. 1A) but attached and extended long neurites when grown in the presence of 500 μmMnCl2 (Fig. 1B). Manganese stimulation of neurite outgrowth was half-maximal at ∼300 μm Mn2+ (Fig.1D) and was completely dependent on β1 integrins, because it could be blocked by the anti-β1 integrin antibody Ha2/5 (Fig. 1C). Similar results were also obtained with cultures of embryonic day 16 mouse retinal neurons (Fig.2A,B). To eliminate the possibility that Mn2+ acts by stimulating the synthesis or deposition of additional ECM molecules, we pretreated the substrata with blocking antibodies to LN-1 or to the long-arm LN-1 fragment E8. In both cases, neurite outgrowth was completely blocked (data not shown). We also examined whether manganese treatment significantly enhanced neuronal survival during the time course of these assays. We find that, at low doses, manganese has little effect on survival, but at the high doses necessary to activate neuronal integrin function fully (e.g., 500 μm), manganese has a slight toxic effect (data not shown). Therefore, the effects we observe on neurite outgrowth are unlikely to be secondary to effects on cell survival.

Effect of manganese on integrin-dependent neurite outgrowth from retinal neurons on LN-1 substrata. A–C, Embryonic day 18 rat retinal neurons were cultured on LN-1 substrata for 18 hr in the absence (A) or presence (B, C) of 500 μm MnCl2.C, Cultures were further treated with the function-blocking anti-β1 antibody Ha2/5 at 10 μg/ml.D, Cultures were incubated overnight with increasing amounts of MnCl2, fixed, and scored for the number of cells or clumps of cells bearing neurites. Scale bar, 75 μm.

Effect of blockade of α6β1 integrin function on Mn2+-stimulated neurite outgrowth on LN-1.A–C, Retinal neurons from embryonic day 16 mice were cultured on LN-1 substrata in the absence (A) or presence (B, C) of 500 μmMnCl2. C, Cultures grown in the presence of 500 μm MnCl2 were further treated with the function-blocking anti-α6 antibody GoH3 at 2 μg/ml. Scale bar, 50 μm.
Because our previous work (Ivins et al., 1998) implicated both α3β1 and α6β1 integrins in neurite outgrowth in response to various LN isoforms (including antibody-activated or cleaved LN-1), we sought to determine whether Mn2+-stimulated neurite outgrowth on LN-1 was primarily mediated by one or both of these receptors. As shown in Figure 2C, treatment with the rat anti-mouse monoclonal antibody GoH3 (2 μg/ml), which blocks α6β1 integrin function, partially inhibited spreading and neurite outgrowth by Mn2+-stimulated mouse retinal neurons plated on LN-1. In contrast, treatment of Mn2+-stimulated rat retinal neurons plated on LN-1 substrata with the mouse anti-rat monoclonal antibody Ralph 3.1 (80 μg/ml), which blocks α3β1 function, did not inhibit neurite growth (data not shown). These data suggest that integrin α6β1, but not α3β1, is activated by Mn2+. The fact that GoH3 did not completely block outgrowth of Mn2+-treated retinal neurons on LN-1 implies either that an additional β1 integrin is involved in that outgrowth or that Mn2+ induces a conformation in α6 such that it is less efficiently recognized by GoH3.
We next asked whether Mn2+ could enhance late-embryonic retinal neurite outgrowth on LN-1 fragments and other LN isoforms (Fig. 3). As shown previously (Ivins et al., 1998), embryonic day 18 rat retinal neurons extended neurites on the LN-1 long-arm fragment E8, as well as on LN-2/4 and LN-5. On both E8 and LN-5 substrata, Mn2+significantly enhanced neurite outgrowth. On LN-2/4, which already promoted the greatest degree of outgrowth in the absence of Mn2+, no additional increase was seen when Mn2+ was added. Neurite outgrowth was also tested on the ECM molecule thrombospondin-1 (TSP-1). As reported previously, TSP-1 promoted modest but detectable neurite outgrowth by embryonic day 18 rat retinal neurons (Ivins et al., 1998); however no increase in outgrowth was seen in the presence of Mn2+ (Fig. 3). Because outgrowth promoted by TSP-1 appears to be mediated by integrin α3β1 (DeFreitas et al., 1995; Ivins et al., 1998), these data further support the idea that Mn2+ does not activate α3β1.

