

Abstract
The pattern of axonal projections early in the development of the nervous system lacks the precision present in the adult. During a developmental process of refinement, mistargeted projections are eliminated while correct projections are retained. Previous studies suggest that during development nitric oxide (NO) is involved in the elimination of mistargeted retinal axons, whereas brain-derived neurotrophic factor (BDNF) may stabilize retinal axon arbors. It is unclear whether these neuromodulators interact. This study showed that NO induced growth cone collapse and retraction of developing retinal axons. This effect was not attributable to NO-induced neurotoxicity. BDNF protected growth cones and axons from the effects of NO. This effect was specific to BDNF, because neither nerve growth factor (NGF) nor neurotrophin-3 (NT-3) prevented NO-induced growth cone collapse and axon retraction. Exposure to both BDNF and NO, but not either factor alone, stabilized growth cones and axons. Stabilized axons exhibited minimal retraction or extension. This response appears to be a new axon “state” and not simply a partial amelioration of the effect of NO, because lower doses of BDNF or NO allowed axon extension. Furthermore, BDNF/NO-induced growth cone stabilization correlated with the appearance of a cytochalasin D-resistant population of actin filaments. BDNF protection from NO likely was mediated locally at the level of the growth cone, because growth cones or individual filopodia in contact with BDNF-coated beads were protected from NO-induced collapse. These findings suggest a cellular mechanism by which some axonal connections are stabilized and some are eliminated during development.
Axonal connections between functionally related groups of neurons have precise, stereotypic patterns. In the visual system, for example, neighboring retinal ganglion cells project to neighboring regions within target nuclei. The initial pattern of connections formed by retinal ganglion cells with the primary visual nuclei, particularly in warm-blooded vertebrates, lacks the topographic precision of mature connections. During development many retinal axons project to inappropriate positions within target regions, to inappropriate nuclei, or to the inappropriate side of the brain (McLoon, 1982, 1985; Cowan et al., 1984; Nakamura and O'Leary, 1989; Holt and Harris, 1998). These targeting errors eventually are corrected during a discrete period of development by a process of refinement. During refinement the axons with inappropriate connections retract, and axons with appropriate connections are stabilized (O'Rourke and Fraser, 1990; Simon et al., 1994).
Recent evidence indicates that the synthesis of nitric oxide (NO) is required for the elimination of some mistargeted axons. The enzyme that synthesizes NO is expressed by cells in the retinorecipient layers of the chick tectum during the developmental period in which the retinotectal projection is refined (Williams et al., 1994), and blocking NO synthesis in chick embryos preserved transient retinotectal projections (Wu et al., 1994; Ernst et al., 1999). A similar disruption of refinement of the retinotectal projection was observed in mice with a double knock-out of endothelial and neuronal NO synthase genes (Mize et al., 1998). Blocking NO synthesis also has been shown to disrupt the segregation of retinal fibers into on–off sublaminae in the ferret lateral geniculate nucleus (Cramer et al., 1996) as well as the targeting of retinal axons within the optic lobe ofDrosophila embryos (Gibbs and Truman, 1998). In vitro, NO has been shown to cause the collapse of growth cones and the retraction of axons (Hess et al., 1993; Renteria and Constantine-Paton, 1996) and to affect the extension of growth cone filopodia (Van Wagenen and Rehder, 1999). These findings suggest that NO may be released by postsynaptic cells during development and function in pruning presynaptic axons or axonal branches that are mistargeted.
It is likely that most axon terminals in the vicinity of a cell that synthesizes NO are exposed to NO, because NO diffuses freely in an aqueous environment and is not impeded by cell membranes (Wood and Garthwaite, 1994; Lancaster, 1997). It is assumed, however, that only mistargeted axon terminals withdraw during refinement. Given that individual target cells are believed to be innervated by both correctly and incorrectly targeted axons before refinement, some mechanism must protect correctly targeted axons from NO, if NO functions as a withdrawal signal. Neurotrophins may play such a role.
Neurotrophins regulate the retention and complexity of axonal projections during development. Excess brain-derived neurotrophic factor (BDNF) was shown to preserve the transient ipsilateral retinal projection to the superior colliculus in rodent (Isenmann et al., 1999), a projection that appears to be pruned by a NO-dependent mechanism (Mize et al., 1998). Geniculocortical projections that normally are eliminated during development also were preserved by the application of BDNF to the cortex (Cabelli et al., 1995). Application of BDNF increased the complexity of retinal ganglion cell arbors in the developing tectum (Cohen-Cory and Fraser, 1995) and of dendritic branches in pyramidal cells in developing cortex (McAllister et al., 1995, 1996). As with NO, peak levels of BDNF expression in the primary visual nuclei correlate with the period of refinement of the retinal projection (Herzog et al., 1994). It is possible, therefore, that target-derived neurotrophic factors, such as BDNF, protect axons with correctly targeted connections from NO-induced withdrawal. This possibility was tested on growing retinal axons in culture.
This study showed that NO induced growth cone collapse and axon retraction, BDNF protected axons from the effects of NO, and exposure to both BDNF and NO, but not either factor alone, stabilized axons, possibly by altering the dynamics of the actin cytoskeleton.
MATERIALS AND METHODS
Preparation of reagents. 3-Morpholino-sydnonimine (SIN-1) (Sigma, St. Louis, MO) was prepared fresh for each experiment in 100 mm NaOH at a concentration 100× the desired final concentration. Stock solutions of 3-[2-hydroxy-1-(1-methylethyl)-2-nitrosohydrazino]-1-propanamine (NOC-5) (Calbiochem, La Jolla, CA) and 3-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-propanamine (NOC-7) (Calbiochem) in 100 mm NaOH were stored at −20°C and diluted to the desired final concentration before each experiment. Recombinant BDNF (a gift of Dr. L. O. Palladino, Regeneron Pharmaceuticals, Tarrytown, NY), nerve growth factor (NGF; R & D Systems, Minneapolis, MN), and neurotrophin-3 (NT-3; a gift of Dr. Palladino) were prepared as stock solutions in 0.15 m NaCl containing 1 mg/ml bovine serum albumin (BSA). The concentrations of neurotrophins stated in Results were based solely on the molecular weight of the neurotrophin monomers, and the actual amount of bioactive dimers was not considered. Cytochalasin D (Sigma) stock was prepared in dimethylsulfoxide (DMSO). 5, 10, 15, 20-Tetrakis (4-sulfonatophenyl) prophyrinato iron (III) chloride (FeTPPS) (Calbiochem) was prepared as a 100 μm stock in F12H medium.
