

Abstract
Spectrins are plasma membrane-associated cytoskeletal proteins implicated in several aspects of synaptic development and function, including presynaptic vesicle tethering and postsynaptic receptor aggregation. To test these hypotheses, we characterizedDrosophila mutants lacking either α- or β-spectrin. The Drosophila genome contains only one α-spectrin and one conventional β-spectringene, making it an ideal system to genetically manipulate spectrin levels and examine the resulting synaptic alterations. Both spectrin proteins are strongly expressed in theDrosophila neuromusculature and highly enriched at the glutamatergic neuromuscular junction. Protein null α- and β-spectrin mutants are embryonic lethal and display severely disrupted neurotransmission without altered morphological synaptogenesis. Contrary to current models, the absence of spectrins does not alter postsynaptic glutamate receptor field function or the ultrastructural localization of presynaptic vesicles. However, the subcellular localization of numerous synaptic proteins is disrupted, suggesting that the defects in presynaptic neurotransmitter release may be attributable to inappropriate assembly, transport, or localization of proteins required for synaptic function.
- spectrin
- Drosophila
- synapse
- neuromuscular junction
- synaptogenesis
- cysteine string protein
- Discs large
- PSD-95
- synaptotagmin
- synapsin
- syntaxin
- glutamate receptor
Spectrin was originally discovered in erythrocytes, in which α- and β-spectrin heterotetramers form part of a submembrane meshwork critical for membrane structural integrity (Bennett, 1990; Bennett and Gilligan, 1993). Closely related spectrin isoforms are found in most other eukaryotic cell types, in which they preferentially associate with plasma membranes at sites of cell–cell contact (Bennett and Gilligan, 1993). Spectrin (also known as “fodrin”) is particularly abundant in mammalian brain, in which it comprises 2–3% of total protein (Davis and Bennett, 1983; Bennett and Gilligan, 1993). However, almost nothing is known about the function of spectrin in neurons.
In neurons, spectrin is often preferentially localized to both central and peripheral synapses (Bloch and Morrow, 1989; Daniels, 1990; Masliah et al., 1991; Bewick et al., 1992, 1996; Goodman et al., 1995; Kordeli, 2000), suggesting a role for spectrin at the synaptic membrane. Calmodulin, sodium channels, munc-13, and synapsin I, which all play important synaptic roles, have all been shown to bind to spectrins (Srinivasan et al., 1988; Steiner et al., 1989; Dubreuil et al., 1991;Sikorski and Goodman, 1991; Iga et al., 1997; Sakaguchi et al., 1998;Wood and Slater, 1998). However, the specific role that spectrins might play at synapses is unknown. Proposed roles for synaptic spectrins include the following: (1) the capture and subsequent tethering (via synapsin I) of synaptic vesicles near the active zone (Landis, 1988;Goodman et al., 1995; Sikorski et al., 2000); (2) the initiation of SNARE vesicle fusion by “dimpling” the cell membrane (Goodman, 1999); and (3) the anchoring of glutamate and/or acetylcholine receptors within the postsynaptic density (Bloch and Morrow, 1989; Daniels, 1990; Bloch et al., 1997; Wechsler and Teichberg, 1998; Hirai and Matsuda, 1999). A recent study using cultured hippocampal cells showed that presynaptic injection of antibodies against the synapsin-binding region of β-spectrin completely blocked synaptic transmission (Sikorski et al., 2000), arguing that presynaptic synapsin–spectrin interactions are essential for synaptic function. No other functional studies of synaptic spectrin have been done.
Drosophila is an attractive system in which to test whether spectrins are required for synaptic development and function.Drosophila contain only three members of the highly conserved spectrin family, each encoded by a single gene (Adams et al., 2000), and previously identified null mutants exist for each spectrin subunit (Dubreuil et al., 1998, 2000; Thomas et al., 1998). Here we show that, in Drosophila protein null mutants lacking α- or β-spectrin, neuromuscular junction (NMJ) morphology is normal, but neurotransmission is severely disrupted. The protein null mutants show a reduction in spontaneous synaptic event frequency with no changes in response to pressure-ejected glutamate or in spontaneous synaptic event amplitude, demonstrating that the neurotransmission defect is presynaptic. Ultrastructural analysis reveals no change in presynaptic vesicle distribution, but immunocytochemistry shows that many classes of synaptic proteins are dramatically mislocalized or absent in both α- and β-spectrin mutants. We propose that spectrin in neuronal synapses is required for capture and tethering of membrane-associated proteins required for presynaptic neurotransmitter release.
MATERIALS AND METHODS
Fly stocks. Molecularly characterized protein null mutants for α- and β-spectrin were used in this study (Lee et al., 1993; Dubreuil et al., 2000). α-specrg41 and β-specem6 do not produce detectable protein, as shown by immunoblots (Lee et al., 1993; Dubreuil et al., 2000). β-specem6produces a truncated protein product according to immunoblots (Dubreuil et al., 2000), but β-spectrin protein is immunohistochemically undetectable in situ (D. E. Featherstone and K. Brodie, unpublished data), presumably because the truncated protein is rapidly degraded and/or fails to localize. α-Spectrin null mutantl(3)drerg41 stocks (Lee et al., 1993) were maintained as heterozygotes using a third chromosome balancer [yw67c23; ru l(3)drerg41st e/In(3LR)TM3, y+Sb Ser]. Homozygousl(3)drerg41 mutants are rescued to adulthood by transgenic expression of an α-spectrinminigene under a ubiquitin promoter (Lee et al., 1993). β-Spectrin null mutants (β-specem21 and β-specem6) stocks (Dubreuil et al., 2000) were maintained as heterozygotes using anFM7[Kruppel-GFP] balancer chromosome (Casso et al., 1999). As with α-spectrin mutants, β-spectrinmutants are rescued to adulthood by transgenic expression of β-spectrin (Dubreuil et al., 2000). Oregon-R (OR) was used for wild-type (WT) controls.
