

Abstract
Loss of nigrostriatal dopaminergic neurons in Parkinson's disease (PD) leads to increased activity of glutamatergic neurons in the subthalamic nucleus (STN). Recent studies reveal that the resultant increase in STN-induced excitation of basal ganglia output nuclei is responsible for the disabling motor impairment characteristic of PD. On the basis of this, it is possible that any manipulation that reduces activity at excitatory STN synapses onto basal ganglia output nuclei could be useful in the treatment of PD. We now report that group II metabotropic glutamate receptors (mGluRs) are presynaptically localized on STN terminals and that activation of these receptors inhibits excitatory transmission at STN synapses. In agreement with the hypothesis that this could provide a therapeutic benefit in PD, a selective agonist of group II mGluRs induces a dramatic reversal of catalepsy in a rat model of PD. These results raise the exciting possibility that selective agonists of group II mGluRs could provide an entirely new approach to the treatment of PD. These novel therapeutic agents would provide a noninvasive pharmacological treatment that does not involve the manipulation of dopaminergic systems, thus avoiding the problems associated with current therapies.
- substantia nigra pars reticulata
- subthalamic nucleus
- group II metabotropic glutamate receptors
- Parkinson's disease
- catalepsy
- presynaptic inhibition
Parkinson's disease (PD) is a common neurodegenerative disorder characterized by disabling motor impairments including tremor, rigidity, and bradykinesia. The primary pathological change giving rise to the symptoms of PD is the loss of dopaminergic neurons in the substantia nigra pars compacta that modulate the function of neurons in the striatum and other nuclei in the basal ganglia (BG) motor circuit. Currently, the most effective pharmacological agents for the treatment of PD include levodopa (l-DOPA), the immediate precursor of dopamine, and other drugs that replace the lost dopaminergic modulation of BG function (Poewe and Granata, 1997). Unfortunately, dopamine replacement therapy ultimately fails in most patients because of loss of efficacy with progression of the disease and severe motor and psychiatric side effects (Poewe et al., 1986). Because of this, a great deal of effort has been focused on developing new approaches for the treatment of PD.
Recent studies reveal that loss of nigrostriatal dopamine neurons results in a series of neurophysiological changes that lead to overactivity of a critical nucleus in the BG motor circuit termed the subthalamic nucleus (STN). The STN contains glutamatergic projection neurons that provide the major excitatory input to the globus pallidus internal segment (GPi) and the substantia nigra pars reticulata (SNr), the major output nuclei of the basal ganglia. Increased activity of STN neurons leads to an increase in glutamate release at STN synapses onto GABAergic projection neurons in the output nuclei. This glutamate-mediated overexcitation of BG output ultimately leads to a “shutdown” of thalamocortical projections and produces the motor impairment characteristic of PD (Wichmann and DeLong, 1997). Discovery of the pivotal role of increased STN-mediated excitation of the BG output nuclei in PD has led to a major focus on surgical approaches for treatment. For instance, lesions or high-frequency stimulation of the STN provides a therapeutic benefit to PD patients (Limousin et al., 1995). In addition, pallidotomy, a surgical lesion of the GP, produces similar therapeutic effects by reversing the impact of increased activity of STN neurons (Laitinen et al., 1992; Baron et al., 1996). Development of these highly effective neurosurgical approaches provides a major advance in our understanding of the pathophysiology of Parkinson's disease. However, surgical approaches are not widely available to Parkinson's patients. Because of their invasive nature, high cost, and the considerable expertise required, such treatment is reserved for patients that are refractory to dopamimetic therapy.
An alternative to surgical approaches to reducing the increased excitation of basal ganglia output nuclei in PD patients would be to use pharmacological agents that counteract the effects of overactivation of the STN neurons by reducing transmission at excitatory STN synapses onto the SNr and GPi neurons. Although antagonists of postsynaptic ionotropic glutamate receptors can improve parkinsonian symptoms in PD patients and in animal models of PD (Klockgether et al., 1993; Kornhuber et al., 1994), these compounds are most effective as adjuncts to dopamine replacement therapy (Starr, 1995). Another approach would be to target metabotropic glutamate receptors (mGluRs), which are often localized presynaptically on glutamatergic terminals where they can inhibit glutamate release. Interestingly, the group II mGluRs (mGluR2 and mGluR3) are expressed in STN neurons (Testa et al., 1994), and these receptors have been shown to regulate glutamate release in other brain regions (Hayashi et al., 1993; Shigemoto et al., 1997). We now report that group II mGluRs are presynaptically localized on STN terminals in the SNr and that activation of these receptors reduces excitatory synaptic responses. Furthermore, activation of group II mGluRs provides a dramatic therapeutic effect in a rat model of Parkinson's disease. If this or related drugs prove to be effective in patients with Parkinson's disease, this could lead to a novel approach for the treatment of this debilitating disorder.