Tissue culture. Fertilized chicken eggs (White Leghorn crossed with Rhode Island Red) were incubated at 37°C. Retinas were harvested from 6-d-old embryos. Dorsal root ganglia (DRG) and hearts were obtained from 12-d-old embryos. Dissociated cells were prepared as described in Gallo et al. (1997, 1998). Retinal and DRG explants and dissociated heart cells were cultured on laminin-coated (25 μg/ml, overnight incubation at 4°C; Life Technologies, Gaithersburg, MD) glass coverslips in F12H medium supplemented with 2 μg/ml insulin, 100 μg/ml transferrin, 5 ng/ml sodium selenite, 20 nm progesterone, 5 mm phosphocreatine, 0.4 mg/ml sodium pyruvate, and 2 mm glutamine (all from Sigma). Dissociated DRG neurons were cultured on fibronectin (25 μg/ml, overnight incubation at 4°C; Life Technologies). Both DRG explants and dissociated cells were raised in F12H medium (as described above) further supplemented with 1 mg/ml BSA (Sigma). Retinal explants were prepared by cutting retinas into squares (∼300 μm across). Four to eight explants were cultured on each coverslip. Similarly, DRG were cut into fourths, and four pieces of tissue were cultured on each coverslip. DRG explants and dissociated DRG cells were cultured in the presence of 0.4 nm BDNF and 0.4 nm NGF, respectively. Dissociated retinal cells were prepared from peripheral retina that first had been cut into 300 μm explants and then triturated mechanically (20×) in Ca2+/Mg2+-free PBS (CMF-PBS). This generated clusters of 10–20 cells that remained viable under these conditions and that exhibited robust retinal axon growth as determined by staining with a retinal ganglion cell-specific antibody (see Cytochemistry). In all experiments that used DRG explants or dissociated cells, the medium was exchanged with F12H medium without BSA on the day of the experiment, and neurotrophin concentrations were either lowered or increased as described in Results.
Video microscopy. After a 24 hr incubation the cultures were placed on an inverted microscope (IM-35, Carl Zeiss, Oberkochen, Germany, or Diaphot 200, Nikon, Tokyo, Japan) under an air curtain incubator, which maintained the medium at a constant 40°C. Time-lapse phase-contrast images were captured with a 16× (Zeiss microscope) or 20× (Nikon microscope) objective, using a Newvicon video camera (NC-65 or NC-68, Dage–MTI, Michigan City, IN). Images were recorded every 1–5 min and stored on digital media. The number of axons with growth cones was counted on a video monitor at 5 min intervals during the 1 hr exposure to a NO donor.
Neurotrophin-coated beads. Neurotrophin-coated (NGF or BDNF) and cytochrome c-coated (Sigma) beads were prepared and used as described in Gallo et al. (1997, 1998). For experiments involving NGF-responsive neurons and NGF-coated beads, the concentration of NGF in the culture medium was lowered to 0.004 nm 2 hr before the introduction of the beads to the cultures. Because retinal explants were cultured in the absence of neurotrophins, BDNF-coated beads were added directly to the cultures. Video microscopic observations of the behavior of retinal axons and growth cones in contact with BDNF-coated beads were initiated 1 hr after the addition of the beads to the cultures. Nitric oxide donors were added to the medium at the start of each 1 hr video recording session. The responses of axons to contact with neurotrophin-coated beads were scored as described in Gallo et al. (1998). Four separate cultures were scored in each experimental group.
Cytochemistry. To visualize the distribution of actin filaments in growth cones, we fixed cultures for 15 min in PHEM buffer (60 mm PIPES, 25 mm HEPES, 10 mm EGTA, and 2 mm MgCl2, pH 6.9) containing 0.2% glutaraldehyde and 5% dextrose and then incubated them for 15 min in CMF-PBS containing 1 mg/ml sodium borohydride (Sigma). Actin filaments were stained with rhodamine-conjugated phalloidin (Molecular Probes, Eugene, OR) as described in Gallo et al. (1998). After phalloidin staining the retinal explants were counterstained with DiOC18(3) (2.5 μg/ml for 30 sec; Molecular Probes). Dissociated retinal cell cultures were double-stained with rhodamine phalloidin and the retinal ganglion cell axon-specific monoclonal antibody RA4 (McLoon and Barnes, 1989). To preserve the RA4 antigen, we fixed cultures for 10 min in 2% paraformaldehyde (in CMF-PBS), extracted them for 15 min in CMF-PBS containing 0.1% Triton X-100, and then treated them for 15 min with 1 mg/ml sodium borohydride in CMF-PBS. Then the cultures were blocked in normal goat serum for 15 min, followed by a 1 hr incubation with RA4 and 1 U of rhodamine phalloidin. Next, the cultures were rinsed with CMF-PBS and then incubated with fluorescein-conjugated goat anti-mouse secondary antibody (Jackson Laboratories, Bar Harbor, ME) for 1 hr. Antibodies were diluted in 1% fish gelatin (Sigma). Cultures raised on glass coverslips were mounted on microscope slides, and cultures raised in video dishes were mounted by placing a glass coverslip on top of the explants. All cultures were mounted with medium containing 10 mg/mlp-phenylenediamine (Sigma).
Procedure for scoring growth cone collapse in fixed preparations.In experiments that used DRG explants and dissociated retinal cells, the frequency of growth cone collapse was determined from phalloidin-stained cultures. Only growth cones that were not contacting other cells were scored. The tips of axons were scored as having a growth cone if they exhibited either a lamellipodium or ≥3 filopodia. For experiments that used dissociated retinal cells, only RA4-positive axons were scored.
Quantitative measurements of F-actin staining. F-actin levels were compared across treatment groups by determining the total staining intensity in growth cones stained with rhodamine phalloidin. Growth cone morphology was visualized via DiOC18(3) staining with a fluorescence microscope (Leitz DM, 100× objective; Leica, Nusslock, Germany), and images were captured. A region-of-interest template outlining the growth cone, including all filopodia and lamellipodia as well as the proximal-most 10 μm of axon shaft, was generated by using Image Pro Plus software (Media Cybernetics, Silver Spring, MD). To measure the total amount of actin filaments present in individual growth cones, we then used the template to obtain the integrated pixel intensity from an in-register image of the phalloidin staining of the same growth cone, using Image Pro Plus software. Data were collected from two or more separate cultures and a minimum of 13 randomly sampled growth cones per experimental condition. Integrated intensity values were compared statistically by the use of the Welch t test for unequal variances.
Measurement of cell death in retinal explants. The trypan blue exclusion method was used to determine the extent of cell death in retinal explants. Explants were stained with 0.4% trypan blue for 3 min. The number of trypan blue-stained cells (dead cells) in each explant was counted by using phase-contrast microscopy (25×). Then images of the explants from which cell counts were performed were acquired and stored digitally. Last, the number of trypan blue-stained cells was divided by the area (μm2) of the explant over which the count was made to determine the density of dead cells.