Embryo preparation and dissection. Homozygous mutant embryos were selected from siblings based on the absence of balancer chromosome markers (green fluorescent protein for β-spectrinmutants, yellow+ for α-spectrin mutants). Homozygous yellow mutants do not show any significant difference in excitatory junctional current (EJC) amplitude compared with Oregon R (y/y, 1548 ± 280 pA; OR, 1476 ± 117 pA; n = 5–13; p = 0.78). For electrophysiology and embryonic immunohistochemistry, morphologically and temporally staged [22–24 hr after egg laying (AEL) at 25°C] embryos were dechorionated with bleach and devitellinated manually. For dissection, embryos were glued (Histoacryl Blue; B. Braun Biotech International GmbH, Melsungen, Germany) to Sylgard (Dow Corning, Midland, MI)-coated coverslips under saline containing (in mm): 135 NaCl, 5 KCl, 4 MgCl2, 1.8 CaCl2, 72 sucrose, and 5 N-Tris[hydroxy-methyl]methyl-2-aminoethane sulfonic acid (TES), pH 7.2. A slit was made manually along the dorsal midline using a glass capillary pulled to a sharp point, and the body walls were glued flat to the coverslip. If electrophysiology was to be performed on the dissected embryos, the exposed muscle sheath was enzymatically removed after dissection using 1–2 min exposure to 1 mg/ml collagenase (type IV; Sigma, St. Louis, MO).
Immunohistochemistry. Dissected embryos or wandering third-instar larvae were fixed in 4% paraformaldehyde for 30–45 min and processed according to standard techniques (White, 1998; Beumer et al., 1999; Featherstone et al., 2000). Mouse monoclonalDrosophila α-spectrin antibody (3A9) (Dubreuil et al., 1997) and rabbit polyclonal Drosophila β-spectrin antibody (Byers et al., 1989) were used at 1:100. These α- and β-spectrin antibodies show no detectable staining in α- or β-spectrin null mutants and/or on immunoblots, confirming antibody specificity. Mouse monoclonal synapsin antibody (Klagges et al., 1996) was used at 1:100. Mouse monoclonal cysteine string protein (CSP) antibody (Zinsmaier et al., 1994) was used at 1:200. Rabbit polyclonal synaptotagmin antibody (Littleton et al., 1993) was used at 1:500. Mouse monoclonal syntaxin 1A (Schulze et al., 1995) was used at 1:500. Rabbit polyclonal Discs large (DLG) (Lahey et al., 1994) was used at 1:1000. Immunoreactivity for all of these antibodies is abolished in the appropriate null mutants, confirming antibody specificity. Fluorescein isothiocyanate and tetramethylrhodamine-conjugated secondary antibodies (goat anti-mouse and goat anti-rabbit; Molecular Probes, Eugene, OR) were used at 1:400. FITC-conjugated anti-HRP (Molecular Probes) was used at 1:100. Images were obtained on a Zeiss (Oberkochen, Germany) LSM510 laser-scanning confocal microscope.
Synaptic/nonsynaptic immunoreactivity ratios. Pixel intensity (0–255) for boutons and nearby extrasynaptic regions (muscle for DLG and nerve for all other proteins) was measured in Zeiss Image Browser software using raw (completely unaltered) confocal fluorescent images. Average background fluorescence intensity (dark areas beyond–in between muscles) was subtracted from these values. To derive the “synaptic/nonsynaptic immunoreactivity ratio,” the background-corrected synaptic fluorescence intensity was divided by the background-corrected nonsynaptic fluorescence intensity. Thus, the ratio was calculated as follows: r = (S− B)/(N − B), wherer is the synaptic/nonsynaptic immunoreactivity ratio,S is the fluorescence intensity in synaptic boutons,B is the background fluorescence intensity measured from dark nontissue parts of the image, and N is the fluorescence intensity in nonsynaptic tissues (nerve for CSP, synapsin, syntaxin, and synaptotagmin; muscle for DLG). This raw ratio represents a measure of both protein localization and antibody quality because poor antibodies might be expected to lower the ratio because of high nonspecific immunoreactivity (high background) and/or reduced specific immunoreactivity. Antibody quality effects can be eliminated by normalizing the raw ratios to wild type. Normalized ratios (see Fig. 7) were calculated by dividing the ratios for wild type and each mutant by these numbers (normalized r =R/RWT).
Morphology. Quantification of NMJ area was performed using the public domain Java-based image processing and analysis program Image/J . Confocal images (see Fig.2A) of wild-type and mutant DrosophilaNMJs, visualized by fluorescently conjugated anti-HRP (which stains all nerve membranes), were manually outlined using Image/J. Once given, pixel dimensions (recorded automatically by the Zeiss confocal software), Image calculated the area of the outlined region (NMJ area). For bouton counting, synaptic varicosities (swellings) were also visualized with fluorescently conjugated anti-HRP (1:100; Molecular Probes).
Electron microscopy. Genotyped embryos were prepared for transmission electron microscopy (TEM) using standard techniques (Prokop et al., 1996; Fergestad et al., 1999). Briefly, mature embryos (22–24 hr after egg laying; AEL at 25°C) were manually dechorionated and injected with fixative (5% glutaraldehyde in 0.05m phosphate buffer). The preparation was then transferred to 2.5% glutaraldehyde in 0.05 mphosphate buffer for 30–60 min. Specimens were washed in buffer, transferred to 1% osmium tetroxide in dH2O for 3 hr, washed again in dH2O, and stained en bloc in 2% aqueous uranyl acetate for 30 min. Embryos were dehydrated in an ethanol series, passed through propylene oxide, and transferred to araldite. Ribbons of thin (∼55 nm) sections were obtained and examined on a Hitachi (Tokyo, Japan) H-7100 TEM. Active zones that were identified in at least two consecutive sections were imaged and analyzed using NIH Image. Vesicles were considered to be “clustered” if they were within 235 nm of the active zone T-bar (Fergestad et al., 1999) and docked if within one-half vesicle diameter of the presynaptic membrane (thus allowing for vesicles that may be in contact with the membrane but were not perfectly bisected in the cross-section).
Electrophysiology. Electrophysiology and data analysis were performed as described previously (Featherstone et al., 2000). Briefly, whole-cell patch-clamp recordings from embryonic muscle 6 were obtained in an extracellular solution containing (in mm): 135 NaCl, 5 KCl, 4 MgCl2, 1.8 CaCl2, 72 sucrose, and 5 TES, pH 7.2. For miniature EJC (mEJC) recordings, calcium was replaced with 5 μm tetrodotoxin (TTX). The patch pipette solution contained (in mm): 120 KCl, 20 KOH, 4 MgCl2, 0.25 CaCl2, 5 EGTA, 4Na2ATP, 36 sucrose, and 5 TES. For EJC measurements, the segmental nerve was stimulated by delivering 5–10 V, 0.1 msec pulses via a glass suction pipette. To assay the glutamate receptor field, 1 mm glutamate was pressure ejected (100 msec pulse) from a small-tipped (∼5 μm opening) pipette directly onto the NMJ. Data were analyzed using Clampfit 8 or 9α (Axon Instruments, Foster City, CA) and/or Minianalysis 4 (Synaptosoft Inc., Decatur, GA).