MATERIALS AND METHODS
Materials.6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX), (R,S)-α-cyclopropyl-4-phosphonophenylglycine (CPPG),d(−)-2-amino-5-phosphonopentanoic acid (d-AP-5), and (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV) were obtained from Tocris (Ballwin, MO). 2R,4R-4-Aminopyrrolidine-2,4-dicarboxylate (2R,4R-APDC), (+)-2-aminobicyclo[3·1·0]-hexane-2,6-dicarboxylate monohydrate (LY354740), and 2S-2-amino-2-(1S,2S-2-carboxycyclopropyl-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495) were gifts from D. Schoepp and J. Monn (Eli Lilly, Indianapolis, IN). All other materials were obtained from Sigma (St. Louis, MO).
Electrophysiology. Whole-cell patch-clamp recordings were obtained as described previously (Marino et al., 1998) except that recordings were made under visual control. Fifteen- to 18-d-old Sprague Dawley rats were used for all patch-clamp studies. Brains were rapidly removed and submerged in an ice-cold sucrose buffer (in mm): sucrose, 187; KCl, 3; CaCl2, 2; MgSO4, 1.9; KH2PO4, 1.2; glucose, 20; and NaHCO3, 26; equilibrated with 95% O2/5% CO2. Parasagittal slices (300 μm thick) were made using a Vibraslicer (WPI). Slices were transferred to a holding chamber containing normal artificial CSF (ACSF; in mm, NaCl, 124; KCl, 2.5; MgSO4, 1.3; NaH2PO4,1.0; CaCl2, 2.0; glucose, 20; and NaHCO3, 26; equilibrated with 95% O2/5% CO2). In some experiments, 5 μm glutathione, 500 μmpyruvate, and 250 μm kynurenate were included in the sucrose buffer and holding chamber. These additional compounds tended to increase slice viability but did not have any effect on experimental outcome. Therefore all of the data from these two groups have been pooled. Slices were transferred to the stage of a Hoffman modulation contrast microscope and continually perfused with room temperature ACSF (∼3 ml/min; 23–24°C). Neurons in the substantia nigra pars reticulata were visualized with a 40× water immersion lens. Patch electrodes were pulled from borosilicate glass on a Narashige vertical patch pipette puller (Tokyo, Japan) and filled with buffer (in mm, potassium gluconate, 140; HEPES, 10: NaCl, 10; EGTA, 0.6; GTP, 0.2; and ATP, 2; pH adjusted to 7.5 with 0.5N NaOH). Biocytin (0.5%; free base) was added just before use. Electrode resistance was 3–7 m[scap]Ω. For measurement of synaptically evoked currents, bipolar tungsten electrodes were used to apply stimuli to the STN. Stimulating electrodes were positioned with one pole slightly penetrating the tissue and the other pole above the slice. Synaptically evoked EPSCs were record from a holding potential of −60 mV, and slices were bathed in 50 μm picrotoxin. IPSCs were evoked in a similar manner but with the electrodes placed in the cerebral peduncle rostral to the recording sight and in the presence of 10 μm CNQX and 10 μmd-AP-5 to block excitatory transmission. IPSCs were recorded from a holding potential of −50 mV. STN-evoked fiber volleys were recorded by placing a low-resistance pipette (0.5–2 m[scap]Ω) filled with 3 m NaCl in the cerebral peduncle and stimulating the STN as described above. Fiber volleys were evoked in the presence of 20 μm CNQX and 20 μm bicuculline. For measurement of kainate-evoked currents, kainate (100 μm) was pressure ejected into the slice from a low-resistance pipette. Kainate-evoked currents were recorded from a holding potential of −60 mV, and slices were bathed in ACSF containing 500 nmtetrodotoxin. For studies of miniature EPSCs (mEPSCs), slices were bathed in standard ACSF with the addition of 50 mmmannitol, 500 nm tetrodotoxin, and 10 μmbicuculline warmed to 25°C. Glutamate-evoked EPSCs were recorded in the presence of 20 μm bicuculline. Glutamate (100 μm in ACSF) was applied by a syringe pump (1 ml/min) through a microapplicator made from a fused silica microtube (MicroFil; WPI). The microapplicator was positioned slightly above the slice and dorsal to the STN. The flow of glutamate was parallel to the bath flow, and the slice was arranged so that glutamate application to surrounding areas was minimized (see Fig. 3). This method was also used to produce a local application of LY345740 for some experiments (see Fig.1A,B). GABAergic projection neurons were identified according to previously established electrophysiological and morphological criteria (Richards et al., 1997). GABAergic neurons exhibited spontaneous repetitive firings, short-duration action potentials (half-amplitude duration = 1.7 ± 0.2 msec), little spike frequency adaptation, and a lack of inward rectification, whereas dopaminergic neurons displayed no, or low-frequency, spontaneous firings, longer-duration action potentials (half-amplitude duration = 7.0 ± 0.5 msec), strong spike frequency adaptation, and a pronounced inward rectification. Light microscopic examination of biocytin-filled neurons indicated that GABAergic neurons had extensive dendritic arborizations close to the cell body, whereas the dendritic structures of dopaminergic neurons were relatively sparse. All of the data presented in these studies are from confirmed GABAergic neurons.