RESULTS
NO donors cause growth cone collapse and axon retraction
Nitric oxide induced retraction of the growing retinal axons. Explants of embryonic chick retina were cultured overnight to allow outgrowth of retinal ganglion cell axons. As the growing axons were monitored continuously via a microscope equipped for video recording, the NO donor SIN-1 was added to the culture medium. The addition of SIN-1 to the culture medium initially induced growth cone collapse, which started with the resorption of filopodia and lamellae (Fig.1). Growth cone collapse was followed by retraction of the axon. Within 15 min of the addition of SIN-1 (0.1 mm), ∼47% of the growth cones had collapsed, and by 40 min, ∼82% of the growth cones had collapsed (Fig.2A). The effect of SIN-1 was dose-dependent (Fig. 2A). Consistent with previous reports (Renteria and Constantine-Paton, 1996), SIN-1 exhausted of NO had no effect. Of the axons, 86% had growth cones 1 hr after the addition of exhausted 0.1 mm SIN-1 (n = 4 cultures), compared with 92% of axons in control cultures (n = 4 cultures). Furthermore, hemoglobin, a NO scavenger, added to the medium prevented SIN-1-induced growth cone collapse and axon retraction. In this case 86% of axons had growth cones 1 hr after the addition of SIN-1, compared with 92% of axons in control cultures (n = 3 cultures), suggesting that the effect was attributable to NO and not other byproducts of SIN-1. Two other NO donors, NOC-5 and NOC-7, had similar effects as SIN-1 (Fig. 2B) and did not collapse growth cones when exhausted of NO. Of the retinal axons, 82% had growth cones 1 hr after the addition of exhausted 3 mm NOC-7 (n = 3 cultures), compared with 92% of axons in control cultures.

Time-lapse micrographs of the growth cones of retinal axons in culture at three time points over a 55 min period. In untreated control cultures the growth cones exhibited considerable motility, and the axons typically extended. In cultures treated with the NO donor SIN-1 (0.1 mm) the growth cones collapsed, and the axons retracted. In cultures treated with 150 nm BDNF the growth cones and axons exhibited normal growth and motility. In cultures pretreated with 150 nm BDNF, followed by the addition of SIN-1 (0.1 mm), the growth cones and axons were stabilized. With stabilization the growth cones exhibited strikingly low motility, and the axons neither extended nor retracted. The0 min time point was arbitrary for the untreated culture and indicates the time of SIN-1 addition and/or 1 hr after BDNF addition for the other cultures. Scale bar, 10 μm.

BDNF prevents NO-induced growth cone collapse. Individual axons extending from retinal explants were monitored by phase-contrast video microscopy. The percentage of axons with growth cones (y-axis) was determined at 5 min intervals during a 1 hr period of exposure to NO donors. Each data set represents at least 25 growth cones in five or more cultures. Time measurements on the x-axis indicate the time after the addition of the NO donor. A, The NO donor SIN-1 caused growth cone collapse in a dose-dependent manner. Control, No treatment; 0.01SIN, 0.01 mmSIN-1; 0.1 SIN, 0.1 mm SIN-1; 1.0 SIN, 1.0 mm SIN-1. B, Pretreatment with BDNF prevented NO-induced growth cone collapse with three different NO donors. BDNF+NOC5, 1 hr pretreatment with 150 nm BDNF, followed by 3 mm NOC-5;BDNF+NOC7, 1 hr pretreatment with 150 nmBDNF, followed by 3 mm NOC-7; BDNF+SIN, 1 hr pretreatment with 150 nm BDNF, followed by 0.1 mm SIN-1; SIN, 0.1 mm SIN-1;NOC5, 3 mm NOC-5; NOC7, 3 mm NOC-7. C, The BDNF protective effect was dose-dependent. 150 BDNF+SIN, 1 hr pretreatment with 150 nm BDNF, followed by 0.1 mm SIN-1; 15 BDNF+SIN, 1 hr pretreatment with 15 nm BDNF, followed by 0.1 mm SIN-1; 1.5 BDNF+SIN, 1 hr pretreatment with 1.5 nm BDNF, followed by 0.1 mm SIN-1; SIN, 1 hr treatment with 0.1 mm SIN-1. D, Neither NGF nor NT-3 prevented NO-induced growth cone collapse. 150 BDNF+SIN, 1 hr pretreatment with 150 nm BDNF, followed by 0.1 mm SIN-1; 150 NGF+SIN, 1 hr pretreatment with 150 nm NGF, followed by 0.1 mm SIN-1;150 NT-3+SIN-1, 1 hr pretreatment with 150 nm NT-3, followed by 0.1 mm SIN-1.E, The BDNF protective effect was time-dependent.cotreat, 150 nm BDNF and 0.1 mmSIN-1 added simultaneously; 15 min pretreat, cultures pretreated with 150 nm BDNF for 15 min, followed by the addition of 0.1 mm SIN-1; 30 min pretreat, cultures pretreated with 150 nm BDNF for 30 min, followed by the addition of 0.1 mm SIN-1; 45min pretreat, cultures pretreated with 150 nm BDNF for 45 min, followed by the addition of SIN-1.
NO-induced growth cone collapse is not attributable to peroxynitrite cytotoxicity
Nitric oxide can be cytotoxic (Dawson, 1995; Roth, 1997). To control for the possibility that the retinal growth cone collapse and axon retraction observed after exposure to NO donors were related to NO neurotoxicity, we observed cultures for an extended period after the addition of SIN-1. In the continuous presence of concentrations of SIN-1 ≤0.1 mm, retinal growth cones recovered and started to reextend in as few as 6 hr after the addition of SIN-1. Cultures returned to the incubator for an additional 24 hr after the addition of 0.1 mm SIN-1 were not distinguishable from control cultures and exhibited a similar percentage of axons with growth cones. Of the axons, 64% had growth cones 24 hr after the addition of 0.1 mm SIN-1 (n = 3 cultures), compared with 59% of axons in 24-hr-old control cultures (n = 3 cultures). Furthermore, measurements of cell death in explants demonstrated that 0.1 mm SIN-1 was not toxic to retinal cells (Table 1). Therefore, concentrations of SIN-1 that reliably produce growth cone collapse and axon retraction were not cytotoxic.