Effect of manganese on other LN isoforms and fragments. Embryonic day 18 rat retinal neurons were cultured on the substrata indicated in the absence (open bars) or presence (solid bars) of 500 μmMnCl2, fixed after 18 hr, and scored for the number of cells or clumps of cells bearing neurites (*p < 0.05; two-way ANOVA with Tukey post hoc tests; 3 independent experiments).
Inside-out integrin activation restores retinal neurite outgrowth on LN-1
The signaling pathways that lead to integrin activation are not fully understood and may vary from one cell type to another. Recent studies, however, suggest that the small GTPase R-ras may be a broad-spectrum activator of integrin function in many cell types (Zhang et al., 1996; Keely et al., 1999). Because there are no known pharmacological activators of R-ras, we turned to amplicon-based HSV vectors to transduce genes into acutely dissociated neurons. These viral vectors use the HSV-1 immediate-early 4/5 promoter to drive high levels of transgene expression in infected cells. In experiments using a control vector, pHSV–lac, which drives expression of β-galactosidase, we found that 50–80% of embryonic day 18 rat retinal neurons could be reliably transduced in vitrowithout any deleterious effects on cell viability or neurite outgrowth. As shown in Figure 4, infection of embryonic day 18 rat retinal neurons with pHSV–lac did not inhibit retinal neurite outgrowth on the LN-1 long-arm fragment E8 (Fig.4A,B), nor did it cause neurite growth on LN-1 (Fig.4C). Identical results were obtained with a control virus that drives expression of green fluorescent protein, rather than β-galactosidase (data not shown).

Effect of R-ras38V on neurite outgrowth from embryonic retinal neurons on LN-1. A, B, Embryonic day 18 rat retinal neurons were plated on the E8 long-arm fragment of LN-1. A, The control uninfected culture was fixed after 18 hr (phase contrast). B, The culture was infected with pHSV–lac at an moi of ∼0.5 at the time of plating, fixed at 18 hr, and processed for β-galactosidase histochemistry. C–E, Embryonic day 18 rat embryonic retinal neurons were plated on LN-1 substrata and infected with viral constructs at the time of plating as indicated. C, Retinal neurons were infected with pHSV–lac at an moi of ∼1. After 18 hr, the culture was fixed and processed for β-galactosidase histochemistry. D, Retinal neurons were infected with pHSV–R-ras38V at an moi of ∼1. After 18 hr, the culture was fixed and processed for anti-myc immunofluorescence.E, Retinal neurons were infected with pHSV–R-ras38V growing on LN-1 (phase contrast).F, Retinal neurons were plated on LN-2/4 (phase contrast). Scale bar, 50 μm.
Next, an HSV vector was generated that drives expression of an epitope-tagged version of R-ras38V, a constitutively active mutant of R-ras (Zhang et al., 1996). As shown in Figure 4, D and E, expression of R-ras38V strongly promoted neurite outgrowth on LN-1. Expression of wild-type R-ras did not promote neurite outgrowth, demonstrating a requirement for the constitutively active form of the protein (Fig. 5). Interestingly, neurites extended by R-ras38V-expressing neurons on LN-1 were significantly longer than were those of the same neurons when stimulated to growth on LN-1 by Mn2+[R-ras38V, 103.4 ± 75.1 μm (mean ± SD); n = 144 neurites; Mn2+, 62.4 ± 41.6 μm (mean ± SD); n = 101 neurites; 2 independent experiments;p < 0.001 by Student's t test].

Quantitative effect of R-ras38Vexpression in retinal neurons. Embryonic day 18 rat retinal neurons were plated on either LN-1 (solid bars) or E8 (open bars) and infected with the viral constructs indicated at an moi of 1. After 18 hr, the cultures were fixed and scored for the number of cells or clumps of cells bearing neurites (*p < 0.05 vs no treatment; **p < 0.01 vs no treatment; two-way ANOVA with Tukey post hoc tests; 2 independent experiments). The data show that expression of activated, but not wild-type, R-ras promoted neurite outgrowth on LN-1 to a level similar to that observed on E8 (an activated form of LN-1). wt, Wild-type.
