

Abstract
Cortical dysplasia is a major cause of intractable epilepsy in children. However, the precise mechanisms linking cortical malformations to epileptogenesis remain elusive. The neuronal-specific activator of cyclin-dependent kinase 5, p35, has been recognized as a key factor in proper neuronal migration in the neocortex. Deletion of p35 leads to severe neocortical lamination defects associated with sporadic lethality and seizures. Here we demonstrate that p35-deficient mice also exhibit dysplasia/ heterotopia of principal neurons in the hippocampal formation, as well as spontaneous behavioral and electrographic seizures. Morphological analyses using immunocytochemistry, electron microscopy, and intracellular labeling reveal a high degree of abnormality in dentate granule cells, including heterotopic localization of granule cells in the molecular layer and hilus, aberrant dendritic orientation, occurrence of basal dendrites, and abnormal axon origination sites. Dentate granule cells of p35-deficient mice also demonstrate aberrant mossy fiber sprouting. Field potential laminar analysis through the dentate molecular layer reflects the dispersion of granule cells and the structural reorganization of this region. Similar patterns of cortical disorganization have been linked to epileptogenesis in animal models of chronic seizures and in human temporal lobe epilepsy. The p35-deficient mouse may therefore offer an experimental system in which we can dissect out the key morphological features that are causally related to epileptogenesis.
Cortical dysplasia, an abnormality of neocortical development associated with disturbed neural migration and/or aberrant neuronal–glial differentation, is recognized as one of the major causes of pediatric epilepsy (Honavar and Meldrum, 1997). Severe forms of cortical dysplasia are often associated with medically intractable seizures, developmental delay, and neurological deficits (Mischel et al., 1995). Misplaced cells (“heterotopia”) are a common feature of cortical dysplasia, which is thought to result from a failure of migration from the site of neuronal genesis to the normal destination of the cells in the cortical plate (Guerrini et al., 1999;Smith et al., 1999; Walsh, 1999). However, mechanisms linking cortical dysplastic malformations to epileptogenesis are still to be elucidated.
Control of the spatial and temporal patterns of neuronal differentiation and migration involves a multitude of factors (e.g., protein molecules, adhesion molecules, channels/receptors, and intracellular cytoskeletal proteins) (for review, see Rakic and Caviness, 1995; Huttenlocher et al., 1995). Defects in developmental processes have been implicated in a large group of genetic disorders associated with cortical dysplasia/heterotopia (Raymond et al., 1995; Barkovich et al., 1996), including early onset epilepsies (Guerrini et al., 1999; Walsh, 1999). To better understand the relationship between these structural malformations and the development of an epileptic state, investigators have turned to animal model systems, including such mouse mutants as reeler (Falconer, 1951;Caviness and Sidman, 1973; Caviness, 1976), Lis-1 (Reiner et al., 1993;Hirotsune et al., 1998; Fleck et al., 2000), doublecortin (Des Portes et al., 1998), and cyclin-dependent kinase 5 (cdk5) (Ohshima et al., 1996; Gilmore et al., 1998) (for review, see Chevassus-au-Louis et al., 1999; Walsh, 1999). Surprisingly, although all of these mutations give rise to disruption of normal cortical development, none is associated with the occurrence of chronic spontaneous seizures.
Recently, a mouse “knock-out” for p35, a neuronal activator of cdk5 that plays an important role in brain development, has been generated (Chae et al., 1997). The protein p35 is highly expressed in postmitotic neurons but completely absent in proliferating neuronal progenitor cells (Tsai et al., 1994; Delalle et al., 1997). Mice with the p35 mutation lack p35/cdk5 kinase activity and exhibit severe cortical lamination disruption similar to that seen in reeler (i.e., an inversion of cortical lamination). In addition, these animals suffer from sporadic adult lethality and spontaneous seizures and display a diminished corpus callosum and abnormal tangential fiber fascicles within the neocortex (Chae et al., 1997; Kwon and Tsai, 1998).