Statistics. All data are presented as mean ± SEM. Eachn represents a different embryo of the stated genotype. Statistics from spontaneous EJCs (sEJCs)and mEJCs are derived from at least 5 min of continuous recording (often 10–20 min in the case of the low-frequency mEJCs). In all figures, statistical significance (compared with wild-type controls) is indicated as *p< 0.05, **p < 0.01, and ***p < 0.001, . Unless otherwise stated, statistical significance was determined using Student's t test. Because spontaneous synaptic event amplitude distributions are skewed rather than Gaussian, we compared these distributions statistically using the Kolmogorov–Smirnov test and do not report variance.
RESULTS
Drosophila spectrins and spectrin mutants
A search of the sequenced Drosophila genome reveals only three members of the highly conserved spectrin family, each encoded by a single gene (Adams et al., 2000; Pinder and Baines, 2000).Drosophila α-spectrin (GenBank accession numberA33733) is 64% identical at the amino acid level to human brain α-spectrin/fodrin (GenBank accession number A35715).Drosophila β-spectrin (GenBank accession numberA46147) is 56% identical to human β-spectrin(GenBank accession number NP003119). DrosophilaβH-spectrin/karst (GenBank accession number CAA37939) is the most divergent, with 31% amino acid identity to the human ortholog β V-spectrin (GenBank accession number AAF65317). Drosophila protein null mutants for α- and β-spectrin are embryonic–early larval lethal, with defects in the structure and function of epithelial cells (Lee et al., 1993, 1997; Dubreuil and Grushko, 1998; Dubreuil et al., 2000). In contrast, null mutants for βH-spectrin are semiviable, with mild defects including rough eyes, disrupted epithelial morphogenesis, tracheal defects, and misshapen wings (Thomas et al., 1998). These results suggest that α- and/or β-spectrin could play vital roles in synaptogenesis and synaptic function, whereas βH-spectrin is unessential. Therefore, we focused our efforts on characterizing the role of α- and β-spectrin subunits in synaptic development and function.
For this study, we used previously identified protein null mutants for α-spectrin (Lee et al., 1993) and β-spectrin(Dubreuil et al., 2000). Homozygous α-spectrin null mutants fail to hatch (∼50%) or die as early first-instar larvae (∼50%). Homozygous β-spectrin protein null mutants fail to hatch (∼90%), and the rest (∼10%) die as early first-instar larvae. Both classes of mutants are lethargic and display limited movement, consistent with a neurophysiological or muscular defect. We chose to study these mutants at the embryonic NMJ for several reasons. First, this synapse is accessible in vivo to a variety of cell biological techniques, including patch-clamp electrophysiology, immunohistochemistry, and electron microscopy. Second, the development, morphology, and function of the Drosophila NMJ is well described and relatively invariant from animal to animal. Like many synapses in the mammalian CNS, the Drosophila NMJ is glutamatergic. These features make the NMJ an excellent place to detect and quantify any changes resulting from spectrin disruption.
Spectrins are present at theDrosophila NMJ
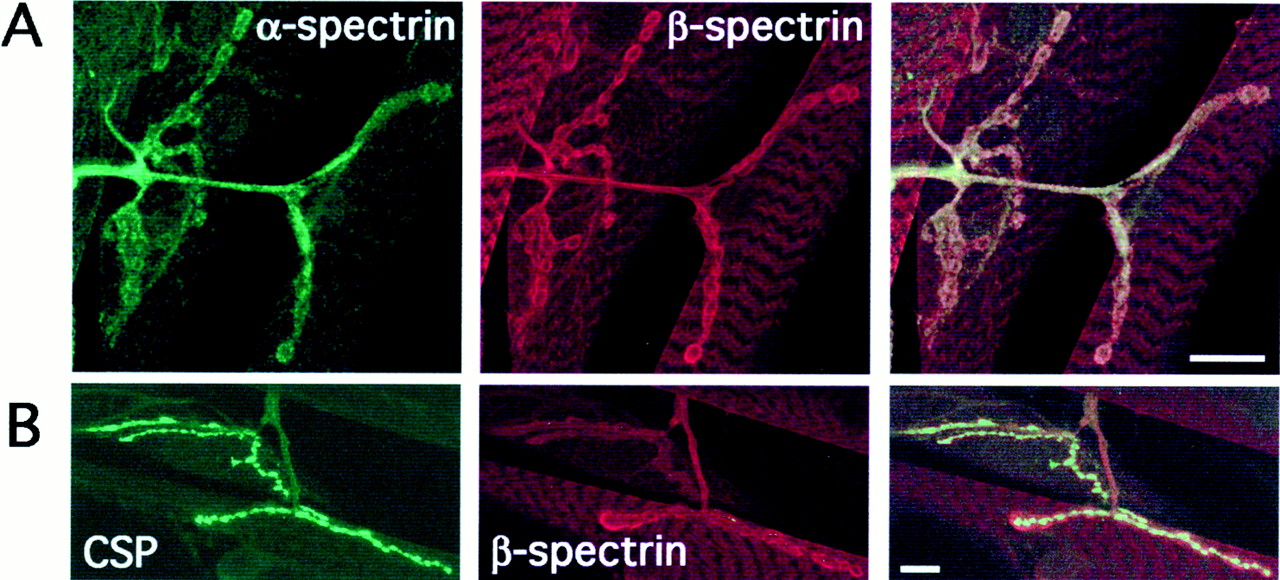
Using antibodies specific for Drosophila spectrins (Byers et al., 1989; Dubreuil et al., 1997), we examined the neuromuscular localization of both α- and β-spectrin (Fig.1). As shown in Figure 1, both α-spectrin (Fig. 1A, green) and β-spectrin (Fig. 1A, red) are found in presynaptic axons proximal to the NMJ and in the periphery of presynaptic boutons. Although α-spectrin staining is typically weaker, most of the α- and β-spectrin staining in the NMJ appears colocalized (Fig. 1A, right panel, α- and β-spectrin overlapping expression appears yellow). α- and β-Spectrin immunoreactivity is also strong throughout muscle (Fig. 1A). We independently confirmed the specificity of both α- and β-spectrin antibodies in null mutant backgrounds (see Materials and Methods).

