

Abstract
Increased levels of glutamate and the subsequent activation of NMDA receptors are responsible for neuronal damage that occurs after an ischemic or hypoxic episode. In the present work, we investigated the relative contribution of presynaptic and postsynaptic blockade of synaptic transmission, as well as of blockade of NMDA receptors, for the facilitation of recovery of synaptic transmission in the CA1 area of rat hippocampal slices exposed to prolonged (90 min) hypoxia. During hypoxia, there was a complete inhibition of field EPSPs, which was fully reversible if released adenosine was allowed to act. When adenosine A1 receptors were blocked with the selective antagonist 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), recovery of synaptic transmission from hypoxia was significantly attenuated, and this impairment could be overcome by preventing synaptic transmission during hypoxia either with tetrodotoxin (TTX) or by switching off the afferent stimulation but not by postsynaptic blockade of transmission with 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) or selective blockade of adenosine A2A receptors. When synaptic transmission was allowed to occur during hypoxia, because of the presence of DPCPX, there was an NMDA receptor-mediated component of the EPSCs recorded in CA1 pyramidal neurons, and blockade of NMDA receptors with AP-5 restored recovery of synaptic transmission from hypoxia. It is concluded that impairment of recovery of synaptic transmission after an hypoxic insult results from activation of synaptic NMDA receptors by synaptically released glutamate and that adenosine by preventing this activation efficiently facilitates recovery.
Hypoxia and/or ischemia induce the release of several neurotransmitters, including excitatory amino acids such as glutamate (Nicholls and Attwell, 1990). Increased levels of glutamate, and the subsequent activation of ionotropic NMDA receptors, are primarily responsible for neuronal damage that occurs as a consequence of ischemic or hypoxic episodes (Simon et al., 1984;Rothman and Olney, 1986; Sattler et al., 2000).
Several neuromodulators are also released during hypoxia–ischemia, and one that has been receiving particular attention is adenosine (Pedata et al., 1993; Dale et al., 2000). This neuromodulator, by activating metabotropic receptors of the A1subtype, decreases the release of excitatory amino acids during ischemia (Phillis et al., 1991; Héron et al., 1993). Indeed, the amounts of adenosine released during hypoxia are enough to decrease the evoked release of glutamate at the synaptic cleft (Katchman and Hershkowitz, 1996), being partially responsible for the hypoxia-induced decrease of excitatory transmission (Fowler, 1989; Canhão et al., 1994). At the postsynaptic level, adenosine efficiently inhibits NMDA receptor-mediated currents (de Mendonça et al., 1995; Costenla et al., 1999). All of these inhibitory actions of adenosine are mediated through inhibitory A1 receptors and might be involved in its neuroprotective role (de Mendonça et al., 2000).
Stroke involves the loss of specific neurological functions caused by reduced blood perfusion in corresponding brain areas, in which neuronal and synaptic electrical activities are silenced (Astrup et al., 1981). Importantly, the neurons at the periphery of the ischemic lesion will be able eventually to recover metabolically, regain electrical activity, and support the partial neurological recovery that hopefully may follow. Recovery of synaptic transmission is thus an essential requisite for functional recovery of the neuronal circuits after an insult. The CA1 area of the hippocampus is highly vulnerable to hypoxia–ischemia (Lipton, 1999), and hippocampal slices have been proving to be a good model to evaluate synaptic transmission in this area. Although evidence has been produced showing that blockade of synaptic transmission may facilitate recovery after a hypoxic insult (Boening et al., 1989; Fowler and Li, 1998), it is not clear whether the protection is attributable to inhibition of glutamate release or to prevention of activation of postsynaptic receptors, including NMDA receptors, or both.
The present work was designed to investigate the relative contributions of presynaptic and postsynaptic blockade of transmission, as well as of blockade of NMDA receptors, for the facilitation of recovery of hippocampal synaptic transmission after a hypoxic insult. We found that synaptic transmission fully recovered, even after prolonged periods of hypoxia providing that adenosine A1 receptors could be activated by released adenosine. During blockade of these adenosine receptors, the recovery from hypoxia was impaired. This impairment could be mainly attributed to the synaptic release of glutamate and selective activation of synaptic NMDA receptors, suggesting a compartmentalization of these receptors during hypoxia-induced excitotoxicity.
MATERIALS AND METHODS
The experiments were performed on hippocampal slice preparations taken from male Wistar rats (4–8 weeks old) handled according to the European Community guidelines and Portuguese law on Animal Care. The animals were decapitated under halothane anesthesia, and the hippocampus was dissected free into ice-cold Krebs' solution of the following composition (mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgSO4, 2 CaCl2, and 10 glucose (previously gassed with 95% O2–5% CO2), pH 7.4. Slices (400 μm thick) were cut perpendicular to the long axis of the hippocampus with a McIlwain tissue chopper and allowed to metabolically recover for at least 1 hr in a chamber within the same gassed medium at room temperature (22–25°C). In the experiments in which field potentials were recorded, the slices were transferred to a recording chamber (1 ml) for submerged slices and continuously superfused with gassed bathing solution at 32°C, at a flow rate of 3 ml/min. In the experiments in which synaptic currents were recorded, the slices were submerged in a 3 ml recording chamber and continuously superfused (4 ml/min) with gassed bathing solution at room temperature, to avoid electric noise from a temperature controller. Drugs were added to the superfusion solution.