Although the morphological disruptions of the cerebral cortex in p35 knock-outs have been studied in detail (Chae et al., 1997; Kwon and Tsai, 1998; Kwon et al., 1999), much less is known about malformations in hippocampus, and little is understood about the relationship of disturbed neuronal organization to neuronal activity and seizure generation. Here we report that significant structural abnormalities appear in the hippocampus (particularly in dentate gyrus) of p35-deficient mice, similar to pathology observed in resected hippocampi of patients with temporal lobe epilepsy (tLE) (Houser, 1990,1999; El Bahh et al., 1999). Furthermore, a high proportion of p35 knock-out mice exhibits spontaneous behavioral and/or electrographic seizures reminiscent of limbic seizures. These observations provide an initial basis for relating epileptogenesis to dysplastic malformations resulting from errors in brain development.
Preliminary data have been reported previously in abstract form (Wenzel et al., 1998; Robbins et al., 1999).
MATERIALS AND METHODS
Animals
A complete description of the gene deletion protocol and intial characterization of these mice has been published (Chae et al., 1997). Morphological analysis of hippocampal neurons was performed in brains of p35 knock-out mice (p35−/−). The p35−/− colony was maintained via brother–sister matings, and wild-type controls (+/+) were derived from the same stock as originally used to generate −/− breeding pairs (129/Sv × C57BL/6 background). Most of the analysis was performed on 2- to 5-month-old mice; animals at ages 2, 10, and 20 d and 14–15 months were included for comparison. Various electrophysiological and morphological methods were used to analyze seizure-related behaviors and related properties of dentate granule cells. All animal care and use conformed to the NIH Guide for Care and Use of Laboratory Animals and were approved by the Animal Care Committee of the University of Washington.
Observations of seizure behavior and EEG monitoring
Threshold testing. Seizure susceptibility was measured at 60 d postnatally in 15 p35 knock-out and 12 wild-type mice, using the volatile convulsant flurothyl (2,2,2-trifluroethyl ether; Aldrich, Milwaukee, WI) according to well established protocols (Samoriski and Applegate, 1997; Rho et al., 1999). The mice were placed individually in a Plexiglas chamber, and flurothyl was infused into the chamber at a rate of 20 μl/min. Latencies were measured from the beginning of infusion to the onset of a first generalized seizure (clonic) and to a second generalized seizure (tonic–clonic) that follows a refractory postictal period. Shorter latencies reflect greater seizure susceptibility (i.e., lower threshold). Exposure to flurothyl was terminated at the onset of the second seizure. Data were analyzed using the Student's ttest (between-group comparison) using the SigmaStat Program (Jandel Scientific).
Video/EEG monitoring. Using ketamine/xylazine anesthesia, we surgically implanted chronic EEG electrodes in 12 mice between 3 and 5 months of age. Microscrews (stainless steel) served as epidural recording electrodes, and a twisted pair of fine stainless steel wires served as a depth electrode, which was stereotactically implanted into the right hippocampus. Electrodes were attached to a microplug, then cemented to the cranium with dental acrylic. A Telefactor Video/EEG monitoring system (Telefactor, W. Conshohocken, PA) was used to record simultaneously both behavior and EEG from freely moving animals. Each animal was monitored for 40–50 hr over the course of 3–4 weeks.
Tissue preparation for light microscopy/immunocytochemistry
General tissue preparation. Forty-five mice used for light microscopic and immunocytochemical studies were anesthetized with Nembutal (100 mg/kg, i.p.), then perfused with isotonic saline with heparin (500 U/ml saline), followed by a solution of 4% paraformaldehyde (PFA) in 0.1 m sodium phosphate buffer (PB), pH 7.4. For electron microscopy, a fixation solution consisting of 2–4% PFA and 0.1–2% glutaraldehyde was used. The brains were immediately removed and placed in the same fixative for 4 hr at 4° C. After post-fixation, the brains were rinsed in PB, cryoprotected in 10% sucrose in 0.1 m PB for 1 hr, followed by 30% sucrose in 0.1 m PB for 24 hr at 4° C, then frozen on dry ice. Thirty micrometer transverse serial sections were cut on a sliding microtome equipped with a freezing stage, then selected for further processing.