The ability of R-ras38V expression to promote neurite outgrowth was blocked by pretreating substrata with either anti-LN-1 antibodies or anti-E8 antibodies (data not shown), demonstrating that the stimulation of neurite outgrowth was not caused by the deposition of another ECM molecule on the substratum. Heat inactivation of the virus at 65°C for 10 min before infection completely eliminated both reporter gene expression and effects on neurite outgrowth, suggesting that the effects of viral treatment require an active virus and are not likely caused by a contaminant in the viral preparation.
To determine whether R-ras38V-stimulated neurite growth on LN-1 is mediated by α3β1 and/or by α6β1 integrin receptors, we treated cultures of both rat and mouse retinal neurons with function-blocking anti-integrin antibodies (Fig.6). All neurite outgrowth was blocked by function-blocking anti-β1 integrin antibodies. Furthermore, neurite outgrowth by R-ras38V-infected mouse retinal neurons on LN-1 was completely inhibited by the function-blocking rat anti-mouse α6 integrin monoclonal antibody GoH3. On the other hand, the function-blocking mouse anti-rat α3 monoclonal antibody Ralph 3.1 had no effect on cultures of R-ras38V-infected rat retinal neurons, suggesting that neurite outgrowth is primarily mediated by α6β1 and not by α3β1 integrins. Thus, like Mn2+, R-ras38V may selectively activate the α6β1 integrin. Also in agreement with this, expression of R-ras38V had no effect on retinal neurite outgrowth on TSP-1 (data not shown), a response that we showed previously to be completely dependent on the α3β1 integrin (Ivins et al., 1998).

Integrin dependence of R-ras38V-stimulated neurite outgrowth on LN-1. Retinal neurons from embryonic day 18 rats (solid bars) or embryonic day 16 mice (open bars) were cultured on LN-1, infected with viral constructs at an moi of 1, and treated with function-blocking anti-integrin antibodies as indicated. After 18 hr, the cultures were fixed and scored for the number of cells or clumps of cells bearing neurites (*p < 0.05 vs no treatment; one-way ANOVA with Bonferroni post hoc tests; 3 independent experiments).
Integrin activation restores retinal neurite outgrowth on collagen type IV