α- and β-Spectrin immunoreactivity in the neuromusculature of Drosophila third-instar larvae.A, Confocal fluorescence images of NMJs stained simultaneously with antibodies raised against α-spectrin and β-spectrin. α-Spectrin immunoreactivity is shown ingreen (left), β-spectrin immunoreactivity is shown in red, (middle), and overlapping α/β-spectrin staining appears yellow (merged image, right). Scale bar, 10 μm. B, Confocal fluorescence images of NMJs stained simultaneously with antibodies raised against CSP and β-spectrin. CSP immunoreactivity is shown in green(left), and β-spectrin immunoreactivity is shown inred (middle); merged image is on theright. Scale bar, 10 μm.
In Figure 1B, we show double-labeling with antibodies against β-spectrin and the presynaptic protein CSP. Much of the β-spectrin and CSP staining is not colocalized, suggesting that the majority of spectrin protein is associated with the periphery of the presynaptic membrane and/or dense membrane foldings of the postsynaptic subsynaptic reticulum. We conclude from this immunohistochemistry that both α- and β-spectrin are present at the wild-typeDrosophila NMJ, in both presynaptic and postsynaptic cells. In subsequent experiments, we used the NMJ as a model synapse to examine the role of spectrins in synaptogenesis and synaptic function.
α- and β-Spectrin null mutants have morphologically normal neuromuscular junctions
We examined gross morphology in protein null mutants of both α- and β-spectrin (Lee et al., 1993; Dubreuil et al., 2000). Light microscope (400×) examination of several dozen acutely dissected mutant embryos reveals that both α- and β-spectrinmutants have normally formed neuromusculature, epidermis, and epidermal specializations (e.g., denticles and mouth parts). The only visible difference is that α-spectrin mutants have slightly thinner muscles, and unhatched (but living) α-spectrinembryos often have uninflated trachea at normal hatch time (22–24 hr AEL). The gut phenotype of these mutants has been described previously (Lee et al., 1993; Dubreuil et al., 1998, 2000).
To examine NMJ anatomy, we visualized embryonic body wall neuroanatomy with fluorescently labeled anti-HRP, which recognizes neural membranes (Fig. 2). We saw no qualitative differences in sites of muscle innervation or presynaptic branching pattern. In Figure 2A, we show confocal fluorescent images of wild-type and mutant embryonic NMJs visualized with fluorescently conjugated anti-HRP. In each panel, four individual NMJs are shown. On the left is the linear NMJ lying between ventral longitudinal muscles 6 and 7, and on theright are the more lateral NMJs on muscles 13 and 12. Quantification of morphology at the muscle 6/7 NMJ showed that there was no significant difference in the number of synaptic boutons (Fig.2B) (WT, 9.9 ± 0.6 boutons; αrg41, 8.4 ± 0.6 boutons; βem21, 9.6 ± 0.8 boutons; βem6, 8.6 ± 0.9 boutons;n = 6–10). Because embryonic boutons are often indistinct (Fig. 2A) and therefore difficult to count, we also quantified NMJ size by measuring muscle 6/7 NMJ area (see Materials and Methods). We detected no significant difference in NMJ area between wild-type and α- or β-spectrin mutants (Fig.2C) (WT, 47.0 ± 1.9 μm2; αrg41, 44.8 ± 4.9 μm2; βem21, 48.1 ± 2.6 μm2; βem6, 49.1 ± 1.9 μm2; n = 6–10). We conclude from this quantification, as well as qualitative observation of several dozen embryos, that NMJ morphology is not detectably altered in either α- or β-spectrin mutants.

Morphology of embryonic NMJs in spectrin mutants is normal. A, Confocal fluorescence images of NMJs on ventral longitudinal muscles 6/7 and 12 and 13 in a single ventral hemisegment of wild-type and spectrin mutant embryos. NMJ morphology was visualized by staining with fluorescently conjugated anti-HRP antibodies. Scale bar, 10 μm. B, Quantification of synaptic bouton number at the muscle 6/7 NMJ shows no significant difference between the genotypes. C, Quantification of muscle 6/7 NMJ area shows no significant difference between the genotypes.
Spectrin mutants are defective in neurotransmitter release
Because spectrins are present at the NMJ and the morphology of spectrin mutant NMJs was normal, we were able to test whether synaptic function was disrupted. To record NMJ function, we voltage clamped (−60 mV) muscle 6 using standard patch-clamp techniques. To evoke synaptic activity, we stimulated (0.5 msec, 5–15 V) the presynaptic segmental nerve using a suction electrode. As shown in Figure 3A, evoked EJCs in both α- and β-spectrin mutants are reduced to approximately one-quarter normal amplitude (WT, 1476 ± 117 pA; αrg41, 473 ± 92 pA; βem6, 453 ± 57 pA; βem21, 334 ± 55 pA;n = 9–13; p < 0.001 vs WT for each allele, using Student's t test). α- and β-Spectrin mutant EJC amplitudes are statistically indistinguishable from each other (Fig. 3A). These results demonstrate that, despite normal morphology, spectrin mutants have severely reduced synaptic transmission.

Patch-clamp electrophysiology from voltage-clamped (−60 mV) muscle demonstrates that spectrin mutant NMJs have severely reduced neurotransmitter release, with no functional alteration of postsynaptic receptor fields. A, EJC amplitude (evoked by nerve stimulation) is significantly reduced in α- and β-spectrin mutants. Representative EJCs are shown on the right.B, Currents triggered by pressure ejection of 1 mm glutamate (100 msec pulse) onto the postsynaptic membrane demonstrate that the spectrin mutant glutamate receptor field function is not significantly different from wild type. Representative glutamate-gated currents are shown on the right.C, Stimulation of the NMJ at increasing frequencies reveals no significant difference in decrement of EJC amplitude between mutants and control.
To determine whether the transmission defect in the spectrin mutants was presynaptic or postsynaptic, glutamate (1 mm) was pressure ejected (5–10 μm tip pipette, 100 msec pulse) directly onto the NMJ of voltage-clamped (−60 mV) postsynaptic muscle 6 (Featherstone et al., 2000). If the defect in synaptic transmission is attributable to an alteration in postsynaptic glutamate receptor function, the resulting glutamate-gated currents should be reduced in spectrin mutants. As shown in Figure 3B, neither α- nor β-spectrin mutants showed any detectable alteration in glutamate response (WT, 1805 ± 248 pA; αrg41, 1763 ± 227 pA; βem6, 1755 ± 178 pA; βem21, 1630 ± 105 pA;n = 7–11). Because the receptor field is functionally normal in spectrin mutants yet transmission is greatly reduced, the striking transmission defect shown in Figure 3A must be presynaptic.