For the quantitative analysis of granule cells, brains were post-fixed for 24 hr, then the hippocampi were isolated and cryoprotected. Before sectioning, hippocampi were slightly extended along the septotemporal axis, then frozen and mounted (oriented transversely to the septotemporal axis) on the freezing stage of a sliding microtome. Serial transverse sections were then cut at 40 μm, collected in PB, and chosen for staining using an unbiased, systematic method (West et al., 1991; Buckmaster and Dudek, 1997); starting from a random position near the septal pole, every 10th section was sampled, yielding an average of 12–15 samples per hippocampus. After they were mounted on slides, sections were stained with cresyl violet. Photomicrographs were taken on 35 mm slide film, scanned, and imported into Adobe Photoshop (Adobe Systems, Mountain View, CA) to assemble the figures.
Timm's histochemistry. The Timm's method for staining heavy metals was used for the detection of synaptic vesicular zinc [particularly enriched in mossy fiber (MF) boutons]. After initial fixation with 4% paraformaldehyde as described above, the brains were transferred to a solution containing 3–4% glutaraldehyde, 0.1% Na2S, and 0.136 mmCaCl2 in 0.12 m Millonig's PB, pH 7.3, for 48 hr at 4° C, followed by cryoprotection in 30% sucrose. Frozen sections were cut at 30 μm and then mounted on slides, air-dried, and transferred to a fresh developer solution containing 30 ml gum Arabic (50%), 5 ml 2 mcitrate buffer, 15 ml hydroquinone (5.76%), and 250 μl silver nitrate (0.73%) for 1 hr in the dark. Sections were counterstained with cresyl violet, dehydrated, cleared in toluene, and coverslipped.
Quantitative analysis of granule cells within the dentate gyrus. The number of granule cells in the granule cell layer, as well as the number of heterotopic granule cells within the molecular layer, were estimated per entire dentate gyrus using the optical fractionator method (West et al., 1991). In Nissl-stained preparations, a granule cell was defined as a neuron with a small cell body and an oval or round nucleus; the Nissl-stained cytoplasm is found at the apical and basal portions of the cell body (Seress and Pokorny, 1981;Wenzel et al., 1981; Seress, 1992). The granule cell and molecular layers also contain the cell bodies of glial cells, basket cells, and other interneurons (for review, see Freund and Buszaki, 1996). The nuclei of glial cells were easily identified and excluded from the counting. The nuclei of basket cells are similar in appearance to those of granule cells, but the cell body of basket cells (and also other interneurons) in these layers is larger and more intensely stained for Nissl substance. On the basis of these criteria, nongranule cells could be identified and excluded from the counting.
Cell counting was performed by an investigator who was blind to the genotype of the animals. Total section thickness was used for dissector height, and only “caps” located within counting frames were counted (Buckmaster and Dudek, 1997); caps were defined as the nuclei of granule cells that came into focus while focusing down through the dissector height. Counting frames (10 × 10 μm) were distributed systematically and randomly over the dentate granule cell layer, according to the method described by West et al. (1991). Using a camera lucida-like microscope/computer interface (Nikon Optiphot-2 Microscope with a Cohu CCD Camera; Nikon, Tokyo, Japan) and an NIH Image 1.62 b4 morphometry software package, the numerical density of granule cells, the volume of the granule cell and molecular layers, and the total number of granule cells for a given hippocampus were estimated. Adequacy of sampling was assessed by determining the intra-animal coefficient of variation (CE) as well as the inter-animal coefficient of variation (CV) for each measure (West and Gundersen, 1990).