Immunocytochemistry. Preparation of the tissue for immunocytochemical analysis at the electron microscopy level followed previously published protocols (Bradley et al., 1996). The avidin–biotin–peroxidase method (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA) was used to detect mGluR2/3 immunoreactivity in rat (n = 2) SNr. The peroxidase reaction was developed in 0.05% diaminobenzidine and 0.01% H2O2. Antibodies that specifically recognize mGluR2 and mGluR3 are from Chemicon (Temecula, CA).
Behavioral studies. Male Sprague Dawley rats 30 d old at the start of experiments were injected intraperitoneally with either haloperidol (2 mg/ml solution dissolved in 8.5% lactic acid, neutralized with 1N NaOH, and diluted to 0.3 mg/ml in saline) or saline and returned to their home cage for 30 min. After 30 min, the animals were injected with either saline or LY354740 (0.6–6 mg/ml dissolved in saline). Catalepsy was measured 1 hr later by placing the animal's forepaws on a bar elevated 4.5 cm. The time to removal of one paw was measured by a stopwatch. Animals were then placed on a vertical mesh ∼6 inches above the ground, and the time to remove one paw from the mesh was measured. Animals were tested once per day, and saline controls were run between each drug test. All animals were habituated to the tasks by 3 consecutive days of saline control treatment before beginning drug testing.
RESULTS
Whole-cell patch-clamp techniques were used to record EPSCs from GABAergic projection neurons of the SNr in midbrain slices. EPSCs were elicited by stimulation of the STN with bipolar stimulating electrodes (0.4–12.0 μA every 60–90 sec) in the presence of 50 μm picrotoxin. EPSCs elicited with this protocol had a constant latency, were monophasic, and were completely abolished with the application of 10 μm CNQX (n = 10; data not shown), suggesting that the synaptic response was a monosynaptic glutamatergic EPSC.
Activation of group II mGluRs inhibits transmission at the STN–SNr synapse
Brief local application of 100 nm LY354740, a highly selective agonist of group II mGluRs (Monn et al., 1997; Schoepp et al., 1997), produced a reversible depression of EPSCs in SNr projection neurons (Fig. 1A,B). It should be noted, whereas LY354740 reduced evoked EPSCs in every cell tested, that longer bath applications resulted in inconsistent washout of the effect of LY354740. This is primarily because we recorded from cells at different depths in the slice. The deeper cells required longer periods of agonist application and exhibited slower washout of effects. However, longer bath applications were used in all additional studies to ensure an equilibrium concentration of drug at the sites of action and a maximal response. A concentration–response curve for LY354740 revealed an EC50 of ∼75 nm (Fig. 1C), consistent with the potency of this compound at group II mGluRs. The steep slope of the concentration–response curve for LY354740 is consistent with the dose–response relationship reported for a number other effects of this drug in both recombinant and native systems (Monn et al., 1997;Schaffhauser et al., 1997; Schoepp et al., 1997). The reduction of EPSC amplitude was mimicked by two other highly selective agonists of group II mGluRs, 2R,4R-APDC (Schoepp et al., 1995) and DCG-IV (Hayashi et al., 1993; Gereau and Conn, 1995a) (Fig. 1D), and was completely blocked by previous application of LY341495 (100 nm) or CPPG (500 μm) (Fig. 1D), both of which are antagonists active at group II mGluRs (Toms et al., 1996; Kingston et al., 1998).