Role of peroxynitrite in SIN-1-induced growth cone collapse and neurotoxicity
NO-mediated neurotoxicity is believed to be attributable to peroxynitrite formed by the reaction of NO with superoxide (Lipton et al., 1993; Dawson, 1995; Bartosz, 1996; Dawson and Dawson, 1996). To separate the effects of NO from that of peroxynitrite, we used FeTPPs, a catalyst of peroxynitrite decomposition (Misko et al., 1998), along with a NO donor to block the effects of peroxynitrite that may be generated downstream of NO. In a previous study FeTPPS inhibited peroxynitrite-induced cell death but had no effect on the NO-mediated acetylcholine-induced relaxation of precontracted aortic rings (Misko et al., 1998). As determined by trypan blue staining of retinal explants, overnight exposure to 1.0 mm SIN-1 was toxic and killed the majority of cells in explants. However, 5 μmFeTPPS fully prevented retinal explant cell death, but not growth cone collapse, in response to 1.0 mm SIN-1 (Table 1). These data indicate that peroxynitrite is responsible for the retinal explant cell death, but not the collapse of growth cones observed in cultures exposed to high levels of SIN-1. Renteria and Constantine-Paton (1996)also concluded that growth cone collapse in response to NO donors is attributable to NO and not to peroxynitrite, because superoxide dismutase did not prevent growth cone collapse in response to NO donors, and peroxynitrite alone failed to elicit growth cone collapse.
BDNF protects growing axons from NO, and BDNF/NO stabilizes growth cones
BDNF protected growing retinal axons from NO-induced growth cone collapse and axon retraction. Retinal cultures were pretreated with BDNF for 1 hr before the addition of a NO donor. BDNF prevented NO-induced growth cone collapse and axon retraction with all three NO donors (Figs. 1, 2B). The effect of BDNF was dose-dependent (Fig. 2C). BDNF-treated growth cones exhibited normal motility (Fig. 3). Whereas growth cones pretreated with low concentrations of BDNF followed by the addition of SIN-1 exhibited mixed responses, including collapse, extension, and retraction during the 1 hr observation period, growth cones pretreated with high concentrations of BDNF rarely collapsed after addition of the NO donor. In the presence of BDNF the addition of the NO donor often resulted in the formation of numerous filopodia, which was followed by growth cone stabilization. Stabilized growth cones exhibited minimal lamellipodial and filopodial activity (see Fig. 1) and underwent neither significant axonal growth nor retraction (Fig. 3). The morphology of stabilized growth cones was often more complex than that of either BDNF-treated or control growth cones. Stabilized growth cones had significantly more filopodia than either BDNF-treated or control growth cones. Stabilized growth cones had on average 7.3 filopodia as compared with 3.8 filopodia on control growth cones (p < 0.0001; two-tailed Student'st test; n = at least 22 growth cones per condition) and 5.7 filopodia on BDNF-treated growth cones (p < 0.005; two-tailed Student's ttest; n = at least 21 growth cones per condition). Growth cone stabilization was not observed in cultures treated with NO alone at any of the concentrations of NO donors that were tested. Growth cone stabilization was reversible in that lamellipodia and filopodia activity restarted as soon as 30 min after washout of NO donor-containing medium, even if BDNF was still present. NO donor-mediated growth cone stabilization was not attributable to peroxynitrite or additional donor decomposition products. FeTPPS (5 μm) did not prevent 1.0 mm SIN-1-induced growth cone stabilization (seven explants tested), and all three NO donors (SIN-1, NOC-5, NOC-7) stabilized BDNF-treated growth cones in a similar manner at concentrations that normally induce growth cone collapse. Furthermore, exhausted SIN-1 did not stabilize BDNF-treated growth cones (five explants tested).

BDNF/NO treatment results in cessation of axonal growth. Cultures were either untreated or treated with 150 nm BDNF, 0.1 mm SIN-1, or 150 nmBDNF plus 0.1 mm SIN-1. Then the percentage of 5 min observation intervals during which growth cones were observed to extend, retract, or remain immotile was determined. As the graph illustrates, growth cones in untreated and BDNF-treated cultures spent most of the time extending. In contrast, growth cones in SIN-1-treated cultures spent little time extending and most of the time retracting. BDNF/NO-treated growth cones underwent neither significant extension nor retraction, indicating stabilization. Numbers inparentheses indicate the number of 5 min observation intervals that were scored.
It is unlikely that BDNF acted as a scavenger of NO, blocked NO release from donors, or had some other nonphysiological effect. BDNF, at 10 times the concentration that was most effective in preventing NO-induced axon retraction, failed to prevent chick heart fibroblasts in culture from retracting lamellipodia in response to SIN-1 (data not shown). Furthermore, neither NGF nor NT-3, neurotrophins similar in size and charge to BDNF, provided protection from NO-induced growth cone collapse and axon retraction (see Fig. 2D). Finally, BDNF-mediated protection from NO was time-dependent. Retinal cultures required a 30–45 min exposure to BDNF before the addition of the NO donor to achieve the full protective effect (see Fig.2E). These results suggest that a cell-specific physiological change induced by BDNF was responsible for the altered response to NO.
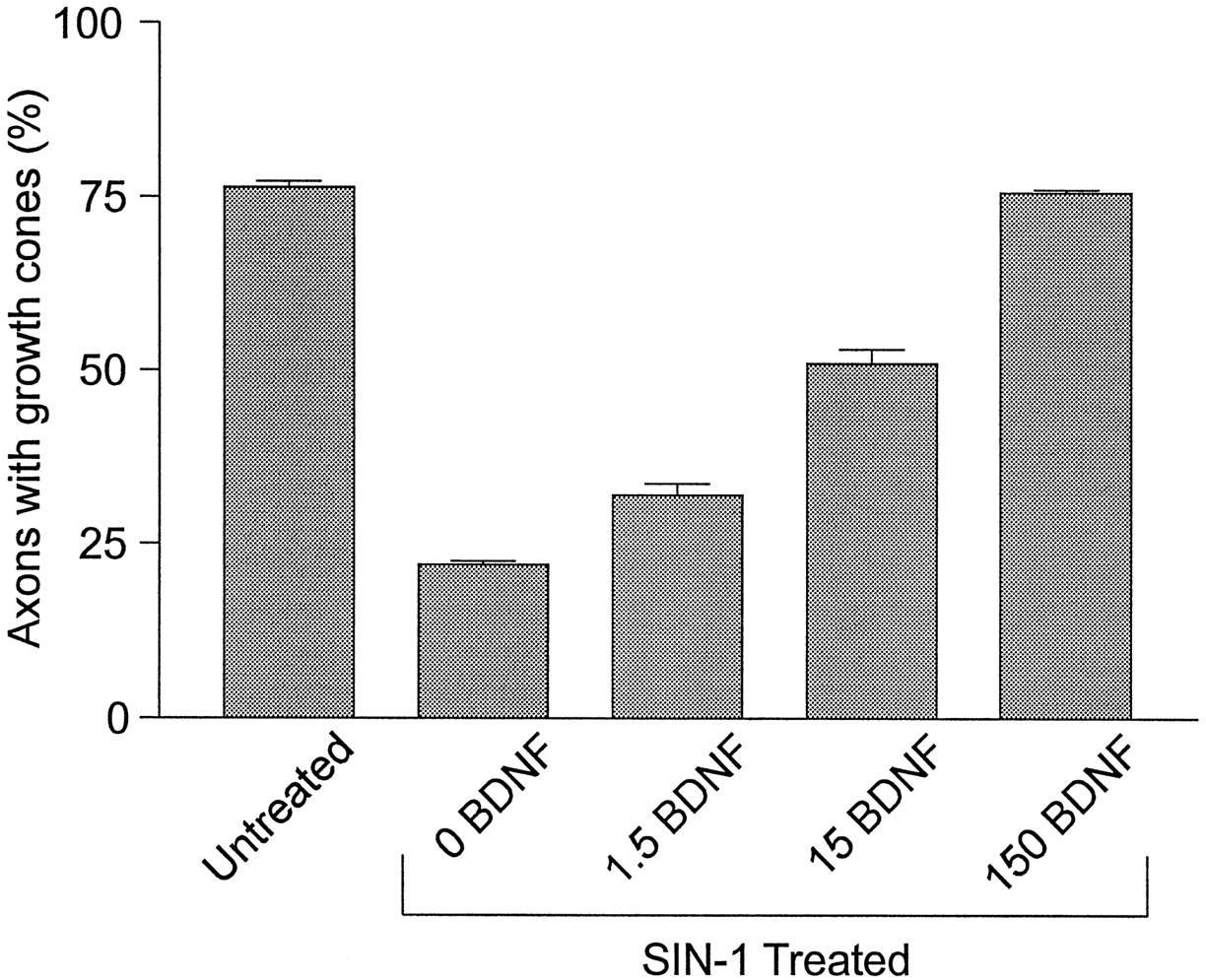