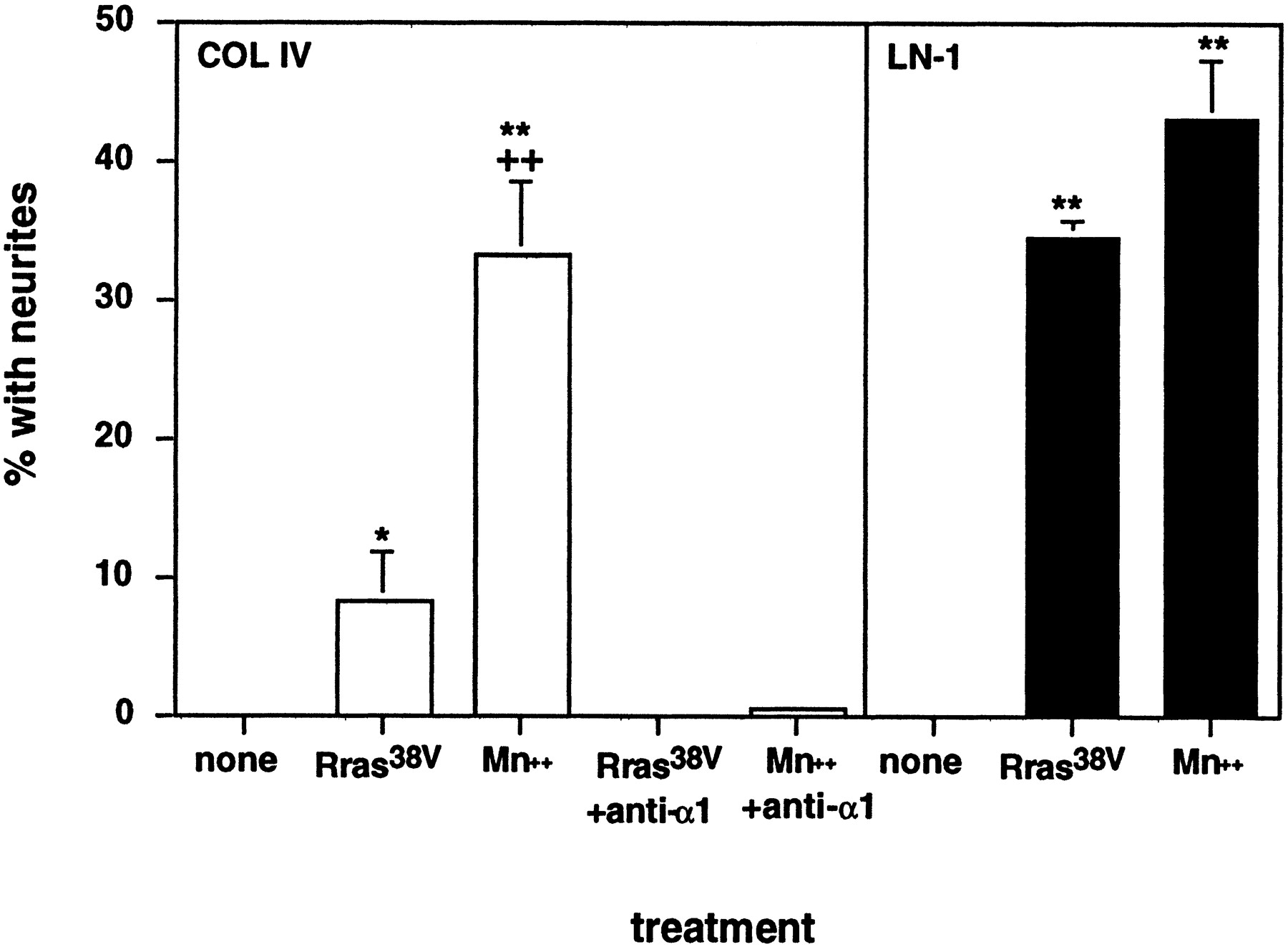
It has been reported in the chick that retinal cells not only lose the ability to respond to LN-1 as they develop but also lose the ability to attach and extend processes on collagens I and IV (Bradshaw et al., 1995). In agreement with this, we failed to observe any neurite outgrowth by embryonic day 18 rat retinal neurons on collagen IV substrata (Fig. 7). We then examined the effects of integrin activators. As shown in Figure 7, Mn2+ promoted outgrowth on collagen IV to an extent similar to the extent that it promoted outgrowth on LN-1 [and with a similar dose–response relationship (data not shown)]. In contrast, R-ras38V promoted a small but significant amount of neurite outgrowth on collagen IV (Fig. 7). In both cases, outgrowth on collagen IV was blocked by anti-β1 integrin antibodies (data not shown). It is interesting to note that the weaker effect of R-ras, as compared with that of Mn2+, on neurite outgrowth on collagen IV cannot simply be explained by a greater potency of Mn2+ in activating integrins because, on LN-1, R-ras had a greater effect on neurite lengths than did Mn2+.

Effect of Mn2+ and R-ras38V on neurite outgrowth on LN-1 and collagen IV. Embryonic day 18 rat retinal neurons were cultured on LN-1 (solid bars) or collagen IV (open bars) with or without integrin activators and anti-α1 integrin antibodies as indicated. After 18 hr, cultures were fixed and scored for the number of cells or clumps of cells bearing neurites (*p < 0.05 vs no treatment; **p < 0.01 vs no treatment;++p < 0.01 vs R-ras38V expression; two-way ANOVA with Tukeypost hoc tests). COL IV, Collagen IV.
Potential integrin receptors for collagen IV include α1β1 and α2β1 (Mendrick et al., 1995); however Bradshaw et al. (1995) report that in the chick, only neuroepithelial cells and not neurons express α2β1. Recently, we showed that α1β1 is strongly expressed in the embryonic day 18 rat retina (Ivins et al., 1998). In agreement with a role for this integrin as a neuronal collagen IV receptor, we find that neurite outgrowth by both Mn2+- and R-ras38V-stimulated embryonic day 18 retinal neurons is completely blocked by a monoclonal blocking antibody to α1β1 (Fig. 7).