Immunocytochemistry. Different sets of sections were processed for immunocytochemistry (ICC), using a modification of the avidin–biotin complex (ABC)-peroxidase technique (Hsu et al., 1981). Immunocytochemical procedures were performed as described previously (Wenzel et al., 1997). Briefly, sections were rinsed in PB, followed by 0.1 m Tris-HCL buffer (TB), pH 7.4; endogenous peroxidases were then inactivated by treatment with 0.5–1% H2O2 in TB for 2 hr. Sections were treated with 3% bovine serum albumin (BSA) (Boehringer Mannheim, Indianapolis, IN), 3% goat or horse serum (Sigma, St. Louis, MO), and 0.25% Triton X-100 (TX) in 0.05m TB, 0.15 m NaCl, pH 7.4 (TBS) for 1 hr to reduce nonspecific staining. Sets of alternating sections were rinsed in TBS for 30 min and incubated for 24 hr at 4°C in the various antisera and dilutions in TBS containing 1% goat or horse serum, 2% BSA, and 0.25% TX: anti-neuron-specific nuclear protein (NeuN) (Chemicon, Temecula, CA), 1:1000; anti-microtubule-associated protein-2 (Boehringer Mannheim), 1:500; anti-glial fibrillary acidic protein (GFAP) (Dako Corporation, Carpinteria, CA) 1:4000; anti-glutamate decarboxylase 67 (GAD67) (Chemicon, Temecula, CA), 1:100; anti-calretinin (Chemicon), 1:4000; anti-parvalbumin (Chemicon), 1:4000; anti-somatostatin (SOM) (Peninsula Laboratories, Belmont, CA), 1:5000; and anti-zinc transporter ZnT3 (provided by Dr. R. D. Palmiter, University of Washington, Seattle, WA), 1:250. After rinses for 2 hr in TBS, sections were incubated in biotinylated goat anti-rabbit IgG or horse anti-mouse IgG (Vector Laboratories, Burlingame, CA), diluted 1:500 for 24 hr at 4°C, rinsed for 2 hr in TBS, and then incubated in ABC (Elite ABC Kit, Vector Laboratories), diluted 1:500 in 1% goat or horse serum, 2% BSA, 0.25% TX, and TBS for 24 hr at 4° C. Sections were rinsed thoroughly in TB, pH 7.6, and then incubated for 15 min in 0.025% 3,3′-diaminobenzidine (DAB; Sigma) in TB. After reaction for 5–10 min in fresh DAB with 0.003% H2O2, sections were rinsed in TB, followed by PB. Specificity of the immunostaining was evaluated by omitting primary antibodies from the regular staining sequence. ZnT3 immunoreactivity in synaptic vesicles of mossy fiber boutons was also localized at the ultrastuctural level using a protocol described byWenzel et al. (1997).
Electrophysiology and intracellular labeling with biocytin
Intracellular recording and labeling. Hippocampal slices were prepared conventionally as described previously (Wenzel et al., 2000) for in vitro experiments. Mice were anesthetized with halothane and decapitated, and the brain was removed quickly, cooled briefly in ice-cold oxygenated [95% O2/5% CO2] artificial CSF (ACSF) containing (in mm): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaCO3, 2 CaCl2, 10 dextrose, and blocked to contain the hippocampus. Using a vibroslicer, 400-μm-thick slices transverse to the longitudinal axis of the hippocampus were cut into a bath of oxygenated ACSF at 4°C. Sections were then transferred to a holding chamber and allowed to equilibrate for at least 1 hr while submerged in ACSF at room temperature. Slices were then individually transferred to a standard interface recording chamber. In the chamber, slices rested on a nylon mesh over a well that was perfused (1 ml/min) with warmed (33–35°C), oxygenated ACSF. In addition, warmed, humidified air was circulated above the slice. Slices remained undisturbed for at least 15 min in the chamber before recording began.
Intracellular electrodes made from borosilicate glass were pulled using a horizontal puller (Sutter Instruments, San Rafael, CA) and filled with 2% biocytin (Molecular Probes, Eugene, OR) dissolved in 1m potassium acetate (80–230 MΩ resistance, pH 7.4). Intracellular potentials were recorded using an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA). Neurons within the granule cell layer were impaled, and biocytin was iontophoretically injected with 700 msec duration and 0.5–1.0 nA hyperpolarizing current pulses delivered every second for 10–30 min. After biocytin injection, the electrode was withdrawn, and the slice was left in the chamber for at least 30 min before removal for fixation.
Retrograde labeling with biocytin. Visualization of multiple dentate granule cells was achieved through retrograde labeling via iontophoresis of 4% biocytin (in 0.05 m Tris HCl, pH 7.3) into extracellular space of hippocampal stratum (s.) lucidum in CA3b subfield (Okazaki et al., 1995). The microelectrode (5–10 μm) was placed ∼200 μm below the surface of the slice, then 600 nA positive current pulses (7 sec on/7 sec off) were passed for 18–20 min. Slices remained incubating in the chamber for an additional 3 hr, then were fixed in 4% paraformaldehyde and further processed as described below.