The concentration of BDNF necessary to provide maximal protection from NO-induced growth cone collapse and axon retraction (150 nm) was significantly higher than expected from the reported KD for neurotrophin binding to either trk or p75 receptors (Barbacid, 1995). If BDNF protection were dependent on access to the cell body, then the high concentration of BDNF required for protection from NO-induced axon retraction might be a consequence of the relative inaccessibility of the cell bodies to BDNF in explant cultures. To test for this possibility, we examined the concentration dependence of BDNF in dissociated retinal cultures. Retinal ganglion cell axons in the cultures were identified by using immunocytochemical staining with a retinal ganglion cell axon-specific antibody, RA4 (McLoon and Barnes, 1989). As with the explant experiments, high concentrations of BDNF were required to protect dissociated retinal ganglion cell axons from NO-induced retraction (Fig. 4). Retinal explant cultures also were treated overnight with lower concentrations of BDNF, reasoning that an increase in the amount of time that BDNF had to diffuse through the explant might result in a situation in which lower concentrations of BDNF were required for protection. Just as in cultures pretreated with BDNF for 1 hr, cultures treated overnight in low concentrations of BDNF (1.5 and 15 nm) were not protected from NO-induced axon retraction (data not shown). These results argue against the possibility that a high concentration of BDNF was required to protect axons from NO because of the physical nature of the explants.

Concentration dependence of BDNF protection in dissociated retinal cultures. Dissociated retinal cell cultures were left untreated (control) or were pretreated with vehicle (0 BDNF), 1.5, 15, or 150 nm BDNF for 1 hr and then exposed to 0.1 mm SIN-1. After a 1 hr exposure to SIN-1 the cultures were fixed and double-stained with RA4 to label retinal ganglion cell axons and with rhodamine-conjugated phalloidin to label growth cones. Then the percentage of retinal ganglion cell axons with growth cones was determined for all treatment conditions.
NO induced DRG growth cone collapse and axon retraction that were virtually identical to those seen with retinal axons. Only 10 nm BDNF was needed to maximally protect axons extending from E12 DRG explants from NO-induced axon retraction (data not shown). This concentration of neurotrophin is similar to that reported to be required for the establishment of neurotrophin-mediated protection from the inhibitory effects of myelin on DRG neuron process growth (Cai et al., 1999) These data demonstrate that the requirement for relatively high concentrations of BDNF to protect retinal axons from NO is retinal axon-specific and not a generalized feature of the neuronal mechanism of BDNF-mediated protection. High concentrations of BDNF may be required for protection against NO-induced axon retraction, because developing retinal ganglion cells express high levels of truncated trkB receptor (Escandón et al., 1994; Garner et al., 1996; Ninkina et al., 1996). Truncated trkB receptors have been shown to inhibit BDNF signaling via full-length trkB receptors and can decrease the response of cells to BDNF by >10-fold (Eide et al., 1996; Fryer et al., 1997). Embryonic DRG neurons, on the other hand, are likely to express low levels of truncated trkB receptor (Escandón et al., 1994). Thus, the difference in the concentration of BDNF required by the two cell types to protect axons from NO-induced retraction may reflect the difference in truncated trkB expression. Further experiments will be required to verify this hypothesis.
The protective effect of neurotrophins relative to NO is local
If neurotrophins are involved in refinement processes during development, it is likely that they act locally, such that axons or portions of axons receiving neurotrophins are stabilized by NO, whereas axons not receiving neurotrophins are susceptible to NO-induced pruning or retraction. This was tested by presenting neurotrophins locally to growing axons. BDNF was conjugated covalently to 10 μm polystyrene beads. Control beads were coated with cytochrome c. Beads were added to overnight cultures of retinal explants for 1 hr (for BDNF-coated beads, n = 12 cultures; for control beads,n = 10 cultures). By chance, some beads contacted growth cones or axons (Fig. 5). After 1 hr incubation with the beads, 1.0 mm SIN-1 was added to the cultures while they were monitored continuously via a microscope equipped for video recording. BDNF-coated beads protected axons from retraction. Fifty-nine percent (n = 28) of the axon tips contacting BDNF beads persisted 1 hr after the addition of the NO donor as compared with only 21% (n = 17) of the axon tips contacting control beads (p < 0.02; Fisher's test, one-tailed). Although growth cones that contacted BDNF-coated beads extended filopodia and lamellipodia on the substratum before SIN-1 treatment, after SIN-1 no further extensions on the substratum were observed. Where growth cones were not in contact with a BDNF-coated bead, in response to SIN-1 they collapsed and the axon retracted up to the point of contact with a bead (Fig. 5A). Five growth cones were observed that only partially contacted BDNF-coated beads (Fig. 5B). In response to SIN-1, the portion of the growth cone in contact with the BDNF-coated bead was retained while the rest of the growth cone collapsed. This indicates that the action of BDNF is local. Furthermore, the anchoring of axons and growth cones at BDNF-coated beads and the cessation of further axon growth after SIN-1 addition indicate that stabilization was induced by a local source of BDNF coupled with NO.