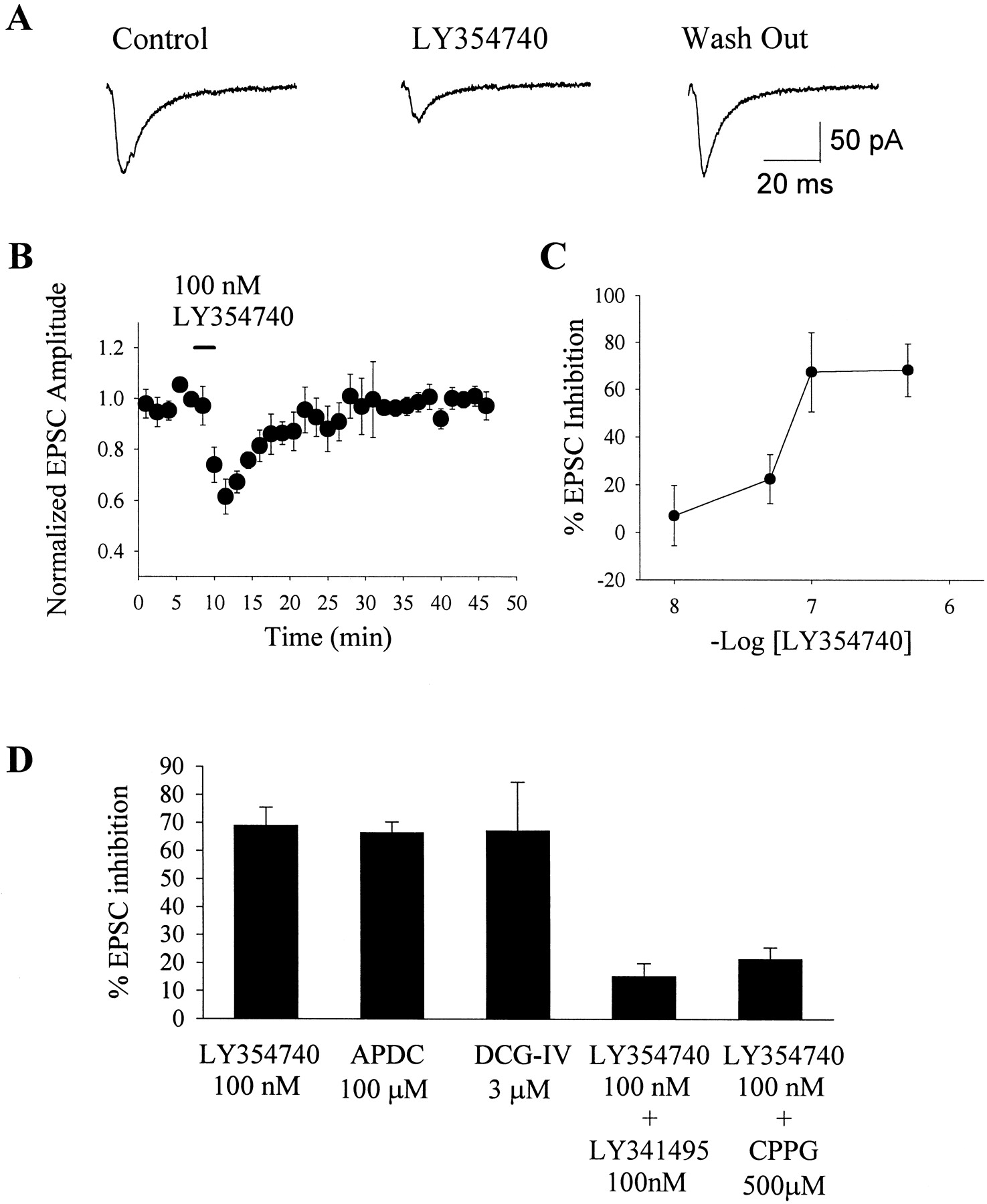
Activation of group II mGluRs reduces EPSCs at the STN–SNr synapse. A, Evoked EPSCs before (Control), during (LY354740), and after (Wash Out) a brief local application of LY354740. Applications of LY354740 dramatically reduce EPSCs, and this effect is reversible. B, Average time course of the effect of 100 nm LY354740 (application indicated byhorizontalbar). Eachpoint represents the mean (± SEM) of data from five cells. C, Dose–response relationship of LY354740-induced inhibition of EPSCs. The effect of inhibition of EPSCs is maximal at 100 nm. Each point represents the mean of three experiments. D, Effects of specific group II mGluR agonists on EPSCs at the STN–SNr synapse and block of the LY354740-induced inhibition of EPSCs by application of group II mGluR antagonists before application of the agonist. Agonists include LY354740 (100 nm), APDC (100 μm), and DCG-IV (3 μm). Antagonists include LY341495 (100 nm) and CPPG (500 μm). Each verticalbar represents the mean (± SEM) of data collected from five cells (*p < 0.01).
Group II mGluRs are localized presynaptically at excitatory terminals in the SNr
Taken together, these data suggest that activation of group II mGluRs reduces transmission at the STN–SNr synapse. We used a combination of immunocytochemical and biophysical approaches to determine whether group II mGluRs elicit this effect by a presynaptic or a postsynaptic mechanism of action. First, we used antibodies that specifically recognize both mGluR2 and mGluR3 for immunocytochemical localization of group II mGluRs in the SNr. Analysis of mGluR2/3 immunoreactivity at the electron microscopic level revealed that group II mGluRs are presynaptically localized (Fig.2). The morphology of the labeled synapses, including their asymmetric nature, was characteristic of STN terminals (Fig. 2A–D) (Bevan et al., 1994). Quantification of the labeling was assessed by counting asymmetric terminals on three randomly selected grids that resulted in an estimated 30% labeling of asymmetric terminals (25 labeled terminal of 82 total). However, it is important to note that quantification of any preembedding immunocytochemical labeling at the electron microscopic level is confounded by the nonhomogeneous penetration of the antibodies through the vibratome sections. The first 5–10 μm on both sides of the sections are usually labeled, whereas the middle remains devoid of immunostaining. This implies that the lack of immunoreactivity in some structures could be attributable either to a genuine lack of antigens or to the inaccessibility of the antibodies to this particular site. Therefore, only the positive immunolabeling can be conclusively interpreted. Because of this, the 30% labeling observed in these studies represents a lower limit to the extent of staining. We also observed labeling of terminals that did not make clear synaptic contact with postsynaptic elements and of fine processes that were reminiscent of previous reports of mGluR2/3 distribution in preterminal axons (data not shown) (Lujan et al., 1997). Furthermore, there was occasional labeling of symmetric synapses (Fig. 2E), although the majority of symmetric synapses were unlabeled. There was no observable staining of dendrites, dendritic spines, or other postsynaptic elements.