Hypoxia was induced by changing to the same solution equilibrated with a 95% N2 plus 5% CO2 mixture. In the experiments in which field potentials were recorded, a hypoxic period of 90 min was used, followed by a reoxygenation period of at least 30 min. In the experiments recording synaptic currents, the time of hypoxia was chosen to obtain a maximum inhibition of the EPSCs for at least 10 min in control conditions, i.e., in the absence of any drugs. The bath pO2 levels were measured with a Dissolved Oxygen Meter (World Precision Instruments, Sarasota, FL). In some experiments, the pO2 levels in the slice were measured through a Clark Style Microelectrode coupled to a Chemical Microsensor (Diamond General, Ann Arbor, MI) while simultaneously recording field EPSPs (fEPSPs). The oxygen measuring microelectrode and the fEPSP recording microelectrode were positioned in the slice under microscopic (∼50×) visual guidance, and care was taken to insert both microelectrodes at approximately the same depth. Each slice was exposed to only one period of hypoxia because the effects of hypoxia may be modified by subsequent episodes in the same slice (Schurr et al., 1986).
Field EPSP recordings. fEPSPs were recorded through an extracellular microelectrode (4 m NaCl, 2–6 MΩ resistance) placed in the stratum radiatum of the CA1 area. Stimulation (rectangular pulse of 0.1 msec applied once every 15 sec) was delivered through a concentric electrode placed on the Schaffer collateral–commissural fibers, in the stratum radium near the CA3–CA1 border. The intensity of the stimulus (80–200 μA) was initially adjusted to obtain a large fEPSP slope with a minimum population spike contamination. Recordings were obtained with an Axoclamp 2B amplifier coupled to a DigiData 1200 interface (Axon Instruments, Foster City, CA). Averages of eight consecutive responses were continuously monitored on a personal computer with the LTP program (Anderson and Collingridge, 1997), kindly supplied by W. W. Anderson (University of Bristol, Bristol, UK). Except when specified, responses were quantified as the slope of the initial phase of the averaged fEPSPs, because slope measures are considered a more accurate measure of fEPSP magnitude than the amplitude attributable to possible contamination by the population spike.
EPSC recordings. Whole-cell patch-clamp recordings were obtained using the “blind” whole-cell recording technique (Blanton et al., 1989) and with a List Biologic (Campbell, CA) EPC-7 patch-clamp amplifier in the voltage-clamp mode. After being filtered (10 and 3 kHz-pole Bessel filters), data were displayed, recorded, and analyzed in a personal computer using the MAP (R.C. Electronics Inc., Santa Barbara, CA) software. Recording electrodes had resistances between 4 and 8 MΩ when filled with the electrolyte solution, which had the following composition (in mm): 140 potassium gluconate, 11 KCl, 0.1 CaCl2, 2 MgCl2, 1 EGTA, 10 HEPES, 2 MgATP, 0.3 NaGTP, and 5 2(triethylamino)-N-(2,6-dimethylphenyl)acetamide [(QX-314) to intracellularly block voltage-dependent sodium channels], pH 7.3. EPSCs were evoked by delivering rectangular pulses (0.15 msec, 200–300 μA) to the Schaffer collateral pathway at 15 sec intervals using a concentric electrode placed on the stratum radiatum near the CA3–CA1 border. Voltage steps (−10 mV) were delivered at 15 sec intervals to monitor input resistance and whole-cell access throughout the course of the experiment. During the experiment, if the neuron showed either a marked change in holding current or a noticeable alteration in the amplitude or shape of the capacitance transients, it was discarded.
Drugs.dl-2-Amino-5-phosphonovaleric acid (AP-5), MgATP, NaGTP, and tetrodotoxin (TTX) were from Sigma (St. Louis, MO). 1,3-Dipropyl-8-cyclopentylxanthine (DPCPX) was from Research Biochemicals (Natick, MA). 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5ylamino]ethyl)phenol (ZM 241385) were from Tocris Cookson (Ballwin, MO). QX-314 was from Calbiochem (La Jolla, CA). 5-Amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (SCH 58261) was generously provided by Dr. E. Ongini (Schering-Plough, Milan, Italy).
DPCPX was made up in a 5 mm stock solution in 99% dimethylsulfoxide (DMSO)–1 m 1% NaOH (v/v). SCH 58261 and ZM 241385 were made up in 5 mm stock solutions in DMSO. Aliquots of these stock solutions were kept frozen at −20°C until use. CNQX was made up in a 20 mm stock solution in DMSO. AP-5 was made up in a 20 mm stock solution in distilled water. TTX was made up into a 3 mm stock solution in citrate buffer, pH 4.3. These stock solutions were kept at 4°C until use. QX-314, MgATP, and NaGTP were added directly to the recording electrode filling solution, which was kept frozen in aliquots at −20°C until use.
Analysis of the data. The data are expressed as mean ± SEM from n number of slices. To allow comparisons between different experiments, the amplitude or slope values were normalized, taking as 100% of the averaged values obtained during 10 or 5 min immediately before applying hypoxia. The significance of the differences between the means was evaluated by one-way repeated-measures ANOVA, followed by the Tukey's multiple comparison test. Values of p < 0.05 were considered to represent statistically significant differences.