Interestingly, on some cells α1β1 also functions as a receptor for LN-1 and LN-2/4 (Hall et al., 1990; Tomaselli et al., 1990; Colognato et al., 1997). The binding site on LN-1 for the α1β1 integrin has been mapped to a domain near the N terminal of the LN α1 chain (Colognato et al., 1997). However, Mn2+did not stimulate outgrowth of retinal neurons on the LN-1 fragment E1′ or on the recombinant LN α1 chain-derived short-arm fragment α1(VI–IVb), both of which contain this binding site (data not shown). Expression of R-ras38V in embryonic day 18 retinal neurons also did not lead to neurite outgrowth on fragment E1′. These results suggest that the α1β1 of rat retinal neurons functions solely as a receptor for collagen and that even activators of integrin function do not change its ligand-binding specificity. Because the cellular context in which α1β1 is expressed can determine its ligand-binding specificity (Wong et al., 1996), such specificity is thought to be determined by the presence ofcis-interacting protein(s). The present data are consistent with the idea that such proteins act independently of the integrin activation state.
Integrin activators stimulate neurite outgrowth on LN-1 by many CNS neurons
Previously, we showed that a poor ability to respond to LN-1 is a common property of many CNS neuronal populations (Ivins et al., 1998). To determine whether integrin activators stimulate neurite outgrowth from other CNS neurons on LN-1, we treated cultures of embryonic day 18 rat hippocampal and embryonic day 18 cortical neurons with 500 μm MnCl2 and asked whether outgrowth on LN-1 was enhanced. In both cases, Mn2+ potently stimulated β1 integrin-dependent neurite outgrowth. Results with embryonic day 18 rat hippocampal neurons are shown in Figure8. We also examined the ability of R-ras38V to promote neurite outgrowth from embryonic day 18 cortical and hippocampal neurons on LN-1. In both cases, expression of R-ras38V also strongly promoted neurite outgrowth relative to uninfected neurons (Fig. 9). As with retinal neurons (Fig.5), infection with virus directing expression of wild-type R-ras was without effect (data not shown).

Effect of manganese on integrin-dependent neurite outgrowth from hippocampal neurons on LN-1 substrata.A–C, Embryonic day 18 rat hippocampal neurons were cultured overnight on LN-1 substrata for 18 hr in the absence (A) or presence (B, C) of 500 μm MnCl2. C, Cultures were further treated with the function-blocking anti-β1 antibody Ha2/5 at 10 μg/ml. D, The culture was grown on LN-2/4. Scale bar, 50 μm.

Effect of R-ras38V expression on neurite outgrowth from embryonic hippocampal and cortical neurons on LN-1. Hippocampal (A, B) and cortical (C, D) neurons were cultured from embryonic day 18 rats on LN-1-coated glass coverslips for a total of 20 hr before fixation. Neurons in B and D were infected with pHSV–R-ras38V 2 hr after plating. Uninfected control cultures are shown in A and C. Cultures were fixed and stained for anti-myc immunoreactivity by indirect immunofluorescence. Little or no outgrowth on LN-1 was seen in control uninfected cultures (A, C; phase contrast). Scale bar, 50 μm.
We next turned to PNS neurons, which generally respond strongly to LN-1, to determine whether integrin activation could enhance outgrowth further. We chose to focus on spinal sensory neurons because, in the chick at least, outgrowth on LN-1 has been shown to depend on the α6β1 integrin (Condic and Letourneau, 1997), a receptor whose activity can, as shown here, clearly be modulated by integrin activators. However, as shown in Table 1, when cultures of dissociated embryonic day 7 chick DRG neurons were exposed to a range of concentrations of Mn2+, no significant change was observed in the numbers of cells with neurites (data not shown) or in the mean neurite length per cell (a more sensitive parameter when high proportions of cells contain neurites). When we tested the effects of Mn2+ on embryonic (embryonic day 14) mouse DRG (Fig. 10), we observed a noticeable decrease in neurite fasciculation but no visually obvious increase in the abundance of neurites (marked fasciculation in these cultures interfered with the ability to quantify neurite outgrowth more precisely). Interestingly, treatment of these cultures with the anti-α6 antibody GoH3 was without effect, suggesting that these neurons use a receptor other than α6β1 for outgrowth on LN-1.