Presynaptic defects can be a result of faulty neurotransmitter release (synaptic vesicle filling and fusion), vesicle recycling defects, or both. Defective synaptic vesicle cycling can be revealed when the nerve is stimulated at high frequencies (Fergestad et al., 1999; Kuromi and Kidokoro, 2000). Under conditions of high demand, neurotransmitter release is reduced because of a reduction in the available pool of neurotransmitter-filled vesicles (Kuromi and Kidokoro, 2000). In α- and β-spectrin mutants, the reduction in synaptic transmission during high-frequency stimulation is slightly, but not significantly, impaired (Fig. 3C) (normalized amplitude at 20 Hz: WT, 0.59 ± 0.06; αrg41, 0.62 ± 0.03; βem21, 0.39 ± 0.12; βem6, 0.51 ± 0.07;n = 4–7). These results suggest that short-term vesicle cycling in the mutants is sufficient to maintain the reduced rate of exocytosis shown in Figure 3A. Because we did not assay endocytosis in the mutants directly (e.g., with FM1–43), we cannot completely rule out defects in endocytosis. However, because of the relatively small (and statistically insignificant) alteration in mutant responses to high-frequency stimulation, we conclude that the functional defect is primarily in exocytosis rather than endocytosis or vesicle cycling.
Together, the data in Figure 3 suggest that the synaptic transmission defect in α- and β-spectrin mutants is attributable to specific disruption in neurotransmitter release, with no functional alteration in the postsynaptic receptors. We confirmed these conclusions using analysis of spontaneous synaptic currents (Fig.4). A reduction in the probability of presynaptic vesicle fusion is revealed by less frequent spontaneous synaptic events, whereas an alteration in receptor localization, receptor number, or receptor biophysics causes changes in the amplitude of spontaneous synaptic events. Figure 4 shows analysis of sEJCs, which are recorded in the presence of calcium (Fig.4A,B, left column), and mEJCs, which are recorded in the absence of extracellular calcium and the presence of TTX (Fig. 4A,B,right column). The frequency of both types of event are lowered in the spectrin mutants (Fig. 4A), suggesting that α- and β-spectrin mutants share a calcium-independent deficit in synaptic vesicle fusion (1.8 mm Ca2+: WT, 11.69 ± 1.59 Hz; αrg41, 6.83 ± 0.74 Hz; βem21, 1.69 ± 0.41 Hz; βem6, 3.52 ± 1.51 Hz;n = 6–11; TTX plus 0 mmCa2+: WT, 0.13 ± 0.03 Hz; αrg41, 0.08 ± 0.02 Hz; βem21, 0.05 ± 0.01 Hz; βem6, 0.07 ± 0.02 Hz;n = 5–14). In contrast, the amplitude of mEJCs is not significantly altered in spectrin mutants compared with wild-type controls (Fig. 4B). These data, like those in Figure3B, suggest that the postsynaptic receptor field is functionally unchanged (mean mEJC amplitudes: WT, 157.9 pA; αrg41, 143.7 pA; βem21, 142.6 pA; βem6, 140.3 pA; n = 4–14 embryos, thousands of events; p > 0.05 by Kolmogorov–Smirnov test). We are unable to confirm this finding qualitatively using antibodies raised against Drosophilaglutamate receptors. Despite success in larvae, we have been unable to visualize embryonic receptors either in vivo or on immunoblot, possibly because of the small number of embryonic receptors (100–200 receptors per NMJ vs tens of thousands of receptors per NMJ in larvae). However, electrophysiology is arguably the most sensitive (able to detect a single functional receptor) and most quantitative method of determining receptor field integrity.

Analysis of spontaneous synaptic events demonstrates that spectrin null mutants have decreased synaptic vesicle fusion rates but no functional alteration of the receptor fields. Currents were recorded in both normal (1.8 mm) calcium saline and saline containing 0 mm calcium plus 5 μm TTX (to block endogenous nerve activity).A, Frequency of spontaneous synaptic currents in voltage-clamped (−60 mV) muscle in both α-spectrin and β-spectrin mutants is reduced in both high- and low-calcium conditions, suggesting disruption of presynaptic vesicle fusion. B, Amplitude histograms (composed of data from multiple recordings) reveal no significant difference (Kolmogorov–Smirnov test) in spectrin mutant event amplitudes in either normal (1.8 mm) calcium or the absence of endogenous activity (0 calcium plus TTX), suggesting that the spectrin mutants have no functional alteration in the postsynaptic glutamate receptor field.
Together, the electrophysiological results show that α- and β-spectrin mutants have severely impaired synaptic transmission and that this impairment is attributable specifically to disruption of neurotransmitter release, without any functional alteration in the postsynaptic receptor field.
Ultrastructure of spectrin mutants is normal
Both α- and β-spectrin mutants have normal NMJ morphology but reduced neurotransmitter release, supporting the idea that spectrins may cluster synaptic vesicles at the active zone. This hypothesis, called “casting the line,” suggests that one end of spectrin is anchored to active zones, whereas the other end captures vesicles via an interaction with synapsin (Landis, 1988; Goodman et al., 1995; Sikorski et al., 2000). Mislocalization and/or absence of synaptic vesicles at the active zone could explain the spectrin mutant electrophysiological phenotype we show in Figures 3 and 4. We tested whether synaptic vesicle clustering is disrupted in spectrin mutants by examining NMJs using electron microscopy (Fig.5). In α- and β-spectrinmutants, presynaptic and postsynaptic membranes are normally structured and spaced, internal organelles appear normal, and the distribution of embryonic T-bars and electron-dense areas associated with active zones are indistinguishable from wild type (Fig.5A,B). Thus, spectrins play no detectable role in the maintenance of gross synaptic morphology.

Ultrastructural analysis of embryonic NMJs shows morphologically normal boutons in spectrin mutants, with no alterations in the distribution of active zones or synaptic vesicles.A, TEM cross-sections through embryonic NMJ boutons showing presynaptic active zones with electron-dense T-bars (surrounded by clustered vesicles) in opposition to a postsynaptic density. Active zones are indicated with arrowheads. Scale bar, 250 nm.B, High-magnification images of active zones from each genotype, showing individual T-bars and clustered vesicles.Arrowheads indicate T-bars. C, Quantification of numbers of docked vesicles (within one-half vesicle diameter of the presynaptic membrane), numbers of clustered vesicles (within 235 nm of T-bar), and vesicle density throughout bouton cross-section.