RESULTS
Adenosine A1 receptor blockade attenuates depression of synaptic transmission during hypoxia and impairs recovery on reoxygenation
The pO2 values in the fEPSPs recording chamber ranged from 490 to 610 mmHg when the perfusion solution was saturated with 95% O2. Switching the perfusion into a 95% N2 saturated solution caused a fast drop of the pO2 levels in the recording chamber, being nearly 20% of the initial value within 5 min after starting the changeover of solutions. The chamber pO2 values reached a minimum within 30 min after changing the solutions and were nearly constant up to the end of the 90 min hypoxia period (range of 8–30 mmHg). In five experiments in which the pO2levels were measured in the slice, the averaged values before applying hypoxia were 461 ± 63 mmHg. These values started to decrease immediately after starting hypoxia and reached nearly zero within 10 min (Fig. 1A). The pO2 levels in the slice recovered toward the levels before hypoxia within 25–30 min (416 ± 55 mmHg) after returning to the 95% O2 saturated solution. As illustrated in Figure 1, the decrease in slice pO2 levels was immediately followed by a decrease in the fEPSPs, which was evident in both the slope and amplitude. In control conditions (i.e., absence of added drugs), the fEPSPs recovered, either measured as slope (Fig. 1) or amplitude. The decrease in the fEPSPs during hypoxia should be mainly attributable to the inhibition of synaptic transmission because the presynaptic volley, which is proportional to the strength of the inputs from the Schaffer collaterals, did not accompany the decrease in the fEPSPs (Fig. 1).

Hypoxia causes a fast and reversible depression of the fEPSPs but not of the presynaptic volley (PV), which parallels the decrease in pO2 levels in the hippocampal slice. A, Time course of the effect of hypoxia [95% N2 plus 5% CO2 applied as indicated (N2) by the horizontal bar] on the slope of fEPSPs, amplitude of the presynaptic volley (all values indicated as percentage of the value before hypoxia;left ordinate axis, 100%: 0.53 mV/msec for fEPSP slope, and 0.28 mV/msec for PV amplitude), and on the slice pO2values (right ordinate axis) recorded simultaneously. Each point represents the computer-averaged value of eight consecutive responses obtained for 2 min. In B are shown the traces obtained in the same experiment (fromleft to right) before hypoxia, at the end of hypoxia, and during recovery from hypoxia. Each traceis the average of eight successive responses and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP.
Averaged results from several experiments (Fig.2) showed that, in control conditions, the slope of the fEPSPs decreased during hypoxia to 8.8 ± 2.1% of its prehypoxia value and recovered up to 111 ± 2.8% within 30 min after starting reoxygenation (n = 12). As previously observed for shorter periods of hypoxia (Canhão et al., 1994), the decrease in the fEPSP slope during hypoxia might be partially attributed to adenosine released during hypoxia because this decrease was nearly 50% attenuated when hypoxia was applied to slices in which the A1 inhibitory adenosine receptors were blocked by a supramaximal concentration (Lucchi et al., 1996) of the selective A1 receptor antagonist DPCPX (50 nm). Interestingly, recovery of fEPSPs was impaired during blockade of A1 receptors (Fig.2). At maximum recovery after hypoxia and in the presence of DPCPX (50 nm), the fEPSP slope was 75 ± 3.5% (n = 12) of its value before hypoxia, a value significantly different (p < 0.05) from that obtained in the absence of the A1 receptor antagonist. This impairment of the recovery after hypoxia could not be attributed to activation of excitatory A2Aadenosine receptors, which are present in the hippocampus (Sebastião and Ribeiro, 2000) and may aggravate hypoxic lesions (Chen et al., 1999), because blockade of A2Areceptors with the selective antagonists (Ongini and Fredholm, 1996) SCH 58261 (50 nm) or ZM 241385 (50 nm) did not overcome the impairment of the synaptic transmission recovery after hypoxia observed during blockade of the A1 receptors with DPCPX (Fig.2C). Similarly, the hypoxia-induced depression of synaptic transmission was not influenced by the A2Areceptor antagonists (Fig. 2B).

Recovery from hypoxia is impaired when adenosine A1 receptors are blocked with DPCPX. A, Comparison of the time course of the effect of hypoxia on the averaged fEPSP slopes recorded from the CA1 area of hippocampal slices in the absence (control, n = 12) or in the presence (n = 12) of DPCPX, which was perfused for at least 45 min before hypoxia and was kept in the bath up to the end of the experiment; hypoxia was applied as indicated by the horizontal bar. In B and C are shown the statistical comparisons of the fEPSP slopes recorded (as indicated below each column) in control slices (absence of drugs), in slices perfused with DPCPX, and in slices in which, in addition to blocking A1 receptors with DPCPX, adenosine A2Areceptors were also blocked with either SCH 58261 (n = 4) or ZM 241385 (n = 4), the A2A antagonist being applied together with DPCPX and at least 45 min before hypoxia; the results obtained at the end of 80–90 min after starting hypoxia are shown in B, and those obtained at 24–30 min after starting reoxygenation are shown inC. All of the values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 min immediately before hypoxia): 0.54 ± 0.04 mV/msec (control), 0.53 ± 0.03 mV/msec (DPCPX), 0.63 ± 0.06 mV/msec (DPCPX plus SCH 58261), and 0.49 ± 0.01 mV/msec (DPCPX plus ZM 241385). *p < 0.05 (one-way repeated-measures ANOVA, followed by the Tukey's multiple comparison test); NS, not statistically significant. Note that the A1 receptor antagonist modified both the hypoxia-induced maximum depression of the fEPSPs and recovery of the fEPSPs, but these were not influenced by additional A2A receptor antagonism.