Group II mGluRs are presynaptically localized at asymmetric terminals in the SNr. A–D, Electron micrographs demonstrating presynaptic mGluR2/3 immunoreactivity at asymmetric terminals in the SNr. Labeled (*) axon terminals (t) are shown synapsing on unlabeled dendrites (d) and dendritic spines (s). E, An example of a labeled terminal forming a symmetric synapse. Synapses are indicated byarrows. Scale bar: A, 301 nm;B, 203 nm; C, 315 nm; D, 263 nm; E, 207 nm.
The group II mGluR-mediated inhibition of synaptic transmission is caused by a presynaptic mechanism
The presence of mGluR2/3 immunoreactivity at presynaptic but not postsynaptic sites in the SNr suggests that these receptors are likely to act by inhibiting glutamate release from presynaptic terminals rather than by modulating the postsynaptic glutamate-gated ion channels. To test this hypothesis further, we determined the effects of maximal concentrations of LY354740 on currents elicited by brief (50–500 msec) pressure ejection of kainate (100 μm) into the slice. In the presence of 500 nm tetrodotoxin, kainate application produced a robust, stable, inward current in SNr GABAergic neurons (Fig. 3A). The kainate-evoked currents were blocked by application of 10 μm CNQX (n = 4; data not shown) indicating that they were mediated by activation of AMPA/kainate receptors. Application of 100 nm LY354740 produced no significant change in kainate-evoked currents (Fig.3A,B).

Activation of group II mGluRs has no effect on the response to exogenously applied kainate. A,Representative traces of kainate-evoked currents in the SNr projection neurons before (Control; left) and during application of 100 nm LY354740 (right).B, Time course of the effect of LY354740 on the amplitude of kainate-evoked currents. C, Mean data demonstrating the lack of effect of group II mGluR activation on kainate-evoked currents (mean ± SEM; p > 0.05; n = 5).
Although the lack of effect of LY354740 on kainate-evoked currents is consistent with a presynaptic mechanism of action, it is conceivable that exogenously applied kainate selectively activates nonsynaptic glutamate receptor channels and that LY354740 selectively modulates channels that are localized at synapses. Thus, we also determined the effect of maximal concentrations of LY354740 on the frequency and amplitude of spontaneous mEPSCs. Recordings were made in the presence of tetrodotoxin (500 nm) to block activity-dependent release and of bicuculline (10 μm) to block GABAA-mediated synaptic currents. LY354740 (100 nm) produced no significant alteration in mEPSC frequency, amplitude, or waveform (Fig.4A–C). This can be observed by a lack of effect of LY354740 on either the amplitude histograms (Fig. 4C) or the cumulative probability plots (Fig. 4D). Furthermore, overlay of an average of all mEPSCs before and after LY354740 application shows identical current amplitudes and kinetics between the two conditions (Fig.4B). The average mEPSC frequency is 4.71 ± 0.79 Hz before drug application and 4.66 ± 0.8 Hz during application of 100 nm LY354740 (p > 0.05; n = 5). The average amplitude of mEPSCs was 9.2 ± 1.3 pA before and 8.4 ± 0.8 pA after LY354740 addition (p > 0.05; n = 5).

Inhibition of EPSCs at the STN–SNr synapse is mediated by a presynaptic mechanism. A,Examples of mEPSCs before (Pre-Drug; left) and during application of 100 nm LY354740 (right).B, Overlayed averages of all mEPSCs recorded before and during LY354740 application, demonstrating the lack of effect on the amplitude and kinetics of mEPSCs. C, Amplitude histograms of mEPSCs before (left) and during application of 100 nm LY354740 (right).D, Cumulative frequency plots illustrating the lack of effect of LY354740 on mEPSC amplitude (left) and inter-event interval (right) (Kolmogorov–Smirnov test;p = 0.99). The data shown are representative of five separate experiments.
The lack of effect on mEPSC amplitude and frequency is consistent with the group II mGluR-mediated inhibition in synaptic transmission having a presynaptic site of action. There are a number of potential mechanisms by which a receptor could act presynaptically to reduce action potential-dependent release without decreasing the frequency of mEPSCs. For instance, mEPSCs are thought to be voltage independent and therefore should be insensitive to modulation of presynaptic voltage-dependent ion channels. If a receptor reduces transmission by inhibiting a presynaptic voltage-dependent calcium channel or increasing conductance through a voltage-dependent potassium channel rather than having some downstream effect on the release machinery, this may reduce evoked responses without affecting mEPSCs. This effect has been demonstrated at a variety of synapses where agents known to act presynaptically, such as cadmium, abolish evoked EPSCs but have no effect on either the frequency or amplitude of mEPSCs (Parfitt and Madison, 1993; Doze et al., 1995; Gereau and Conn, 1995b; Scanziani et al., 1995).