The location of active zones and synaptic vesicles are readily visible, allowing us to determine whether vesicle clustering is altered in either α- or β-spectrin mutants. We quantified the number and distribution of synaptic vesicles around each active zone, and these results are graphed in Figure 5C. Spectrin mutants show no consistent alteration in the number of docked [within one-half vesicle diameter of the presynaptic membrane (Broadie et al., 1995)] or clustered [within 235 nm of T-bar (Fergestad et al., 1999)] vesicles (docked: WT, 1.74 ± 0.16; αrg41, 1.58 ± 0.17; βem21, 1.79 ± 0.22; βem6, 1.42 ± 0.76;n = 19–27; clustered: WT, 21.3 ± 1.1; αrg41, 22.1 ± 1.31; βem21, 19.7 ± 1.23; βem6, 17.95 ± 0.73;n = 19–27). Similarly, when synaptic vesicle density throughout the entire bouton cross-section is quantified, both α- and β-spectrin null mutants are comparable with wild type (vesicle density: WT, 74.4 ± 6.9; αrg41, 69.4 ± 8.3; βem21, 96.6 ± 22.9; βem6, 49.7 ± 9.1;n = 19–27). β-spectrinem6 shows a slight (but statistically significant) reduction in clustered vesicles and vesicle density, but this change is unlikely to explain the synaptic transmission defect for two reasons: (1) the change is too small to explain the severe decrease in vesicle release, and (2) the change is not shared by either α-spectrinrg41 or β-spectrinem21, which otherwise have identical phenotypes. We conclude that spectrins do not play a substantial role in synaptic vesicle tethering at active zones.
Synaptic protein localization is disrupted in both α- and β-spectrin mutants
NMJ morphology in the spectrin mutants is normal by light and electron microscopy, yet neurotransmitter release is severely disrupted. In other (non-neuronal) cell types, spectrins have been proposed to capture and maintain proteins in distinct membrane-associated domains, especially at sites of cell–cell interaction (Drubin and Nelson, 1996; Pinder and Baines, 2000). At synapses, proper function requires precise assembly and alignment of the molecular machinery required for synaptic vesicle fusion and recycling. If this machinery is mislocalized or incorrectly assembled, it would not be surprising to find a synaptic defect such as we observe in α- and β-spectrin mutants. Although there is no method by which we can test whether the in vivosubmicrometer assembly of proteins is appropriate in spectrin mutants, we can determine whether synaptic proteins are polarized and properly localized to the NMJ. In epithelial cells, disruption of protein polarization attributable to the absence of spectrin is visible by immunohistochemistry and confocal light microscopy (Dubreuil et al., 2000). We used the same techniques to determine whether spectrins play a similar role in protein compartmentalization at synapses.
Figure 6 shows representative staining in wild-type and spectrin mutant embryos for two of the bestDrosophila NMJ markers available: presynaptic anti-CSP and postsynaptic anti-DLG. CSP is present in both vesicular membrane-associated and cytosolic fractions of presynaptic boutons; CSP staining normally appears as tightly localized presynaptic puncta (Zinsmaier et al., 1994). DLG is a plasma membrane-associated PDZ [postsynaptic density-95(PSD-95)/DLG/zona occludens-1] domain protein with 60% homology to PSD-95 that is tightly localized to both presynaptic and postsynaptic membranes (Lahey et al., 1994; Budnik et al., 1996). Each panel in Figure 6 shows the body wall neuromusculature of two to three embryonic hemisegments stained with anti-CSP (green) and anti-DLG (red). The (out of focus) ventral ganglion (CNS) is visible in thetop left of each panel, from which segmental nerves (SN) extend into the body wall musculature on the right. The CNS serves as a positive control for overall image intensity. In wild-type embryos, CSP and DLG staining in the body wall neuromusculature is restricted to tightly defined puncta at the NMJ (Fig. 6, left column); little or no staining is visible in either the presynaptic nerve axon or nonsynaptic muscle membrane. Thus, neither the segmental nerves nor the majority of muscle tissue is visible in the fluorescence image (Fig. 6, left column). In both α- and β-spectrin mutants (Fig. 6,middle and right columns), the synaptic localization of both presynaptic CSP and postsynaptic DLG is dramatically perturbed. The segmental nerves are now visible (because of CSP immunoreactivity), as are the muscles (because of DLG immunoreactivity). We conclude that, in both α- and β-spectrin mutants, CSP is distributed abnormally throughout presynaptic axons, and DLG is distributed abnormally throughout muscle cells. Neither protein appears properly polarized and localized to the NMJ boutons in spectrin mutants.
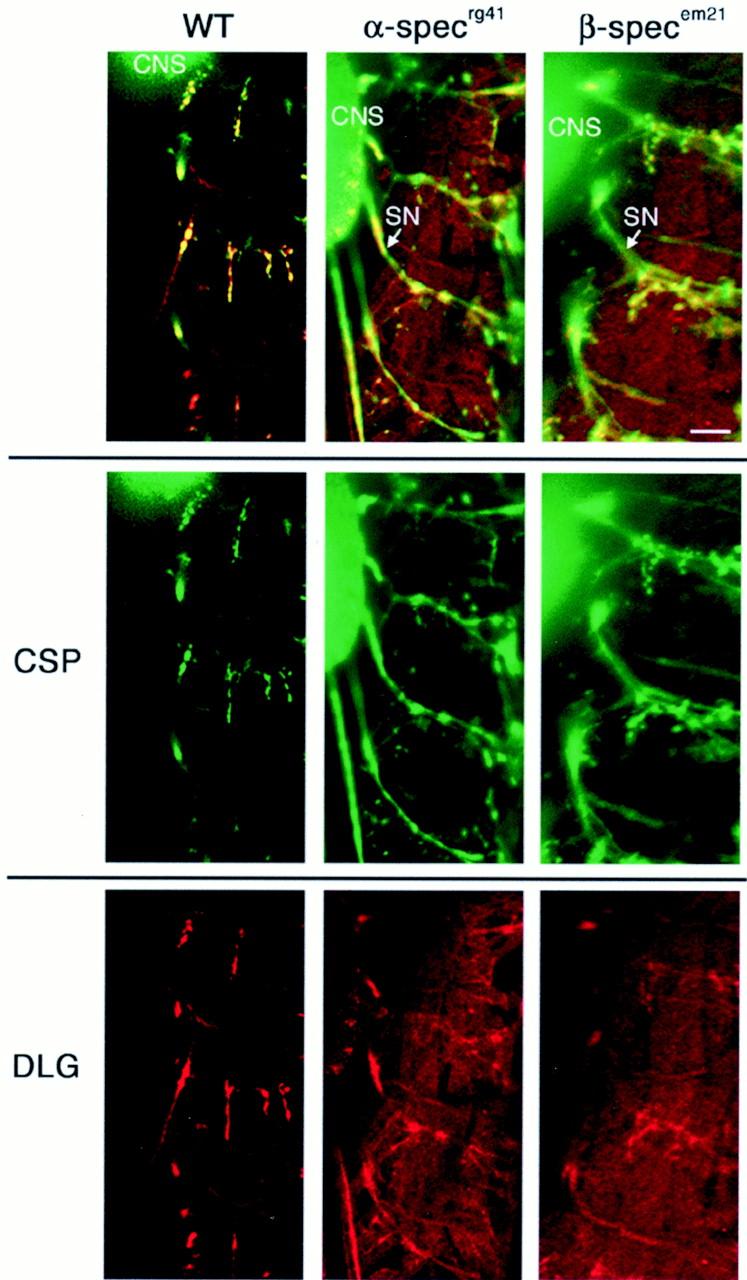