Presynaptic blockade of transmission during hypoxia restores recovery on reoxygenation
The impaired recovery of the fEPSPs from hypoxia when adenosine A1 receptors are blocked suggests that activation of these receptors by released adenosine facilitates recovery of synaptic transmission during reoxygenation. To evaluate whether the ability of adenosine to depress synaptic transmission during hypoxia could be related to its ability to facilitate recovery on reoxygenation, experiments were designed in which A1 receptors were blocked with DPCPX, and synaptic transmission during hypoxia was prevented by either switching off stimulation of the Schaffer collaterals during hypoxia (Fig.3A) or perfusion of the sodium channel blocker TTX (100 nm) (Fig.3B). Under both conditions, there was a full recovery of synaptic transmission from hypoxia (Fig. 3C), despite the presence of DPCPX (50 nm). Thus, in the experiments in which stimulation was stopped during hypoxia, the fEPSP slope recovered up to 99 ± 2.3% (n = 5) of its value before hypoxia. TTX fully prevented both the presynaptic volley and the EPSPs; when it was present during hypoxia, the fEPSP slope after reoxygenation returned to 95 ± 4.8% (n = 5) of the pre-TTX value. These values were not significantly different (p > 0.05) from those obtained in the absence of any drugs.

Prevention of synaptic transmission by procedures that preclude evoked release of glutamate facilitates recovery from hypoxia. The hippocampal slices were in the presence of the selective A1 receptor antagonist DPCPX (50 nm) throughout the experiments and for at least 45 min before addition of any other drug or experimental condition.A, Synaptic transmission during hypoxia was prevented by switching off the stimulation of the Schaffer collaterals, as indicated by the horizontal bar. B, Synaptic transmission was prevented by perfusion of the sodium channel blocker TTX, as indicated by the horizontal bar; stimulation was continued throughout. In both A andB, each point corresponds to averaged data from five experiments. In C is shown the statistical comparison of the fEPSP slopes recorded at maximum recovery from hypoxia in the experiments in which the stimulation was “on” during hypoxia (st. on; n = 12), the stimulation was stopped during hypoxia (st. off;n = 5), and in the experiments with TTX (n = 5) during hypoxia, as indicated below eachcolumn. All the values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 min immediately before hypoxia): 0.53 ± 0.03 mV/msec (st. on), 0.48 ± 0.03 mV/msec (st. off), and 0.73 ± 0.11 mV/msec (TTX). *p < 0.05 (one-way repeated-measures ANOVA, followed by the Tukey's multiple comparison test); NS, not statistically significant. Note that recovery of the fEPSPs in the experiments in which synaptic transmission was allowed to occur during hypoxia was significantly lower that the recovery obtained when synaptic transmission was prevented during hypoxia.
Postsynaptic blockade of transmission during hypoxia does not restore recovery on reoxygenation
Inhibition of synaptic transmission during hypoxia, with either TTX or by absence of stimulation, prevents synaptic release of glutamate and as a consequence precludes the activation of postsynaptic glutamate receptors by synchronously (i.e., action potential dependent) released glutamate. To evaluate whether postsynaptic prevention of synaptic transmission during hypoxia could overcome the impairment of synaptic transmission recovery when adenosine A1receptors were inoperative, we blocked fast synaptic transmission with the AMPA–kainate receptor antagonist CNQX in the presence of DPCPX and compared the recovery of synaptic transmission under normoxia and after a 90 min hypoxia period. CNQX (4 μm) caused a virtually complete inhibition of the fEPSPs within 20 min after starting its perfusion (Fig. 4). In slices not exposed to hypoxia, when CNQX was removed from the bath, the fEPSP slope gradually recovered up to 73 ± 12% (n = 3) of the values before CNQX. This recovery from CNQX in normoxic conditions was taken as reference and, as illustrated in Figure 4, was significantly (p < 0.05) greater than that occurring in the slices subjected to a 90 min hypoxia period (fEPSP slope during recovery, 38 ± 10% of pre-CNQX values;n = 5). In slices subjected to 90 min hypoxia in the presence of CNQX (4 μm) but in the absence of DPCPX, the fEPSP slope recovered up to 68 ± 19% (n = 3) of its value before CNQX, which was significantly different (p < 0.05) from recovery of synaptic transmission under similar conditions but in the presence of DPCPX (fEPSP slope, 38 ± 10%; n = 5) and was not significantly different (p > 0.05) from the fEPSP slope values obtained after washing out CNQX in slices not exposed to hypoxia (73 ± 12%; n = 3). Thus, together these results show that postsynaptic blockade of fast EPSPs did not overcome the impairment of synaptic transmission recovery observed in the presence of DPCPX.