Although the analysis of the effects of LY354740 on mEPSCs is consistent with a presynaptic site of action, one concern with studies of mEPSCs is that it is impossible to identify the source of afferent fibers. This issue is particularly important in cases in which there is no observable effect on mEPSC frequency because of the possibility that the majority of mEPSCs arise from a separate population of afferents than those stimulated to produce evoked release. Although the majority of glutamatergic input to the SNr arises from the STN, several other regions including the pedunculopontine nucleus (Charara et al., 1996) and the nucleus raphe (Corvaja et al., 1993) provide a sparse projection accounting for a small percentage of asymmetric terminals in the SNr that could release glutamate. To address this issue, we applied glutamate directly to the STN to produce a selective activation of STN cell bodies without exciting fibers of passage (Fig.5A). Application of glutamate (100 μm) to the STN produced an increase in the frequency of spontaneous EPSCs recorded in SNr neurons (basal, 4.8 ± 1.6 Hz; glutamate, 12.4 ± 5.7 Hz; n = 5) without affecting spontaneous EPSC amplitude (basal, 9.6 ± 1.2 pA; glutamate, 9.4 ± 1.4 pA; n = 5). In agreement with a selective activation of cell bodies, movement of the glutamate application pipette slightly out of the STN to the cerebral peduncle had no effect on spontaneous EPSC frequency (ratio of glutamate/basal, application to STN, 3.0 ± 1.3; application to cerebral peduncle, 0.92 ± 0.1; n = 3) (Fig. 5A). To test for group II mGluR-mediated inhibition of transmission at the STN–SNr synapse, we determined the effects of maximal concentrations of LY354740 on the frequency and amplitude of glutamate-evoked EPSCs. In agreement with the electrical stimulation results, we found that activation of group II mGluRs significantly reduced the frequency of glutamate-evoked EPSCs without affecting the amplitude or kinetics of the response (Fig. 5B–F).

Activation of group II mGluRs reduces the frequency of EPSCs evoked by glutamate application to the STN.A, A demonstration of the experimental paradigm used. Direct application of glutamate (100 μm; 1 ml/min; 30 sec) to the STN produces approximately a threefold increase in EPSC frequency without affecting EPSC amplitude. Moving the microapplicator to a position above the cerebral peduncle (cp) produced no change in the frequency of EPSCs, indicating that the glutamate effect is caused by selective activation of STN neurons and not by fibers of passage. B, Examples of glutamate-evoked EPSCs both before (left) and during the application of 100 nm LY354740 (right). C, Overlayedtraces of average glutamate-evoked EPSCs before and during 100 nm LY354740 application indicating no change in the amplitude or kinetics of the responses. D,Cumulative frequency plots illustrating a lack of effect of LY354740 on amplitude (left; Kolmogorov–Smirnov test;p > 0.05) and a significant increase in interevent interval (right; Kolmogorov–Smirnov test;p < 0.01), indicating that LY345740 selectively reduces the frequency of glutamate-evoked EPSCs. E,Frequency–amplitude histograms demonstrating a decrease in the frequency but no change in the mean amplitude of glutamate-evoked EPSCs. F, Mean (± SEM) data demonstrating that glutamate induces approximately a threefold increase in frequency over basal values without altering amplitude. This glutamate-evoked increase is significantly reduced by LY345740. Each verticalbar represents the mean (± SEM) of data collected from five cells (*p < 0.05).
Taken together, these data strongly support the hypothesis that activation of group II mGluRs decreases transmission at the STN–SNr synapse by a presynaptic mechanism. However, it is possible that a group II mGluR agonist could reduce evoked EPSCs by a mechanism that does not directly involve regulation of synaptic transmission, such as inducing a decrease in the excitability of the STN neurons or decreasing axonal conductance. To examine the mechanism of this presynaptic modulation further, we assessed the effects of maximal concentrations of LY354740 on the excitability of STN neurons. Whole-cell current-clamp recordings from STN neurons during application of 100 nm LY354740 indicate that activation of group II mGluRs has no effect on membrane potential (control infusion ΔVm = −0.93 ± 0.34 mV;n = 4; LY354740 infusion ΔVm = −0.99 ± 0.83 mV; n = 7) (Fig.6A,D) or input resistance (control, 671 ± 123.6m[scap]Ω; LY354740, 665 ± 129.4m[scap]Ω; n = 3) (Fig.6B,D). We also applied a series of small depolarizing current injections to obtain an approximate estimate of the action potential threshold. Application of 100 nmLY345740 did not effect the lowest potential at which action potentials were observed (control infusion, −48.3 ± 1.28 mV; LY354740, −48.7 ± 1.55 mV; n = 4) (Fig.6C,D). Therefore, these data indicate that the group II mGluR-mediated inhibition of transmission at the STN–SNr synapse cannot be explained by a decrease in the somatic excitability of the presynaptic neurons. We also recorded presynaptic fiber volleys by placing an extracellular recording electrode in the cerebral peduncle, the point of entry of STN fibers into the SNr, and electrically stimulating the STN. In the presence of blockers of fast glutamatergic (20 μm CNQX) and GABAergic (20 μm bicuculline) transmission, we recorded a robust negative inflection in the field that was sensitive to tetrodotoxin (500 nm), indicating that this is a measure of the firing of presynaptic axons (Fig.6E—G). Application of 100 nmLY354740 had no effect on the presynaptic fiber volley, indicating that activation of group II mGluRs does not alter STN axonal excitability. Taken together, these data indicate that the group II mGluR-mediated reduction in transmission at the STN–SNr synapse is caused by a modulation of the presynaptic terminal or the preterminal axon.