Effect of manganese treatment on neurite outgrowth from embryonic chick DRG growing on LN-1

Effect of manganese on embryonic mouse spinal sensory neurons. Examples of embryonic day 14 mouse DRG neurons cultured on LN-1 substrata overnight in the absence (A) or presence (B) of 300 μm MnCl2. Scale bar, 100 μm.
We also examined DRG neurons at a much later stage, in this case from the adult mouse (no adult dorsal root ganglion culture method exists for the chicken). In this case, the treatment of cultures plated on LN-1 with 300 μm Mn2+ did significantly enhance neurite outgrowth, increasing the number of neurites per cell by ∼50% and the mean neurite length by ∼25% (Fig. 11). All neurite outgrowth by dorsal root ganglion neurons on LN-1, both in the absence and presence of Mn2+, was completely blocked by anti-β1 antibodies (data not shown).

Effect of manganese on adult mouse spinal sensory neurons. A, B, Examples of adult mouse DRG neurons cultured on LN-1 substrata for 8 hr in the absence (A) or presence (B) of 300 μm MnCl2 are shown. C, Within such cultures, 30 (control; open bars) and 37 (Mn2+-treated; solid bars) neurons were evaluated for the mean neurite length, the number of neurites per cell, and the total neurite length per cell (±SD). The effect of Mn2+ was statistically significant (p < 0.005) for each metric (Student's t test). Higher concentrations of Mn2+ were also tested but appeared to be toxic to these cells (data not shown). Scale bar, 100 μm.
DISCUSSION
Age-related decreases in neurite outgrowth ability have been observed in many neuronal populations. This phenomenon has been most extensively studied in the vertebrate retina, which loses responsiveness to LN-1 almost entirely, and rather abruptly, in late embryogenesis (Cohen et al., 1986; Hall et al., 1987). Here we show that this loss can be reversed by either of two reagents—extracellular Mn2+ and virally expressed R-ras38V—that increase the activation state of integrins (Figs. 1, 4, 5). Increasing integrin activation also restored the ability of late-embryonic retinal neurons to extend neurites on type IV collagen (Fig. 7) and enhanced outgrowth on substrata such as LN-5 and the LN-1 fragment E8 (Fig. 3). All outgrowth elicited by Mn2+ and R-ras38V was dependent on β1 integrins, primarily α6β1 [on LN-1 substrata (Figs. 2, 6)] and α1β1 [on collagen IV (Fig. 7)]. Neither Mn2+ nor R-ras38V promoted outgrowth mediated by the integrin α3β1, despite the fact that late-embryonic retinal neurons express this integrin and can use it to interact either with LN-1 that has been conformationally or proteolytically modified or with other LN isoforms (Ivins et al., 1998).
Mn2+ and R-ras38V also strongly enhanced neurite outgrowth on LN-1 by late-embryonic CNS neurons cultured from the hippocampus and cerebral cortex (Figs. 8, 9). In contrast, the consequences of integrin activation on PNS neurons were more subtle. Neurite outgrowth by embryonic dorsal root ganglion neurons from both mouse and chick was little affected by Mn2+ (Fig. 10, Table 1), and even outgrowth by adult mouse dorsal root ganglion neurons was elevated only modestly (Fig. 11).
These results raise the possibility that differences in ECM responsiveness among neurons from different regions and developmental stages may stem, at least in part, from differences in the intrinsic activation state of β1 integrins. Such a view provides an explanation for why late-embryonic retinal neurons that no longer respond to LN-1 continue to express the LN-1 receptor α6β1 (de Curtis et al., 1991) and can use this receptor for neurite outgrowth on other LN isoforms, or even LN-1 that has been appropriately modified (Ivins et al., 1998). This explanation is supported by previous work with late-embryonic chick retinal neurons in which a monoclonal antibody (TASC) that binds and activates β1 integrins increased adhesion to LN-1 substrata (Neugebauer and Reichardt, 1991). Apparently, late-embryonic retinal neurons possess integrin receptors for LN-1 that are functional but exist in a less than fully activated state.
Do changes in integrin activation underlie developmental changes in axonal growth potential?