Postsynaptic blockade of synaptic transmission does not facilitate recovery from hypoxia. The hippocampal slices were in the presence of the selective A1 receptor antagonist DPCPX (50 nm) throughout the experiments and for at least 45 min before addition of any other drug or experimental condition. A, Time course of averaged results taken from experiments in which the AMPA–kainate receptor antagonist CNQX was applied to slices exposed to hypoxia (filled squares, hypoxia applied as indicated by the horizontal bar; n = 5) or to slices not exposed to hypoxia (open squares; n = 3). The period of CNQX application is indicated by the horizontal bar. In B is shown the statistical comparison of the fEPSP slopes corresponding to 168–174 min in A in the experiments in which hypoxia was applied (N2) and in the experiments in which hypoxia was not applied (O2 ), as indicated below each column. All of the values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 min immediately before hypoxia): 0.58 ± 0.06 mV/msec (N2 ) and 0.46 ± 0.01 mV/msec (O2 ). *p < 0.05 (one-way repeated-measures ANOVA, followed by the Tukey's multiple comparison test).
Blockade of NMDA receptor-mediated synaptic transmission during hypoxia restores recovery on reoxygenation
While decreasing synaptic release of glutamate by a presynaptic inhibition of transmission, the synaptic activation of NMDA receptors might also be reduced. Because synaptic NMDA receptors are particularly relevant (compared with extrasynaptic NMDA receptors) for excitotoxicity processes involving oxygen–glucose deprivation (Sattler et al., 2000), we evaluated the influence of NMDA receptor blockade on recovery of synaptic transmission under conditions of adenosine A1 receptor blockade. As expected (Collingridge et al., 1983), the NMDA receptor antagonist AP-5 (50 μm) did not affect the fEPSPs in normoxic conditions and, also, it did not virtually influence the fEPSPs in hypoxic conditions (Fig.5A). In slices with adenosine A1 receptors blocked with DPCPX (50 nm), the degree of depression of synaptic transmission caused by hypoxia was similar (p > 0.05) in the presence or in the absence of AP-5 (50 μm) (Fig. 5C). However, despite the persistence of synaptic potentials during hypoxia, the fEPSPs fully recovered from hypoxia (Fig. 5B). Indeed, during reoxygenation, the averaged fEPSP slopes in the presence of DPCPX plus AP-5 were significantly different (p < 0.05) from those obtained in the presence of only DPCPX and not significantly different (p > 0.05) from those obtained in the absence of any drugs (Fig. 5D).

Blockade of NMDA receptors facilitates recovery from hypoxia without influencing hypoxia-induced depression of synaptic transmission. A, Absence of modification of fEPSPs by the NMDA receptor antagonist AP-5 (50 μm) in either normoxic (top row) or hypoxic (bottom row) conditions; the adenosine A1 receptors were blocked with DPCPX (50 nm). Each trace is the average of eight successive fEPSPs preceded by the presynaptic volley and stimulus artifact. B, Time course of the effect of hypoxia (applied as indicated by the horizontal bar) on the slope of fEPSPs recorded from hippocampal slices in the presence of AP-5 (50 μm) and DPCPX (50 nm) (n = 6). In C andD are shown the statistical comparisons of the fEPSP slopes recorded (as indicated below each column) in control slices (absence of drugs), in slices perfused with DPCPX, and in slices in which, in addition to blocking A1 receptors with DPCPX, NMDA receptors were also blocked with AP-5; the results obtained at the 80–90 min after starting hypoxia are shown inC, and those obtained at 24–30 min after starting reoxygenation are shown in D. All of the values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 min immediately before hypoxia): 0.54 ± 0.04 mV/msec (control;n = 12), 0.53 ± 0.03 mV/msec (DPCPX;n = 12), and 0.61 ± 0.05 mV/msec (DPCPX plus AP-5; n = 6). *p < 0.05 (one-way repeated-measures ANOVA, followed by the Tukey's multiple comparison test); NS, not statistically significant.
Together, the results so far obtained point toward the possibility that a relevant consequence of blocking synaptic transmission during hypoxia is to prevent activation of NMDA receptors by synchronously and synaptically released glutamate. If so, one could anticipate that NMDA receptors might be activated during hypoxia and contribute to the total ionic charge carried by synaptic currents. To directly assess this point and because fEPSP recordings are not sensitive enough to evaluate NMDA receptor activation without pharmacological blockade of fast glutamatergic and GABAergic transmission (Collingridge et al., 1983;Bashir et al., 1991), we recorded evoked EPSCs in hippocampal neurons and looked for an NMDA-mediated component of the EPSCs in hypoxic conditions. The NMDA receptor-mediated component of the EPSC was evaluated by subtraction of EPSCs obtained in the same neuron in the absence and in the presence of a supramaximal concentration (100 μm) of AP-5 (Hestrin et al., 1990). We preferred this procedure to the pharmacological isolation of the NMDA receptor-mediated currents (Clark and Collingridge, 1995) because this involves the use of a cocktail of drugs, some of them (e.g., GABAA receptor blockers) being able to influence synaptic responses to hypoxia, namely in conditions of A1 receptor blockade (Lucchi et al., 1996).