Extracellular field recordings. Field EPSPs (fEPSPs) were recorded in the dentate gyrus of both p35−/− and wild-type mice with extracellular electrodes (filled with ACSF, 5–10 MΩ resistance) in response to perforant path stimulation. A laminar profile of evoked fEPSPs was constructed by recording sequentially from several sites through the molecular layer, perpendicular to the granule cell layer (superior blade of the dentate gyrus).
Tissue processing and morphological analysis of biocytin-labeled granule cells (light and electron microscopy). After the intracellular recording procedure and iontophoretic injection of biocytin, slices were removed from the recording chamber and immersion-fixed in a solution of 4% paraformaldehyde and 0.1–1% glutaraldehyde in 0.1 m sodium PB, pH 7.4, for 2–4 hr at 4°C. The slices were rinsed in 0.1 mPB, then infiltrated for cryoprotection with 10% sucrose in 0.1m PB for 1 hr, followed by 30% sucrose for 8–12 hr. Frozen sections were cut (60 μm) and further processed with a histochemical procedure. Sections of a total of 99 slices with biocytin-filled granule cells were subsequently processed for light and electron microscopy, as follows.
Sections were rinsed in 0.1 m PB, pH 7.4, and then in 0.1m TB, pH 7.4. Endogenous peroxidases were suppressed with 0.5–1% H2O2 in 0.1m TB for 2 hr. Sections were pretreated with 2% BSA, 0.25–0.4% dimethylsulfoxide (DMSO) (Sigma), and 0.05 mTBS, pH 7.4, for 1 hr to reduce nonspecific background staining and to permeabilize membranes. Sections were rinsed in 0.1 m TBS for 30 min and then incubated in ABC (Elite ABC Kit, Vector), diluted 1:500 in 2% BSA, 0.25–0.4% DMSO, and 0.05 m TBS for 36–48 hr at 4°C. Sections were then rinsed throughly in 0.1m TBS followed by 0.1 m TB, pH 7.6, and preincubated in 0.025% DAB with 0.005% NiNH4SO4 added to increase the density of stain for 15 min. Subsequently, the sections were reacted with fresh DAB/NiNH4SO4 solution containing 0.01% H2O2 for 15–60 min. The reaction was stopped by rinses in 0.1 m TB. Sections were further processed for electron microscopy (EM) using a method that included post-fixation in 1% osmium tetroxide in 0.15m PB, pH 7.4, for 1 hr at room temperature, alcohol dehydration, and flat-embedding in Eponate 12 Resin (Ted Pella, Redding, CA) between two aclar sheets. The biocytin-filled granule cells (and their dendrites and axon arborizations) were visualized at the light microscopical level and photographed before remounting and further sectioning for EM. Camera lucida drawings were made of each biocytin-filled granule cell, and its dendrites and axon arborizations were reconstructed by superimposing all sections of the hippocampal slice. Serial thin sections from various slices containing different portions of the biocytin-filled granule cells (e.g., mossy fiber axon collaterals in the dentate inner molecular layer) were stained with uranyl acetate and Reynold's lead citrate and examined on a Philips 410 electron microscope.
RESULTS
The gene-targeting strategy and generation of p35−/− mice have been reported previously (Chae et al., 1997). The initial histological study demonstrated a general disorganization of the forebrain in these mice, with a major defect in the development of the normal lamination pattern of the neocortex (Kwon and Tsai, 1998). In the present study, histological examination of the neocortex of p35−/− mice reconfirmed this observation of severe defects in the lamination of neocortex, associated with formation of aberrant fiber fascicles. Our observations also revealed structural abnormalities within the hippocampus, particularly associated with the appearance of dispersed granule cells and abnormal mossy fiber localization. We have focused on these hippocampal abnormalities and their functional correlations, with respect to the occurrence of spontaneous seizures.