Fluorescent confocal micrographs of embryonic neuromusculature showing distribution of the presynaptic protein CSP and postsynaptic protein DLG. Each panelshows CSP and DLG immunoreactivity in three or more hemisegments. In wild-type embryos, DLG (red) and CSP (green) tightly associate with NMJ boutons, which appear as immunoreactive puncta at NMJs in the body wall neuromusculature (left column). Note that, in wild-type embryos, anti-CSP and anti-DLG antibodies detect only the NMJ and not the preterminal axon or extrasynaptic regions of the muscle. In both α- and β-spectrin null mutants, however, CSP (green) is abnormally distributed throughout distal axons (extending horizontally from the CNS on theleft into the musculature on the right). In both α- and β-spectrin null mutants, DLG staining (red) is scattered throughout postsynaptic muscles. In each image, a portion of the ventral ganglion (CNS) is shown (out of focus) as a positive control. Scale bar, 15 μm.
In addition to CSP and DLG, we examined the staining patterns of several other synaptic proteins, including synaptotagmin, synapsin, and syntaxin. Synaptotagmin is a transmembrane protein normally restricted to synaptic vesicles (Littleton et al., 1993; Marqueze et al., 2000). Synapsin is a spectrin-interacting phosphoprotein that is associated with the presynaptic actin cytoskeleton at synaptic boutons (Klagges et al., 1996; Iga et al., 1997; Hilfiker et al., 1999; Turner et al., 1999). Syntaxin is a transmembrane protein normally present in presynaptic membrane, including both axons and synaptic boutons (Schulze et al., 1995; Gerst, 1999). All of these proteins, like CSP and DLG, showed severely disrupted subcellular localization in both α- and β-spectrin mutant embryos.
We quantified protein distribution (measured immunocytochemically) by comparing staining intensity in NMJ boutons with staining intensity outside the synapse (see Materials and Methods). In wild-type embryos, fluorescence intensity from each synaptic marker was significantly higher in NMJ boutons than elsewhere. Specifically, the synaptic/nonsynaptic fluorescence intensity for each marker (in wild-type embryos) was as follows: 30.75 ± 6.85 (CSP), 8.56 ± 1.65 (DLG), 5.51 ± 1.19 (synapsin), 7.46 ± 1.88 (synaptotagmin), and 3.41 ± 0.74 (syntaxin). In other words, wild-type embryos showed anti-CSP fluorescence that was 30.75 times higher in boutons than in nerve. In contrast, anti-syntaxin fluorescence in wild-type embryos was only 3.41 times higher in boutons than in nerve. These observations are consistent with the fact that CSP is strongly restricted to synaptic boutons, whereas syntaxin is present throughout the neuronal membrane and only weakly polarized to boutons (Schulze et al., 1995). This raw ratio represents a measure of both protein localization and antibody quality because poor antibodies might be expected to lower the ratio because of high nonspecific immunoreactivity (high background) and/or reduced specific immunoreactivity.
For ease of comparison and to eliminate effects on the ratios from antibody quality, we normalized all of these ratios to wild type. Normalized ratios for wild-type and spectrin mutant embryos are shown in Figure 7. The synaptic/nonsynaptic immunoreactivity ratios in both α- and β-spectrinmutants for CSP, DLG, synaptotagmin, synapsin, and syntaxin were each significantly reduced compared with wild type (p< 0.05; t test). The normalized ratios (WT is 1) for α-spectrin mutants were as follows: 0.04 ± 0.004 (CSP), 0.14 ± 0.017 (DLG), 0.21 ± 0.036 (synapsin), 0.36 ± 0.782 (syntaxin), and 0.47 ± 0.060 (synaptotagmin) (n = 4–13; mean of 10). The normalized ratios (WT is 1) for β-spectrin mutants were as follows: 0.03 ± 0.003 (CSP), 0.14 ± 0.017 (DLG), 0.23 ± 0.051 (synapsin), 0.25 ± 0.065 (syntaxin), and 0.12 ± 0.006 (synaptotagmin) (n = 4–13; mean of 10). We conclude from these results that synaptic proteins are improperly polarized and localized in both α- and β-spectrin mutants.

Spectrin mutants have mislocalized synaptic proteins. Protein distribution was quantified by comparing staining intensity in NMJ boutons with staining intensity outside the synapse (see Materials and Methods). In wild-type embryos, fluorescence intensity from each synaptic marker was much higher in NMJ boutons than elsewhere. In α- and β-spectrin mutants, however, immunoreactivity of CSP, DLG, synaptotagmin, synapsin, and syntaxin were all reduced in boutons and simultaneously increased in nonsynaptic nerve (CSP, synaptotagmin, synapsin, and syntaxin) or muscle (DLG) membrane. Thus, the relative amount of synaptic protein at the synapse relative to other tissues was significantly reduced in spectrin mutants.
DISCUSSION
Spectrins have been known for over a decade to be present at both central and peripheral synapses in a variety of organisms (Lazarides et al., 1984; Bloch and Morrow, 1989; Goodman et al., 1989; Masliah et al., 1991; Bewick et al., 1992, 1996; Goodman et al., 1995; Gelot et al., 1996; Bloch et al., 1997; Sakaguchi et al., 1998; Wechsler and Teichberg, 1998; Wood and Slater, 1998; Goodman, 1999; Hirai and Matsuda, 1999; Dunaevsky and Connor, 2000; Hammarlund et al., 2000;Kordeli, 2000; Sikorski et al., 2000; Sunderland et al., 2000). The role that spectrins might play at synapses has been the subject of intense speculation. The Drosophila genome contains only one α-spectrin and one conventional β-spectringene, making it an ideal system to genetically manipulate spectrin levels and examine the resulting synaptic alterations. Using protein null mutants for α- and β-spectrin, we tested whether spectrins are required for development and/or function of theDrosophila neuromuscular junction.