Under control conditions, i.e., in the absence of any drugs, hypoxia induced a progressive decrease of the EPSCs, which were virtually abolished within 17 min after starting the perfusion with the N2 saturated solution. When similar experiments were performed in the presence of DPCPX (50 nm), the hypoxia-induced inhibition of the EPSCs was much smaller (Fig.6A). The amplitude of the EPSCs after 30 min hypoxia under these conditions was 68 ± 12% (n = 4) of the amplitude value before hypoxia. AP-5 (100 μm) was then applied, and, as illustrated in Figure 6B, it did not influence the initial phase of the inward current but inhibited a slow component of the EPSC, so that an NMDA receptor-mediated EPSCs could be put into evidence by subtraction of the EPSC recordings obtained without and with AP-5 (Fig. 6B, bottom panels). An NMDA receptor-mediated component of the EPSC in hypoxic conditions in the presence of DPCPX was observed in four of four cells from different experiments.

Influence of hypoxia on fast EPSCs and on the NMDA receptor-mediated slow component of the EPSCs. A, Time course of the effect of hypoxia (applied as indicated by thehorizontal bar) on the amplitude of EPSCs recorded from pyramidal CA1 cells in hippocampal slices in the absence (open circles) and in the presence (filled circles) of the adenosine A1 receptor antagonist DPCPX. Data shown were obtained in the absence of AP-5 and in three (control) to five (DPCPX) cells from different experiments. In each experiment, averages of four consecutive EPSCs were used to measure the EPSC amplitude. B, Individual recordings of EPSCs, preceded by the stimulus artifact, obtained during hypoxia in the presence of DPCPX (50 nm), without (left top panel; recording obtained after 30 min hypoxia), or with the NMDA receptor antagonist AP-5 (100 μm) (right top panel; recording obtained after 50 min hypoxia and 20 min AP-5). In the left bottom panel are shown the superimposed traces for better comparison; the difference between bothtraces (right bottom panel; note change in amplitude calibration) reveals an NMDA receptor-mediated slow component of the EPSC. Resting membrane potential, −55 mV; input resistance, 128 MΩ. In both A and B, the cells were clamped at −40 mV, at which inward NMDA currents are larger in the presence of Mg2+ (Hestrin et al., 1990). Note that, in the absence of DPCPX, the EPSCs were virtually abolished by hypoxia (A), preventing the occurrence of any NMDA receptor-mediated component.
DISCUSSION
Action potential-dependent, thus synchronous, release of glutamate is depressed during hypoxia, which might be at least in part responsible for the hypoxia-induced inhibition of excitatory synaptic transmission (Lipton and Whittingham, 1979; Khazipov et al., 1993). This decrease in evoked glutamate release at the synaptic level contrasts with the hypoxia–ischemia-induced global enhancement of glutamate release, which occurs through reversal of the glutamate transport system (Nicholls and Attwell, 1990). The present results showing that conditions that prevent the synaptic release of glutamate (i.e., TTX or absence of stimulation), but not conditions that prevent the postsynaptic action of glutamate at AMPA–kainate receptors (i.e., with CNQX), facilitate the recovery of synaptic transmission from hypoxia constitute a strong indication that, in our experimental conditions, impairment of synaptic recovery is related with synaptic release of glutamate. This implies a compartmentalization of the glutamate release processes as well as of the glutamate receptors responsible for the impairment of the recovery from hypoxia. In fact, without compartmentalization, the glutamate released through the transport system should be able to reach glutamate receptors equally well, because this form of glutamate release might not be influenced by inhibitors of synaptic transmission as a consequence of its independence from action potential and extracellular calcium (Sanchez-Prieto and Gonzalez, 1988).
The glutamate receptors responsible for the impairment of recovery of synaptic transmission are probably of the NMDA subtype, because activation of these receptors contributes to synaptic currents during hypoxia (Fig. 6) and their blockade facilitates recovery even in conditions in which synaptic transmission is maintained during hypoxia (Fig. 5). It thus emerges that, to facilitate recovery from hypoxia, it is necessary to prevent synaptic release of glutamate and/or to preclude activation of NMDA receptors. Together, these findings strongly suggest that an important way to facilitate synaptic transmission recovery from hypoxia is to prevent activation of synaptic NMDA receptors, which implies distinct roles for synaptic and extrasynaptic NMDA receptors (Sattler et al., 2000). This compartmentalization of NMDA receptors may be related with their differential association with other proteins or subcellular organelles, which may be critical for triggering excitotoxicity (Furukawa et al., 1997; Sattler et al., 2000; Sinor et al., 2000).
Glutamate transporters are highly temperature sensitive (Bergles and Jahr, 1998), and a decrease of the carrier-mediated release of glutamate during hypoxia–ischemia may account for the mild hypothermia (32–34°C)-induced neuroprotection in vivo (Illievich et al., 1994; Zornow, 1995). Hippocampal slices maintained in vitro suffer hypoxic injury at temperatures above 33°C, even under perfusion of oxygen saturated solutions, probably because of insufficient oxygen delivery into the slice core (Schiff and Somjen, 1985). To minimize the slice injury before inducing the hypoxic stimulus and to maximize the effects of hypoxia on synaptic responses, which are more pronounced at higher slice temperatures (Taylor and Weber, 1993), we kept the slices at 32°C while recording the fEPSPs. These experimental constraints could favor recovery from prolonged hypoxia and would lead to an underestimation of the contribution of the nonsynaptic release of glutamate. Furthermore, the presence of glucose may also lead to a decrease in the amounts of glutamate that accumulate extracellularly during the hypoxic period (Swanson et al., 1994). Clearly, the experiments now reported do not intend to mimic conditions of severe ischemia with pronounced release of glutamate and scarce neuronal recovery but instead intend to reflect conditions in the ischemic penumbra, in which the presence of some glucose can prevent the massive uncontrolled glutamate release and neuronal death.