BDNF locally protects axons and growth cones from NO-induced collapse and retraction. Neurotrophins were presented locally to growing axons by adding polystyrene beads with covalently conjugated neurotrophins to retinal cultures. A, Growth cones that did not contact BDNF-coated beads directly were not protected from SIN-1. By 20 min after the addition of SIN-1 the growth cone collapsed, and during the next 40 min the axon retracted.B, Only the portions of growth cones in direct contact with BDNF-coated beads were protected from the effects of SIN-1. The regions of the growth cones making contact with beads (arrows in Before SIN-1) are retained, whereas the filopodia and lamellipodia not in contact with beads (arrowheads) collapsed after the addition of SIN-1. Beads have a diameter of 10 μm.
Additional retinal cultures incubated with BDNF-coated beads or control beads were fixed and stained with rhodamine-labeled phalloidin to allow visualization of the growth cone morphology via its actin cytoskeleton. Without SIN-1 treatment the growth cones contacting BDNF-coated beads spread over the surface of the bead but also extended filopodia and lamellipodia onto the substratum (Fig.6A). After 1 hr incubation with SIN-1, 83% of axon tips in direct contact with BDNF-coated beads exhibited growth cones (n = 138, from four cultures), compared with 18% of axon tips still in contact with control beads (n = 114, from four cultures;p < 0.0001; Fisher's test, one-tailed). The filopodia and lamellipodia of growth cones after SIN-1 treatment were predominantly in contact with the surface of BDNF-coated beads and generally did not extend onto the substratum (Fig. 6B). This provides further evidence that a local source of BDNF can protect growth cones from NO-induced collapse.

A localized source of BDNF protects retinal growth cones and axonal sprouts from NO-induced collapse. Shown are confocal images of retinal axon growth cones in contact with BDNF-coated beads. Because the beads do not show up on confocal images, the approximate locations of beads are indicated by arrowheads.A, In the absence of NO donor the filopodia extend from the growth cone onto the substratum. B, After a 1 hr treatment with 1.0 mm SIN-1 the axon tips are associated with BDNF-coated beads and do not extend processes onto the substratum.C, BDNF-coated beads elicited axonal sprouting locally from the axon shaft. D, Bead-associated axonal sprouts were unaffected by SIN-1 treatment. Scale bar, 10 μm.
Filopodia and lamellipodia sprouts appeared along the shaft of retinal axons near the point of contact with BDNF-coated beads (Fig.6C), as previously described for DRG axons contacted by neurotrophin-coated beads (Gallo and Letourneau, 1998). These sprouts appeared to be unaffected by SIN-1 treatment (Fig. 6D). However, the high degree of fasciculation in explant cultures prevented a quantitative analysis. Therefore, the effects of SIN-1 treatment on NGF-coated bead-induced sprouts along the axons was examined with dissociated DRG neurons. Similar to the results obtained with BDNF-coated beads contacting retinal ganglion cell growth cones, NGF-coated beads locally protected DRG growth cones from SIN-1-induced collapse (Fig. 7A–C). Also, SIN-1 did not decrease the percentage of axon–bead contacts that exhibited sprouts (NGF = 50%, n = 257; NGF+SIN-1 = 48%, n = 295; p > 0.3; Fisher's test, one-tailed) (Fig. 7D,E) or the mean number of filopodial tips associated with individual beads (p > 0.05; Welch t test, one-tailed). These data demonstrate that local sources of neurotrophin protect both growth cones and axonal sprouts from SIN-1-induced collapse.

NGF locally protects DRG axons from NO-mediated growth cone collapse and disruption of axonal sprouts.A, Schematic representation of the effects of axon–bead contact location on growth cone response to NO (0.1 mmSIN-1). Growth cones in contact with NGF-coated beads were protected from NO, whereas growth cones collapsed in response to NO when bead contact with the axon was >10 μm distant from the growth cone.B, C, Confocal images of growth cones directly contacting beads that did not collapse in response to NO. As in Figure4, the arrowheads denote the locations of beads. InB, three separate beads (bd 1–bd3) contacted the growth cone. The growth cone in C had spread underneath the bead. D, Confocal image of an axon with a bead located ∼30 μm behind the most distal position of the axon that is collapsed and appears to be undergoing retraction. At the site of axon–bead contact, however, filopodial sprouts developed, and these NGF-induced structures persisted although the growth cone collapsed. E, Confocal image of filopodial sprouts at the site of axon–bead contact persisted after SIN-1 treatment. Scale bars, 10 μm.
Growth cone stabilization in response to BDNF/NO is accompanied by changes in the cytoskeleton
Both the growth cone collapse induced by NO and the growth cone stabilization induced by BDNF and NO are accompanied by changes in the filamentous actin (F-actin) component of the growth cone cytoskeleton. Control cultures stained for F-actin that used rhodamine-labeled phalloidin showed F-actin broadly distributed across the growth cones of retinal axons (Fig.8A). F-actin filaments were lost from the growth cone and distal axon, as it collapsed and retracted in response to NO (Fig. 8B), consistent with previous studies that showed NO causes F-actin depolymerization in non-neuronal cells (Clancy et al., 1995; Bonfoco et al., 1996). This indicates that NO affects the actin cytoskeleton of growth cones. Other factors that induce growth cone collapse result in a similar loss of F-actin (Y. Luo et al., 1993; Kuhn et al., 1999). F-actin staining of retinal explant cultures treated with BDNF alone did not differ significantly from the F-actin staining observed in control cultures. The addition of SIN-1 to BDNF-pretreated cultures, which resulted in growth cone stabilization, decreased growth cone F-actin levels by 68% relative to those levels of cultures exposed only to BDNF (p < 0.0001; Welch t test, two-tailed) (Fig. 8C). The F-actin loss was primarily from the inner regions of lamellipodia. Interestingly, a subpopulation of F-actin filaments was retained in BDNF/NO-treated growth cones. The F-actin that remained in BDNF/NO-stabilized growth cones was concentrated in filopodia and along the edges of the growth cone (Fig.8C).

A population of actin filaments is stabilized in retinal growth cones treated with BDNF and NO. Micrographs show growth cones of retinal axons stained with rhodamine phalloidin (red) to show F-actin distribution and counterstained with DiO (green) to show the complete growth cone. A, In untreated cultures F-actin was distributed throughout the growth cone and was organized in bundles in filopodia and in a meshwork in lamellipodia. B, In cultures treated for 30 min with 1.0 mm SIN-1, the growth cones collapsed and F-actin staining was greatly diminished, indicating F-actin depolymerization. C, In cultures pretreated for 1 hr with 150 nm BDNF, followed by 30 min with 1.0 mm SIN-1, growth cone morphology was maintained, but the normally pronounced meshwork of actin filaments in lamellipodia was lost, allowing the DiO signal to show through (thin arrows), whereas actin bundles in filopodia or along the edges of the growth cone profile were maintained (thick arrows). D, In cultures pretreated for 1 hr with 150 nm BDNF, followed by 30 min with 1.0 mmSIN-1 and then treated with 0.1 μg/ml cytochalasin D for 30 min, the distribution of F-actin filaments was relatively unaffected as compared with growth cones similarly treated with BDNF and SIN-1, but not cytochalasin D. Scale bar, 10 μm.