There is no biochemical marker that allows the quantification of β1 integrin activation states independently of assaying integrin-dependent cell behaviors. Because of this, we can say that increasing integrin activation reverses a developmental loss in neurite outgrowth ability, but we cannot definitively show that a developmental decrease in integrin activation was the cause of that loss. It is also possible that a decline in the expression level of certain integrins (de Curtis et al., 1991) or changes in the levels of integrin-modulatory proteins [e.g., members of the tetraspanin family (Hemler, 1998)] cause integrin function to dip below some threshold, such that function can be restored by increasing integrin activation via exogenous means.
Regardless of the cause of the loss of integrin function, promoting integrin activation in CNS neurons does not simply result in increased cell–substratum adhesion as might be expected but instead results in enhanced or restored neurite outgrowth. This effect is observed with both outside-in and inside-out integrin activation and suggests that all of the events necessary for neurite extension downstream of integrin–ligand binding must be intact. Thus, integrin–ligand binding is likely to be the limiting step for interactions between CNS neurons and LN-1, and perhaps other ECM components as well. These results suggest a model in which integrin function is either downregulated or maintained at low levels in CNS neurons relative to peripheral neurons in the mature nervous system (Fig. 12) and raise the possibility that such low levels of integrin activity, by failing to support neuron–ECM interactions, contribute to the regenerative failure that is characteristic of the injured adult CNS. Our results with adult spinal sensory neurons further suggest that, even in neurons that have the potential to regenerate their axonsin vivo, integrin activation might enhance such regeneration.

Model relating integrin activation states with neurite outgrowth. The data presented here, together with those reported previously (Ivins et al., 1998), suggest that the ability of neurons to respond to ECM ligands by extending neurites depends on a combination of the activation state of neuronal integrins and the features of ECM molecules that are themselves subject to regulation.Curve A depicts the proposed relationship between neurite outgrowth and integrin activation for LN-1; curve B depicts the same relationship for LN-2/4, LN-5, and LN-1 that has been activated via antibody binding or proteolytic cleavage. By attributing different degrees of integrin activation to different types of neurons (early and late embryonic, other CNS, and PNS), it is possible to explain the responses of these neurons to such ECM molecules, as well as the effects of Mn2+ and R-ras38V on those responses.
Selectivity of integrin activators
The effects of integrin activators on retinal neurite outgrowth on LN-1 were blocked by antibodies to α6 but not α3 integrins, suggesting that such effects are primarily mediated by α6β1 and not α3β1. This was unexpected because we have shown previously that α3β1 is primarily responsible for neurite outgrowth by retinal neurons on activated LN-1 and other LN isoforms, whereas α6β1 primarily mediates attachment and cell spreading (Ivins et al., 1998). It is possible that the α3β1-binding site on native LN-1 is cryptic (Ivins et al., 1998); however this cannot explain our observation that neurite outgrowth on TSP-1—a ligand recognized in its native state by α3β1 on late-embryonic retinal neurons (Ivins et al., 1998)—was not stimulated by Mn2+. Overall, the data support recent observations in non-neural cells suggesting that α3β1 is activated poorly, if at all, by extracellular reagents (Bazzoni et al., 1998).
Interestingly, in the present study, inside-out integrin activation also failed to activate neuronal α3β1 function on either LN-1 or TSP-1. In addition, R-ras38V only weakly activated α1β1 function on collagen IV, whereas Mn2+ appeared to be a potent activator of α1β1. Together these results argue that inside-out integrin activators can be selective for integrins with particular α chains. Recently, activated R-ras and the closely related GTPase TC21 were reported to enhance migration and invasion of breast carcinoma cells through collagen (which is mediated by integrin α2β1) and yet not to affect migration on LN-1, even though these cells express α6β1 (Keely et al., 1999). The apparent lack of effect of R-ras on α6β1 is at odds with the data in the present study and raises the possibility that R-ras acts on different integrins in different cell types. Alternatively, because R-ras is thought to exert its effects at the level of integrin cytoplasmic domains, these discrepancies may reflect the expression of different cytoplasmic domain splicing variants in different cells. Indeed, most carcinoma cell lines express varying proportions of α6A and α6B isoforms (Tamura et al., 1991), whereas neurons predominantly express α6B (Tamura et al., 1991; de Curtis and Reichardt, 1993). The α6A cytoplasmic domain is highly dissimilar to that of α6B and yet is related to that of α3A, the predominant α3 isoform in neural cells (Tamura et al., 1991;DeFreitas et al., 1995; de Melker et al., 1997).