Previous studies have shown that TTX enhances recovery of potentials in situations of oxygen and/or oxygen plus glucose (i.e., ischemic conditions) deprivation (Boening et al., 1989; Taylor et al., 1995;Fowler and Li, 1998). At least under ischemic conditions, the neuroprotective action of TTX may result from inhibition of sodium entry through non-inactivating TTX-sensitive sodium channels (Taylor, 1993) during ischemic depolarization. Alternatively, and as pointed out previously (Boening et al., 1989; Zhu and Krnjevic, 1999), the beneficial action of TTX may result from a TTX-mediated depression of action potentials with subsequent inhibition of calcium influx at nerve terminals and exocytotic transmitter release. In our experimental conditions, TTX fully inhibited the presynaptic volley, indicating a blockade of action potentials in the Schaffer collaterals. Furthermore, our results showing that facilitation of recovery of synaptic transmission could be obtained just by switching off stimulation to the Schaffer collaterals during hypoxia, i.e., by preventing action potential generation without addition of sodium channel blockers, suggest that, at least under reduced temperature, inhibition of action potential-dependent release of glutamate during hypoxia is a major mechanism to facilitate recovery. Also in favor of this interpretation is the finding that a very efficient way to facilitate recovery of synaptic transmission is to allow released adenosine to operate A1 receptors. Indeed, in the absence of DPCPX, there was a full blockade of transmission without marked modification of the presynaptic volley, indicating that the action of endogenous adenosine during hypoxia is mainly on synaptic transmission rather than on the conduction of the nerve impulse. In fact, the concentrations of adenosine or adenosine receptor agonists required to inhibit voltage-dependent sodium channels (Simões et al., 1988) or action potentials (Ribeiro and Sebastião, 1984; Swanson et al., 1998) are much higher than those required to inhibit synaptic transmission (Ribeiro and Sebastião,1987; Dunwiddie and Miller, 1993).
Through activation of A1 receptors, adenosine is able to simultaneously inhibit transmission at the presynaptic level (Ginsborg and Hirst, 1972; Lupica et al., 1992; Prince and Stevens, 1992), even in hypoxic conditions (Khazipov et al., 1993), and to postsynaptically inhibit NMDA receptors (de Mendonça et al., 1995; Costenla et al., 1999) by a process not related to adenosine induced postsynaptic hyperpolarization because it requires much lower concentrations of A1 receptor agonists (for discussion, see de Mendonça et al., 1995). From the present findings, it is evident that these two types of adenosine action (presynaptic inhibition of transmission and NMDA receptor inhibition) contribute in a convergent way to its ability to facilitate recovery from hypoxia and, therefore, to neuroprotection. An intense inhibitory tonus by released adenosine during hypoxia may explain why we could only observe an impairment of the recovery of synaptic transmission when adenosine A1 receptors were blocked.
Adenosine A2A receptors are generally known to mediate excitatory actions in the nervous system and are able to aggravate hypoxic–ischemic injury in vivo (for review, seede Mendonça et al., 2000). In the present work, blockade of adenosine A2A receptors did not influence recovery from hypoxia, at least in conditions in which adenosine A1 receptors were blocked. This suggests that putative synaptic actions of A2A receptors (Sebastião and Ribeiro, 2000) do not influence adenosine-mediated neuroprotection but does not preclude a role of A2A receptors in hypoxia–ischemia (Chen et al., 1999) that might be exerted at nonsynaptic and/or non-neuronal levels, including vascular and glial actions (Phillis, 1989; Rudolphi et al., 1992; von Lubitz, 1999). There is also the possibility that the blockade of adenosine A1 receptors used in the present work to impair recovery of synaptic transmission from hypoxia might obscure any synaptic action of adenosine A2A receptors during hypoxia. Other models of synaptic injury not requiring blockade of the strong adenosine A1 receptor-mediated inhibitory tonus are required to further elucidate this point.
In conclusion, the present results strongly suggest that, at least under moderately low temperature, impairment of recovery of synaptic transmission after an hypoxic insult results from activation of synaptic NMDA receptors by glutamate released during hypoxia and that adenosine, by efficiently preventing this activation, facilitates recovery from hypoxia (Fig. 7). These findings stress the importance of the regulation of the synaptic levels of glutamate and highlight the synaptic mechanisms operated by adenosine A1 receptors under hypoxic conditions, providing evidence for a close relationship between synaptic actions of adenosine and neuroprotection. Whether nonsynaptic release of glutamate assumes a major importance at temperatures closer to body temperature and whether the now discussed mechanism of action of adenosine to protect neurons from hypoxia will prove particularly relevant in the design of strategies aiming a potentiation of the neuroprotective actions of mild hypothermia awaits further investigation.