The F-actin bundles in BDNF/NO-treated growth cones were highly stable. Treatment of control cultures for 30 min with 0.1 μg/ml cytochalasin D, a drug that inhibits the polymerization of actin by capping the barbed ends of filaments (Cooper, 1987), resulted in growth cone collapse and axon retraction (data not shown). Whereas cytochalasin D treatment reduced F-actin levels in BDNF-treated cultures by 51% (p = 0.0006; Welch t test, one-tailed), cytochalasin D had no effect on F-actin levels in BDNF/NO-stabilized growth cones (Fig. 8D) (p > 0.3; Welch t test, one-tailed), and the axons did not retract. Although BDNF partially stabilized actin filaments against cytochalasin D-induced depolymerization, the combined treatment of BDNF and NO resulted in complete resistance of the remaining F-actin filaments to cytochalasin D treatment. Furthermore, the F-actin retained in BDNF/cytochalasin D-treated growth cones was distributed homogeneously across the growth cone and was not concentrated in filopodia or along the edges of the growth cone, as was observed in BDNF/NO-treated growth cones.
DISCUSSION
NO induces retraction of axons
This study showed that chick retinal ganglion cell growth cones collapse and the axons retract in response to NO, a finding consistent with previously reported data on amphibian retinal axons (Renteria and Constantine-Paton, 1996) and rodent DRG neurons (Hess et al., 1993). This in vitro phenomenon complements previous in vivo studies that showed NO is involved in the elimination of a transient ipsilateral retinotectal projection in chick and mouse (Wu et al., 1994; Mize et al., 1998; Ernst et al., 1999) and the elimination of inappropriately targeted axonal branches in the ferret lateral geniculate nucleus (Cramer et al., 1996), and that it is necessary for correct targeting of retinal axons in the optic lobe ofDrosophila (Gibbs and Truman, 1998). The present results in combination with previous studies suggest that one role of NO in the refinement of neuronal connections is to cause the withdrawal of inappropriately targeted axons.
BDNF prevents NO-induced axon retraction
Why is it that appropriately targeted axons in vivo do not withdraw in the presence of NO, if NO functions as a withdrawal signal? This is a crucial question, given that NO can diffuse tens of microns from a source cell (Lancaster, 1997) and, therefore, potentially can interact with multiple presynaptic axons, presumably those that are appropriately and inappropriately targeted. This study showed that growing retinal axons in cultures pretreated with BDNF were protected from NO-induced axon retraction. This result suggests that axons appropriately connected in vivo may be protected from NO-induced retraction by neurotrophins. This possibility is supported by previous in vivo studies. Inhibition of endogenous BDNF resulted in a loss of retinal axon branches in the tectum (Cohen-Cory and Fraser, 1995), suggesting the existence of tectum-derived signals that initiate axonal withdrawal and that normally are blocked by tectum-derived BDNF. Adenovirus-mediated overexpression of BDNF in superior colliculus neurons resulted in the retention of the transient ipsilateral retinal projection to the superior colliculus in rat (Isenmann et al., 1999). This last result is of particular relevance to the present study, because NO has been shown to mediate the elimination of the ipsilateral retinal projection in both chick (Wu et al., 1994;Ernst et al., 1999) and mouse (Mize et al., 1998). These data support the hypothesis that the elimination of some neuronal projections depends on the balance between positive (e.g., BDNF) and negative (e.g., NO) target-derived signals.
The intracellular signal transduction mechanisms responsible for BDNF protection against NO-induced axon retraction remain to be determined. This study showed that retinal axons must be primed with BDNF for 30–45 min to achieve maximum protection from NO-induced collapse. The time dependence indicates that a physiological change in the retinal ganglion cell induced by BDNF is responsible for the altered response to NO. Recently, it was shown that neurotrophins can protect regenerating spinal axons from the growth-inhibiting effects of myelin and myelin-associated glycoprotein (MAG) (Cai et al., 1999) and alter the collapse response of DRG neurons to collapsin (Tuttle and O'Leary, 1998). The neurotrophin-mediated protection of regenerating spinal axons from the growth-inhibiting effects of myelin and MAG was also time-dependent and required the activation of the cAMP-protein kinase A (PKA) pathway. In preliminary experiments the treatment of cultures with PKA inhibitors blocks BDNF-mediated protection from NO-induced growth cone collapse (Gallo et al., 1999). In the case of spinal neuron regeneration, however, 6 hr of priming with BDNF was required to prevent MAG-mediated inhibition of neurite growth. This difference in time course between the two systems presumably reflects some difference in intracellular signaling pathways.
BDNF together with NO stabilizes growth cones
Perhaps the most surprising result of this study was that growth cones of retinal axons exposed to both BDNF and NO, but not either factor alone, became strikingly immotile. BDNF/NO-stabilized growth cones exhibited minimal activity of their lamellipodia and filopodia and showed neither axon extension nor retraction. This is unusual, given that growth cones are normally highly motile. Stabilization is not simply an ameliorated NO-induced retraction but rather appears to be a specific reaction by the cell to the combination of BDNF and NO. With concentrations of BDNF that do not result in NO-induced stabilization, axons often continue to extend in the presence of NO. Thus, BDNF can protect growing axons from NO without inducing growth cone stabilization. Stabilization is accompanied by a reorganization of the actin cytoskeleton of the growth cone, indicating a specific physiological response to the combined signals. Actin filaments had a different distribution in BDNF/NO-treated growth cones than seen in control growth cones or growth cones treated with either BDNF or NO alone. The F-actin bundles in BDNF/NO-treated growth cones were highly stable as demonstrated by their resistance to the effects of cytochalasin D, a drug that normally causes depolymerization of dynamic F-actin. These findings may have implications for the mechanisms of BDNF and NO in regulating synaptic transmission. Previous studies established that aspects of synaptic transmission are dependent on the dynamics of the actin cytoskeleton (Wang et al., 1996; Allison et al., 1998; Halpain et al., 1998; Kim and Lisman, 1999). Furthermore, BDNF and NO have been shown to modulate synaptic activity (Schuman and Madison, 1991; Haley et al., 1993; Lohof et al., 1993; Wang et al., 1995a; Akaneya et al., 1997; Liou et al., 1997; Wang and Poo, 1997;Martı́nez et al., 1998; Vicario-Abejón et al., 1998), and BDNF has been demonstrated to promote the maturation of synapses (Wang et al., 1995b) and regulate hippocampal synaptogenesis (Martı́nez et al., 1998; Vicario-Abejón et al., 1998). These changes could all be linked to changes in the cytoskeleton induced by these neuromodulators.