Mechanisms underlying integrin activation
A number of mechanisms by which integrin activators can work have been documented. These include causing conformational changes that mimic conformations induced by ligand binding (Bazzoni and Hemler, 1998; Bazzoni et al., 1998), clustering integrins on the cell surface to increase avidity (Yauch et al., 1997), altering the cytoskeleton to limit integrin diffusion or promote clustering (Pfaff et al., 1998), recruiting signaling molecules to sites of integrin engagement (Schaller and Parsons, 1994; Hannigan et al., 1996), and enhancing interactions of integrins with other membrane proteins (Porter and Hogg, 1998).
Although Mn2+ most likely stabilizes a conformation that mimics the ligand-bound state, the mechanism by which R-ras acts is less clear. Some clues have emerged from recent studies of the closely related ras family member H-ras. Activated H-ras and its kinase effector Raf-1 inhibit integrin activity (Hughes et al., 1997), an action opposite to that of activated R-ras (Zhang et al., 1996). Both H-ras and R-ras bind to the common effectors Raf-1 (Spaargaren et al., 1994), Ral-GDS (Urano et al., 1996), and phosphoinositide 3-kinase (PI3-kinase) (Marte et al., 1997), but R-ras does not activate Raf-1 or Ral-GDS and may instead inhibit their activation by H-ras. Unlike H-ras, R-ras does not activate the MAP kinase pathway but does activate PI3-kinase, which may in turn activate PKB/Akt (Marte et al., 1997). PI3-kinase can stimulate rac-dependent effects on the cytoskeleton (Rodriguez-Viciana et al., 1997) and promote increased α5β1 integrin-dependent binding of mast cells to fibronectin (Kinashi et al., 1999). Thus, R-ras may promote integrin activation indirectly by organizing the cytoskeleton, by activating signaling cascades potentially involving PI3-kinase (Khwaja et al., 1998; Kinashi et al., 1999), or by inhibiting the ability of H-ras to bind and activate Raf-1, thus inhibiting an integrin-suppression pathway (Sethi et al., 1999).
Although R-ras has also been implicated in cell survival pathways in some systems (Fernandez-Sarabia and Bischoff, 1993; Troppmair and Rapp, 1997), the stimulation of neurite outgrowth observed in the present study is too specific for particular integrins and ECM ligands (and is also relatively rapid) to be attributable to general effects on neuron survival.
Integrin function in the normal and injured nervous system
Little is currently known about the function of integrins in the mature brain, even though the expression of both integrins and components of the ECM is subject to complex regulation (Chen and Strickland, 1997; Hoffman et al., 1998; Pinkstaff et al., 1998, 1999). Although expression of many integrin ligands in the brain declines at the end of neural development, several ECM molecules are upregulated after injury to either the PNS or CNS, among them, both laminins and collagens (Zak et al., 1987; Giftochristos and David, 1988; Carbonetto, 1991; Brodkey et al., 1993; Frisen et al., 1995; Fu and Gordon, 1997;Weidner et al., 1999). Low levels of integrin activation in mature CNS neurons may be among the factors that contribute to the failure of such neurons, when confronted with ECM ligands, to respond with vigorous outgrowth. If this view is correct, then activated R-ras may be an attractive candidate for gene transfer into the injured CNS to promote neuronal regeneration.
Footnotes
This work was supported by National Institutes of Health Grant NS 36049 and American Paralysis Association Grant LA2-9603 to A.D.L. We gratefully acknowledge the gifts of plasmids from Dr. Alan Hall (University College, London, UK), viral amplicons from Dr. Filip Lim (Universidad Autonoma de Madrid, Madrid, Spain), and viral packaging cells from Dr. Rozanne Sandri-Goldin (University of California at Irvine, Irvine, CA).
Correspondence should be addressed to Dr. J. K. Ivins, Department of Developmental and Cell Biology, BioSci II, 4132, University of California at Irvine, Irvine, CA 92697. E-mail: jkivins{at}uci.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}