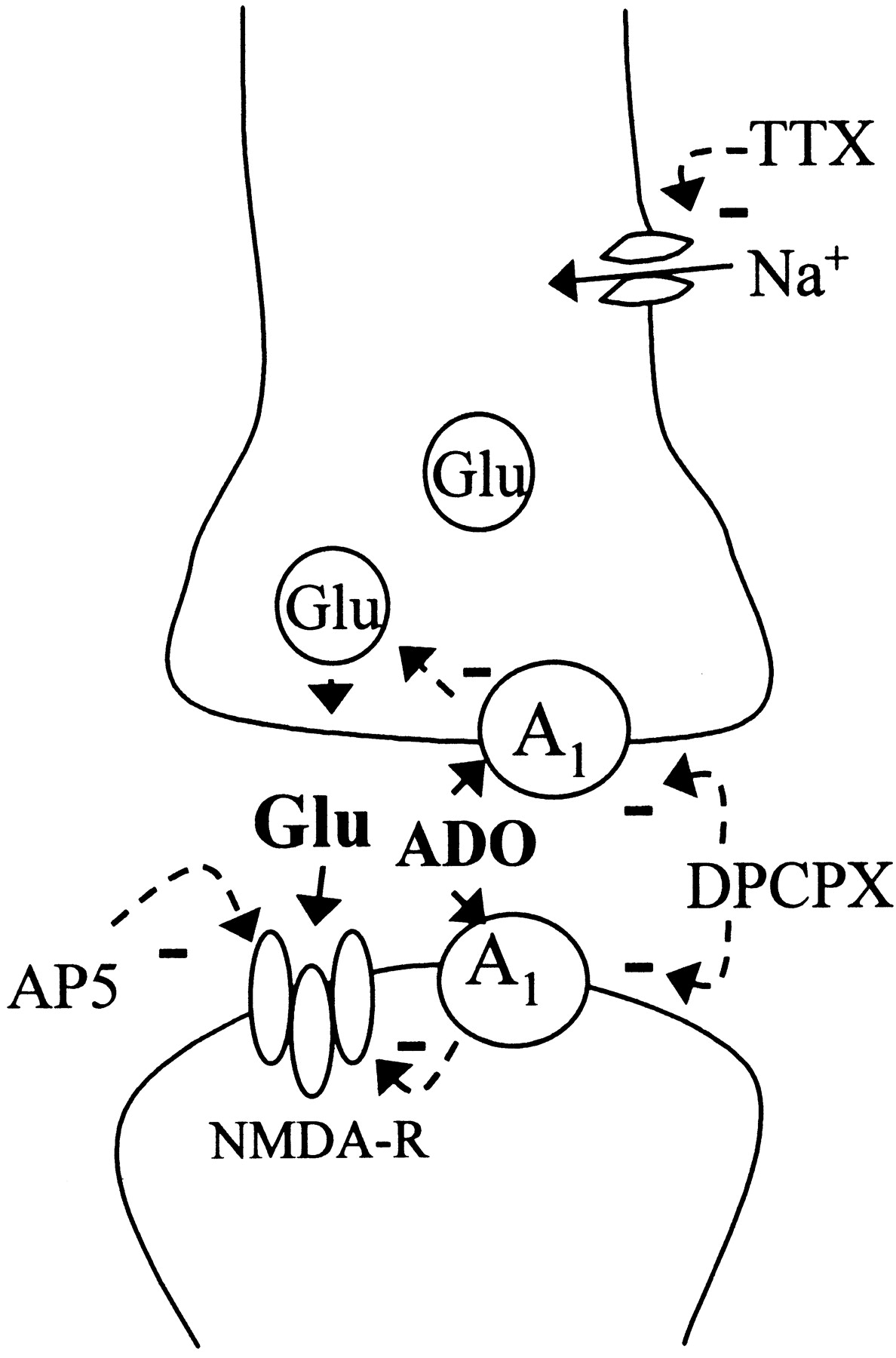
Schematic depiction of the synaptic mechanisms that influence recovery of synaptic transmission after a hypoxic insult. Action potential-dependent release of glutamate (Glu) at the synaptic cleft leads to activation of synaptic NMDA receptors responsible for neuronal damage. By preventing presynaptic action potentials with TTX or absence of stimulation, the synaptic release of glutamate is decreased during hypoxia, leading to less activation of synaptic NMDA receptors. Adenosine (ADO) released during hypoxia activates A1receptors and inhibits synaptic release of glutamate as well as postsynaptic NMDA receptors. When adenosine A1 receptors are blocked with DPCPX, higher synaptic levels of glutamate are attained and more intense activation of synaptic NMDA receptors occurs. AP-5 prevents activation of NMDA receptors and compensates the deleterious action of DPCPX during recovery of synaptic transmission.
Footnotes
This work was supported by Fundação para a Ciência e Tecnologia. We thank Dr. E. Ongini and Shering-Plough Research Institute (Milan, Italy) for the gift of SCH58261, and Dr. W. W. Andersen (University of Bristol, Bristol, UK) for the kind gift of the data analysis (LTP) program. The animal housing facilities of the Institute of Physiology of the Faculty of Medicine of Lisbon are also acknowledged.
Correspondence should be addressed to Ana M. Sebastião, Laboratory of Neurosciences, Faculty of Medicine of Lisbon, Avenida Prof. Egas Moniz, 1649-028 Lisbon, Portugal. E-mail:anaseb{at}neurociencias.pt.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}