P35−/− mutation is associated with low seizure threshold and with occurrence of spontaneous electrographic and behavioral seizures
Seizure susceptibility was compared in p35−/− and wild-type mice through flurothyl seizure testing. In all animals, exposure to flurothyl elicited seizure activity. Statistical analysis of the latencies to the flurothyl-induced seizures revealed that the p35−/− mice had significantly shorter latencies than wild-type to both the first (352.2 ± 1.6 vs 423.7 ± 12.0 sec, respectively) and second seizures (590.6 ± 14.3 vs 688.4 ± 13.8 sec) (Fig.1A). All statistical data are presented as means ± SEM. These results represent a 16% reduction in mean latency to the first seizure and a 14% reduction to the second seizure in p35 −/− mice.
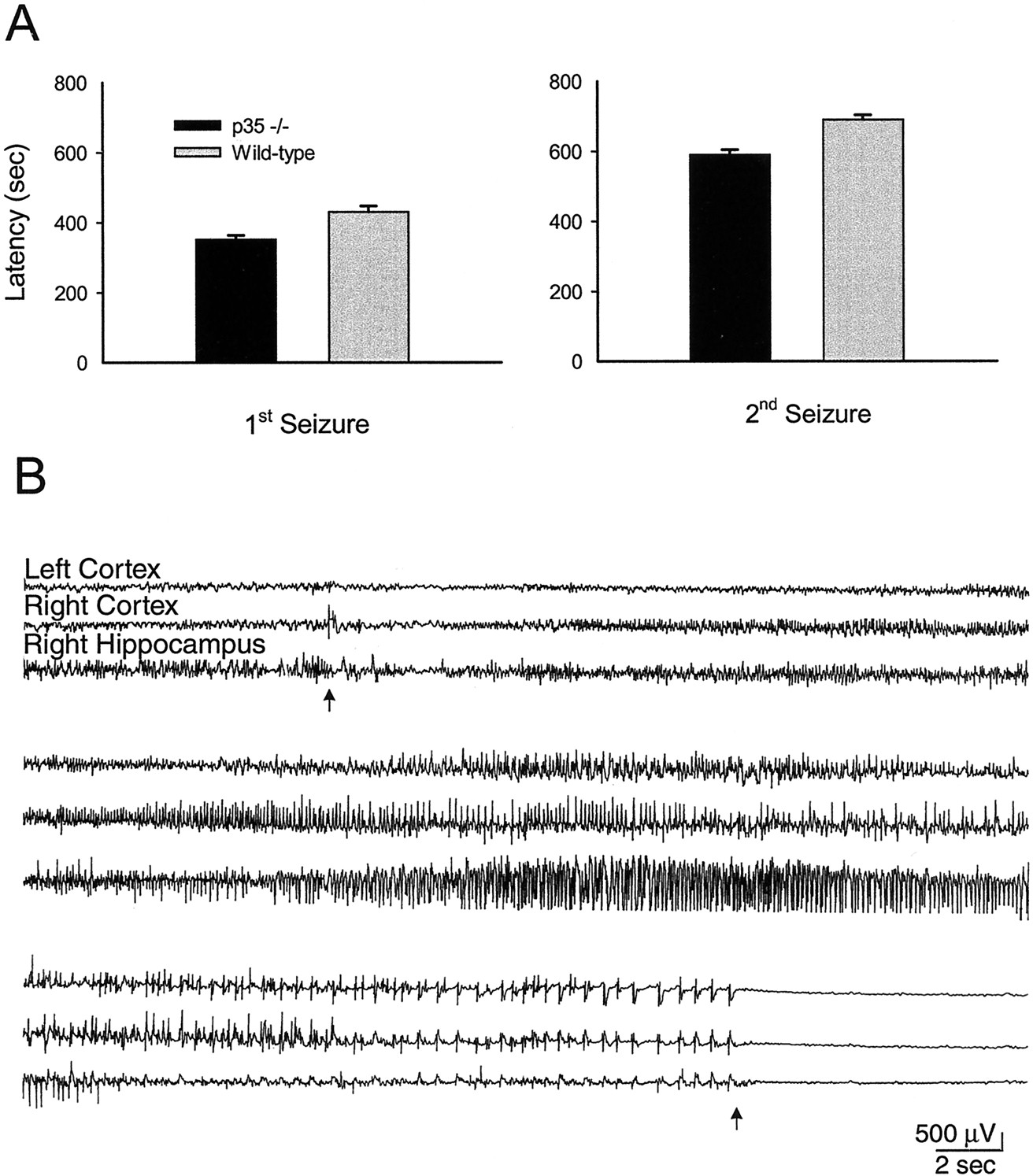
A, Flurothyl seizure threshold testing demonstrated significantly shorter latencies, to both first and second seizures, in p35 knock-out mice in comparison with control (wild-type) mice. B, Representative EEG recording from a p35−/− mouse during a spontaneous tonic–clonic seizure. Ictal episodes often began (top arrow) with a decrease in EEG voltage, followed by fast spiking activity that increased in amplitude and proceeded into a clonic pattern; seizure activity was followed by an isoelectric postictal period (starting at bottom arrow). The top two traces were recorded epidurally from the left and right cortex, respectively; thebottom trace was recorded with a depth electrode in the right hippocampus.