First, we showed that both α- and β-spectrin are present at theDrosophila NMJ (Fig. 1). This observation supports the synaptic localization of spectrins observed in other systems (Bloch and Morrow, 1989; Daniels, 1990; Masliah et al., 1991; Bewick et al., 1992,1996; Goodman et al., 1995; Kordeli, 2000). Second, we showed that, in α- and β-spectrin mutants, synaptic morphology is normal (Fig. 2). This result contrasts with the severe morphological defects observed in Caenorhabditis elegans spectrin mutants (Hammarlund et al., 2000). The normal morphological development inDrosophila spectrin mutants may be possible because of a maternal contribution. Third, because NMJ morphology was normal, we were able to undertake a detailed electrophysiological analysis of synaptic function in spectrin mutants (Figs. 3, 4). This analysis showed that both α- and β-spectrin mutants have equal and severe disruptions in synaptic transmission. Using pressure-ejected glutamate to directly measure postsynaptic glutamate receptor function, we were able to rule out the possibility that the transmission defect was attributable to any alteration in glutamate receptor function. This conclusion was confirmed using analysis of spontaneous synaptic events, which showed reduced probability of vesicle fusion yet normal event amplitudes. Thus, we concluded that the source of the synaptic function defect in spectrin mutants was presynaptic. Based on immunohistochemical localization and biochemistry, spectrins have been proposed to play an important role in development and/or function of postsynaptic receptor fields (Bloch and Morrow, 1989; Daniels, 1990;Bloch et al., 1997; Wechsler and Teichberg, 1998; Hirai and Matsuda, 1999). Our data strongly suggest that this is not true at theDrosophila NMJ, although we cannot rule out the possibility that maternal spectrin contributes to the initial development of the postsynaptic receptor field.
What is the cause of the presynaptic defect in spectrin mutants? Spectrins have been proposed to capture and tether (via synapsin I) synaptic vesicles near the active zone (Landis, 1988; Goodman et al., 1995; Sikorski et al., 2000). In support of this hypothesis, it has been shown that disruption of spectrin–synapsin binding, via antibodies raised against the synapsin binding site of spectrin, eliminate synaptic transmission in cultured hippocampal cells (Sikorski et al., 2000). However, synaptic vesicle localization was never examined in that study. To test whether the defective neurotransmitter release in Drosophila spectrin mutants was attributable to altered synaptic vesicle localization, we examined the ultrastructure of wild-type and spectrin mutant synaptic terminals using electron microscopy (Fig. 5). We found no changes in synaptic vesicle distribution in the spectrin mutants. This observation is in agreement with recent data from C. elegans β-spectrinmutants (Hammarlund et al., 2000). Thus, data from bothDrosophila and C. elegans spectrin mutants argue that spectrins are not required for synaptic vesicle clustering or docking. We are unable to visualize, in either wild type or mutants, any electron-dense “rods” connecting synaptic vesicles to the active zone. These rods, which are visible in some other preparations, have been suggested to be spectrin based on their size (Landis, 1988;Goodman et al., 1995).
In epithelial cells, spectrins are required for polarization and localization of a variety of membrane-associated proteins, especially at sites of cell–cell contact (Bennett, 1990; Bennett and Gilligan, 1993; Drubin and Nelson, 1996; Brown and Breton, 2000; Dubreuil et al., 2000; Pinder and Baines, 2000). Because synapses are highly polarized sites of cell–cell interaction between ectodermally derived cells, it stands to reason that the function of neuronal spectrin might be similar to that shown in epithelia. We used methods similar to those used in studies of epithelia to show that indeed this is the case; several classes of synaptic proteins fail to properly polarize and localize in spectrin mutants (Figs. 6, 7). Because both α- and β-spectrin are distributed widely, spectrin alone cannot be sufficient for organization of functional synaptic domains. Synaptic spectrins must be “activated” via a local synaptic cue or work in conjunction with other molecules to capture and accumulate synaptic proteins. In this regard, neuronal spectrin appears to be different from epithelial spectrin, which has a polarized distribution that matches its site of activity precisely (Dubreuil et al., 2000). Because neurotransmitter release requires precise organization of presynaptic protein machinery, it is not unreasonable to conclude that the defects in synaptic release measured in Drosophila mutants are attributable to alterations in synaptic protein localization. However, we cannot rule out another, less likely, direct role for spectrin in synaptic vesicle fusion, as has been proposed by Goodman (1999).
In summary, we have shown spectrins are, as in other organisms, present in Drosophila synapses. Electrophysiological analyses showed that neurotransmitter release in Drosophila α- and β-spectrin protein null mutants is severely impaired. However, contrary to current models, this synaptic impairment is not attributable to defects in receptor field function or synaptic vesicle localization. We conclude, based on immunolocalization of several classes of synaptic proteins, that proper polarization and localization of synaptic proteins does not take place in the absence of spectrin. We propose, based on these results and the synaptic localization of spectrin, that a spectrin-based scaffold is formed early in synaptic development, and this scaffold is subsequently required for proper assembly, transport, or localization of synaptic proteins during development. Future work will aim to understand the time course and mechanisms by which synaptic spectrin is specifically activated and/or localized to capture and accumulate synaptic proteins.
Footnotes
This work was supported by a National Institutes of Health (NIH) National Research Service Award postdoctoral fellowship to D.F., NIH Grant GM49301 to R.R.D., and an ELJB Foundation fellowship, grants from the Muscular Dystrophy Association, and NIH Grant GM54544 to K.B. We thank L. S. Goldstein for β-spectrin antibodies, E. Buchner for synapsin antibodies, K. Zinsmaier for CSP antibodies, T. Littleton for synaptotagmin antibodies, H. Bellen for syntaxin 1 antibodies, and V. Budnik for DLG antibodies. We also thank T. Fergestad for confocal assistance and M. Hammarlund, R. Weimer, and C. Rodesch for critical review of this manuscript.
Correspondence should be addressed to Kendal Broadie, University of Utah, Department of Biology, 257 South 1400 East, Salt Lake City, UT 84112-0840. E-mail: broadie{at}biology.utah.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}