Activation of group II mGluRs does not effect the excitability of STN neurons. A,Representative current-clamp recording demonstrating that application of 100 nm LY345740 does not alter membrane potential.B, Overlayed traces of responses to the injection of hyperpolarizing current demonstrating that LY354740 has no effect on input resistance. C, Representativetraces of experiments in which small depolarizing current injections were used to determine the lowest potential at which an STN neuron would produce an action potential. Application of 100 nm LY354740 has no effect on this potential. D, Mean (± SEM) of data demonstrating the lack of effect of group II mGluR activation on membrane potential, input resistance, or lowest spike potential. Data are from three to seven cells per condition. E, F, Representativetraces (E) and time course (F) demonstrating that LY354740 does not alter presynaptic fiber volleys evoked by stimulation of the STN.G, Mean (± SEM) of data from four independent experiments demonstrating that activation of group II mGluRs has no effect on presynaptic fiber volleys. The fiber volley is blocked by the application of 500 nm TTX indicating that the volley is a measurement of presynaptic axonal action potential.
Activation of group II mGluRs has no effect on inhibitory synaptic transmission in the SNr
If group II mGluRs selectively regulate transmission at STN synapses without altering transmission at inhibitory synapses in the SNr, agonists of these receptors would have a net inhibitory effect on excitatory drive through this portion of the basal ganglia circuit. The immunocytochemical data presented above suggest that mGluR2/3 immunoreactivity is not present on the majority of inhibitory synapses in the SNr, suggesting that group II mGluRs are not likely to modulate IPSCs in this region. To test this hypothesis directly, we determined the effect of LY354740 on evoked IPSCs recorded in SNr projection neurons. Consistent with previous reports (Radnikow and Misgeld, 1998), stimulation of the cerebral peduncle produced a robust, bicuculline-sensitive IPSC (Fig. 7). Application of a concentration of the group II mGluR agonist LY354740 that is maximally effective in reducing EPSCs had no effect on IPSC amplitude. These results suggest that agonists of group II mGluRs will selectively inhibit excitatory transmission through the indirect pathway to the SNr without impacting direct GABA-mediated inhibition of SNr neurons.

Activation of group II mGluRs has no effect on inhibitory transmission in the SNr. A, Representativetraces of evoked IPSCs before (Pre-Drug; left) and during the application of 100 nm LY354740 (right). B, Time course of the effect of LY354740 on IPSC amplitude. C, Mean data demonstrating the lack of effect of group II mGluR activation on IPSC amplitude. Data represent the mean (± SEM) of seven separate experiments (p > 0.05).
Activation of group II mGluRs exhibits antiparkinsonian effects
The preceding data clearly demonstrate that group II mGluRs mediate a presynaptic inhibition of transmission at the STN–SNr synapse. Because overactivity at this synapse is thought to contribute to the motor dysfunction associated with PD and other hypokinetic disorders, we tested the hypothesis that activation of group II mGluRs would increase mobility in a rat model of parkinsonism using haloperidol-induced catalepsy (Ossowska et al., 1990; Schmidt et al., 1997). Two standard behavioral measures were used to assess catalepsy in rats treated with the dopamine receptor antagonist haloperidol. First, the front paws of control and experimental rats were placed on a horizontal bar (4.5 cm high), and the latency to remove a paw from the bar was measured. Second, rats were placed on a vertical grid, and the latency to remove a paw from the grid was measured (Kronthaler and Schmidt, 1996). Consistent with previous reports (Ossowska et al., 1990; Schmidt et al., 1997), haloperidol (1.5 mg/kg) induced a robust catalepsy that could be observed as an increase in latency with both behavioral measures (Fig. 8). Interestingly, haloperidol-induced catalepsy was reversed in a dose-dependent manner by intraperitoneal injection of the group II mGluR agonist LY354740. Injection of LY354740 alone had no significant effect on these behavioral measures.

Activation of group II mGluRs reverses catalepsy in an animal model of Parkinson's disease. The degree of haloperidol-induced catalepsy was measured as either latency to the first paw movement when the animal was placed on a vertical grid (A) or latency to remove a paw from a bar (B). Haloperidol (1.5 mg/kg, i.p.) induces a pronounced catalepsy that was reversed in a dose-dependent manner by LY354740 (3–30 mg/kg, i.p.) (*p < 0.05). LY354740 alone had no effect on either measure of catalepsy. Data shown represent the mean (± SEM) of data collected from eight animals.
DISCUSSION
We have found that group II mGluRs are presynaptically localized on STN terminals in BG output nuclei where they reduce transmission at STN–SNr synapses. Furthermore, a selective agonist of group II mGluRs has behavioral effects in rats that are consistent with an antiparkinsonian action. These data suggest that activation of group II mGluRs restores the normal function of BG circuits by acting at a point downstream of the striatum where dopamine receptor blockade occurs.