Video/EEG recording confirmed the occurrence of spontaneous seizures in many p35−/− mice. A typical example of EEG recorded from a p35−/− mouse during a tonic–clonic seizure is shown in Figure1B. Spontaneous epileptiform EEG activity was verified in 75% of the p35−/− mice (8 of 12 animals). Twenty-five percent of p35−/− animals exhibited spontaneous tonic–clonic seizures with behavioral manifestations, whereas an additional 50% displayed intermittent interictal electrographic activity. The spontaneous seizures had durations ranging from 59 to 84 sec and were followed by a prolonged isoelectric postictal period lasting 3–5 min. Ten percent of the p35−/− mice died during seizure. No epileptiform activity was observed either behaviorally or electrographically in wild-type mice.
Most of the recorded spontaneous seizures were strong generalized tonic–clonic episodes that appeared to occur simultaneously in cortex and hippocampus. Seizures frequently began from a state of sleep or quiet rest, with a sudden decrease in amplitude of the EEG, sometimes preceded by a spike corresponding to the animal awakening; the mouse then lifted and turned its head back and forth. As the seizure evolved, both electrographic and behavioral activity reflected the tonic phase of a seizure; spiking gradually increased in amplitude and frequency, and the animal's head turned rigidly upward and back concurrent with bilateral forelimb extension. This tonic phase was frequently prolonged and usually progressed into a clonic EEG pattern. The animal's behavior often progressed into vigorous bouncing, produced by rapid bilateral hindlimb thrusting. The seizure ended with the onset of a protracted isoelectric post-ictal period; behavioral inactivity was seen when the cortical EEG flattened. Occasionally the hippocampus would continue to spike for several seconds longer than neocortex.
Histological abnormalities are characteristic features of the hippocampus in p35−/− mice
Histological examination of the neocortex revealed striking differences between wild-type and p35−/− mice. Figure2, A1 and A2, shows the typical lamination of the wild-type neocortex with six layers distinguished by neuronal morphology, cell density, and general cytoarchitecture. This normally laminated cytoarchitecture is not apparent in p35−/− mice (Fig. 2B1,B2). With the exception of lamina I containing a few Cajal-Retzius cells, laminas II–VI represent an inversion of the normal neuronal patterning; i.e., large pyramidal neurons normally localized in lamina V are now present underneath lamina I, and small pyramidal cells are distributed in the deep cortical zone [further details are described by Chae et al. (1997)]. In addition, aberrant fiber fascicles course through the neocortex, leading to further interruptions in the cytoarchitecture.