The intracellular link between BDNF/NO and the actin cytoskeleton remains to be identified, but it appears that this link operates very locally. Beads coated with BDNF or NGF were used to present neurotrophin locally to growing axons. Only regions of axons and growth cones in direct contact with the beads were protected from the effects of NO. This demonstrates that different regions of the same axon can be rendered differentially susceptible to the effects of NO. Such a local response could allow BDNF/NO to preserve specific branches of axons during refinement in vivo. It is interesting that the potentiating effects of postsynaptically derived NT-4 on the developing neuromuscular junction are synapse-specific (Wang et al., 1998).
Growth cone stabilization observed after exposure to BDNF and NO could not have been predicted from the behavior of growing axons exposed to either signaling molecule alone. Although NO alone caused retraction of filopodia, growth cones treated with both BDNF and NO exhibited more filopodia than growth cones either untreated or exposed only to BDNF. This indicates that the effect of NO is switched by previous exposure to BDNF from causing a loss of filopodia to promoting the formation and long-term retention of filopodia. Recent studies showed that growth cones could be made to turn toward or away from a source of chemoattractant such as BDNF or netrin by modulating internal levels of cAMP (Ming et al., 1997; Song et al., 1997, 1998). It could be that, in the case of the present study, BDNF alters cAMP levels in the growth cone, which in turn alters the response to NO. These data emphasize that growth cone behavior is ultimately dependent on multiple signaling molecules (Winberg et al., 1998; Rose and Chiba, 1999).
The stabilization of axon terminals signaled via a combination of molecules, such as NO and BDNF, may be important during the development of neuronal circuitry (Holt and Harris, 1998). Axon branches that are retained through the refinement process must be stabilized to prevent both retraction and overgrowth. BDNF and NO are produced by postsynaptic neurons in response to activity (Lindefors et al., 1992; D. Luo et al., 1993; Lindholm et al., 1994; Mowla et al., 1999); thus, it is reasonable to expect that retinal axon branches and synapses would be exposed to both BDNF and NO. Importantly, NMDA receptor activity has been demonstrated to be necessary for the stabilization of presynaptic retinal axons in vivo (Rajan et al., 1999) and is necessary for the generation of NO by tectal cells (Ernst et al., 1998). Thus, the stabilization of growth cones treated with BDNF and NO seen in vitro may reflect a process that occurs in vivo when an axon establishes synaptic contact with an appropriate target cell.
The potential role of BDNF and NO in development of the retinotectal projection
These findings have implications for the mechanisms that could account for the selective elimination and retention of different retinal axon branches on a tectal neuron during development. Initially, in the development of retinotectal connections both correctly and incorrectly targeted axons connect with individual tectal neurons. Coordinated activity in correctly targeted retinal axons is postulated to result in activation of NMDA receptors on postsynaptic tectal cells. Activation of NMDA receptors has been linked to the production of NO (D. Luo et al., 1993; Ernst et al., 1999) as well as to an increase in neurotrophin expression (Lindefors et al., 1992). Similar to the NO-induced retraction of retinal axons in tissue culture, which is blocked by BDNF, NO released by tectal cells in vivocould initiate the retraction of retinal axons that do not receive adequate levels of BDNF (i.e., mistargeted axons whose firing does not correlate with activation of the postsynaptic cell). An axon most likely would have multiple branches. NO could induce retraction of the incorrectly targeted branches, whereas correctly targeted branches may be stabilized, possibly by the combined signaling of BDNF and NO. Axons without stable connections may continue to grow. Cells that fail to establish stable axonal connection by some critical stage in their development may be eliminated by cell death (Hughes and McLoon, 1979).
A number of models for the interactions of BDNF and NO signaling during retinotectal development can be postulated. For instance, release of BDNF from an activated tectal cell may be targeted specifically to recently active synapses. In support of this hypothesis, BDNF mRNA and protein are localized selectively to dendritic branches in an activity-dependent manner in hippocampal neurons (Tongiorgi et al., 1996, 1997). Interestingly, the overexpression of BDNF in superior colliculus neurons results in the retention of the ipsilateral retinocollicular projection (Isenmann et al., 1999), suggesting that cellular sources of neurotrophins can protect axons from pruning during refinement. Alternatively, recently active synapses may be more sensitive to BDNF than inactive synapses. Thus BNDF could be distributed homogeneously within the target tissue and yet only preserve appropriate synapses. This possibility is supported by a recent study demonstrating that presynaptic depolarization facilitates BDNF-induced synaptic potentiation (Boulanger and Poo, 1999). If a similar mechanism applies to retinotectal synapses in vivo, then by either model the combined effect of NO and BDNF could stabilize the branches of correctly targeted axons. In summary, the adult pattern of connections between the retina and the tectum could depend in part on intricately balanced levels of BDNF and NO. The present findings also could have implications for other forms of neuronal plasticity (e.g., LTP and LTD) that have been shown to be modulated by both NO and neurotrophins and could serve to highlight the possible importance of combinatorial signaling during both neurodevelopment and adult plasticity.
Although models involving interactions between BDNF and NO are consistent with our current understanding of retinotectal development, they may not hold for every system. The refinement of some systems does not appear to depend on NO (Cramer et al., 1996; Ruthazer et al., 1996;Finney and Shatz, 1998; Renteria and Constantine-Paton, 1999), and some neurons are responsive to neurotrophins other than BDNF (Reichardt and Farinas, 1997). It could be that other NO-like molecules, possibly carbon monoxide, serve a role similar to that of nitric oxide during the refinement of connections in other systems. Furthermore, even for the retinotectal system, NO-independent mechanisms also must be active in refinement (Ernst et al., 1998; Mize et al., 1998). It might be expected that such a fundamental process as the development of the proper pattern of connections is mediated by a number of redundant mechanisms.
Footnotes
This work was supported by National Institutes of Health Grants EY11926 and HD19950, by a grant from the Minnesota Medical Foundation, and by National Institutes of Health and National Science Foundation training grants. We are grateful for gifts of BDNF and NT-3 from Dr. L. O. Palladino (Regeneron Pharmaceuticals, Tarrytown, NY).
A.F.E. and G.G. contributed equally to this work.
Correspondence should be addressed to Dr. Steven C. McLoon, Department of Neuroscience, University of Minnesota, 6-145 Jackson Hall, 321 Church Street SE, Minneapolis, MN 55455. E-mail:mcloons{at}lenti.med.umn.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}