The finding that LY354740 alone had no effect on measures of catalepsy is interesting because of previous studies demonstrating that nonselective mGluR agonists can induce catalepsy (Kronthaler and Schmidt, 1996). Because LY354740 is highly selective for group II mGluRs, it is likely that this mGluR-induced catalepsy is caused by activation of another mGluR subtype. Consistent with this, LY354740 produces no effect on motor activity when administered alone (Helton et al., 1998) but reduces haloperidol-induced muscle rigidity (Konieczny et al., 1998). Furthermore, agonists of group I mGluRs have physiological and behavioral effects that suggest that agonists of these receptors are likely to have catalepsy-inducing effects (Sacaan et al., 1991; Kaatz and Albin, 1995).
Other potential sites of action of group II mGluR agonists
Taken together with previous studies revealing a critical role of the STN in parkinsonian states (Guridi and Obeso, 1997; Wichmann and DeLong, 1998), the results of the present anatomical and physiological studies suggest that the behavioral effects of LY354740 are at least partially attributable to an mGluR2/3-mediated reduction in glutamate release from STN terminals. However, it is possible that actions of group II mGluR at other sites could also contribute to this effect. Although the distribution of group II mGluRs in other basal ganglia structures is somewhat limited, previous studies reveal that these receptors are present in the striatum (Testa et al., 1998) where they are involved in regulating transmission at corticostriatal synapses (Lovinger and McCool, 1995; Pisani et al., 1997). If group II mGluRs are preferentially involved in inhibiting synaptic excitation of striatal projection neurons that give rise to the indirect pathway, this could contribute to the overall behavioral effects of group II mGluR agonists. Also, it is possible that group II mGluRs present in motor regions outside of the basal ganglia, such as the cortex (Neki et al., 1996) and thalamus (Ohishi et al., 1993), could contribute to the motor effects of group II mGluR agonists.
It is interesting to note that, in addition to projecting to basal ganglia output nuclei, STN neurons also project to the dopaminergic neurons of the SNc (Kita and Kitai, 1987; Iribe et al., 1999). Furthermore, glutamate has been implicated as an excitotoxic agent in PD (Albin and Greenamyre, 1992; Rodriguez et al., 1998), suggesting that increased excitatory drive to the SNc may contribute to the progressive loss of SNc dopaminergic neurons in PD. On the basis of this, if group II mGluRs are also involved in inhibiting transmission at STN synapses in the SNc, it is possible that agonists of these receptors could reduce the component of SNc neuronal death that is mediated by STN-induced excitotoxicity. Interestingly, previous immunocytochemical studies reveal that mGluR2/3 immunoreactivity is present in the SNc (Testa et al., 1998). Furthermore, physiological studies reveal that agonists of group II mGluRs inhibit evoked EPSPs in this region (Wigmore and Lacey, 1998). Although the source of the excitatory afferents regulated by group II mGluRs in the SNc was not defined, it is possible that these EPSCs are mediated in part by activity at STN terminals. These data raise the exciting possibility that group II mGluR agonists have the potential not only for reducing the symptoms of established PD but also for slowing the progression of PD. Future studies will be needed to define clearly the role of increased STN activity in contributing to progression of the disorder and to define rigorously the mGluR subtypes involved in regulating transmission at STN–SNc synapses.
Summary
The data presented suggest that group II mGluRs are presynaptically localized on STN terminals in the SNr and that activation of these receptors selectively reduces transmission at excitatory STN synapses in this region. Taken together with the behavioral data presented, these studies raise the exciting possibility that agonists of group II mGluRs may provide a novel, nonsurgical approach to the treatment of PD that bypasses the problems inherent with dopamine-replacement therapy. Furthermore, because group II mGluR agonists act downstream from nigrostriatal dopaminergic neurons, these compounds could be useful for the treatment of drug-induced parkinsonism in patients treated with haloperidol and other dopamine receptor antagonists that are used as antipsychotic agents. Finally, it is important to note that pallidotomy and inactivation of the STN are being explored as having therapeutic potential in other movement disorders, including dystonia and tardive dyskinesias (Vitek et al., 1998), and that increased activity in the STN is implicated in some forms of epilepsy (Deransart et al., 1996, 1998, 1999; Vercueil et al., 1998). Thus, it is conceivable that inhibition of excitatory transmission at the STN–SNr synapse with group II mGluR agonists could have broader therapeutic potential than that of l-DOPA and other drugs used for dopamine replacement in PD patients.
Footnotes
This work was supported by grants from the National Institutes of Health National Institute of Neurological Disorders and Stroke, the United States Army Medical Research and Material Command, and the National Parkinson Foundation. We would like to thank Dr. Dieter Jaeger (Emory University) for helpful advice on basal ganglia physiology, Drs. Steve Holtzman (Emory University) and Werner Schmidt (University of Tuebingen) for helpful advice on haloperidol-induced catalepsy, and Stephanie Carter and Sean Stoy for valuable technical assistance. Also, we thank Drs. Darryle Schoepp and James Monn (Eli Lilly) for supplying LY354740 and for helpful conversations regarding the use of this drug.
S.R.B. and M.J.M. contributed equally to this work.
Correspondence should be addressed to Dr. P. J. Conn, Department of Pharmacology, Rollins Research Building, Atlanta, GA 30322. E-mail:pconn{at}emory.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}