A1, B1, Coronal brain sections from wild-type (A1) and p35−/− knock-out (B1) mice, stained with cresyl violet. Neocortex (N), hippocampus (H), and dentate gyrus (DG) are indicated. A2, Higher magnification of neocortex and hippocampus from a wild-type mouse shows normal neocortical laminae (I–VI) and hippocampal subregionsCA1 and CA3 with s. oriens (OR) and radiatum (RAD), pyramidal cell layer (PCL), molecular layer (ML), and dentate gyrus (DG) with hilus (H), granule cell layer (GCL), and molecular layer (ML).B2, Coronal section of neocortex and hippocampus of a p35−/− mouse demonstrates heterotopic pyramidal neurons in CA1/CA3 (arrows) and dispersed dentate granule cells localized within the inner molecular layer (ML;arrows). Note the abnormal cortical lamination, the presence of aberrant fiber fascicles within the neocortex (arrowheads), and the absence of callosal fibers crossing the midline (white arrow). Scale bars:A1, B1, 1000 μm; A2,B2, 300 μm.
The most striking morphological finding in the hippocampal formation of adult p35−/− mice was a severe disarrangement of its neuronal architecture (Figs. 2, 3). The principal cell layers (CA1–3 pyramidal and dentate granule cell layers) are disorganized and show frequent interruptions and aberrantly localized cell somata (Figs. 2B2,3B1,B2,C1,C2). In particular, pyramidal cells of the medial CA1 subfield and subiculum tend to be dispersed into s. oriens, where they form a distinct cell layer (Fig. 2B2). In the CA3 subfield, pyramidal neurons are found outside the pyramidal cell layer, in both s. oriens and s. lucidum/radiatum (Figs. 2B2,3C1,D,E). To determine the neuronal nature of these heterotopic cells, the neuron-specific NeuN antibody was used. In hippocampal sections immunoreacted against NeuN, a large proportion of the heterotopic cell population was neuronal.

Transverse sections (cresyl violet) of the hippocampus (A1) and dentate gyrus (A2) of the wild-type mouse. B1, B2, Transverse sections of the dorsal hippocampus of a p35−/− mouse demonstrate heterotopic CA3 pramidal cells (arrows) and dispersed granule cells within the dentate inner and middle molecular layer (IML and MML) (arrows). C1, C2, Transverse sections of the ventral hippocampus of a p35−/− mouse show extensive granule cell dispersion in the dentate gyrus (arrows) and a gap within the CA3 pyramidal cell layer (arrow). Higher magnification of the granule cell layer (C2) shows displaced cells in both the molecular layer and the hilus. D, E, Comparison of the CA3 subfield of wild-type and p35−/− mice, showing pyramidal neurons heterotopically localized within s. oriens (OR) and lucidum/radiatum (LU, RAD) (arrows). Scale bars:A1–C1, 400 μm;A2–D2, 100 μm.
Most p35−/− mice also show a severe disorganization of the granule cell layer, particularly affecting the superior blade. Granule cells are dispersed and heterotopically localized into the molecular layer and hilus, blurring the borders of the granule cell layer with adjacent layers or forming separate cell clusters deep in the hilus (Fig.3B2,C2,4A). Although no statistical comparison was performed, there appeared to be a strikingly higher degree of granule cell dispersion into the molecular layer (and particularly into the hilus) in the ventral than in the dorsal hippocampus (Fig. 3, compare B1–2, C1–2). NeuN ICC confirmed the neuronal nature of these cells, and their shape suggested that most of these neurons are displaced dentate granule cells (Fig. 4B).

Transverse sections of the dentate gyrus from p35−/− mice, showing cresyl violet staining (A) and NeuN immunoreactivity (B). Most of the dispersed cells within the inner molecular layer (IML) are NeuN-positive. C, D, Timm's histochemistry for zinc in mossy fiber boutons shows the normal staining pattern in the dentate hilus (H) of a wild-type mouse (C). In p35−/− mice (D), Timm stain can be seen in the hilus (H), the granule cell layer (GCL), and inner and middle molecular layers (IML and MML), indicating the abnormal distribution of mossy fibers associated with heterotopically dispersed granule cells (arrows). E, Immunocytochemistry for GFAP shows a relatively normal astrocytic border between hilus and granule cell layer of the inferior blade (A, arrows). In contrast, note the absence of GFAP-positive astrocytes at the hilus/granule cell layer border in the superior dentate blade.F1–F3, Sections immunoreacted against calretinin antibody show normal distribution of the calretinin-positive fiber plexus (CRP) within the IML of the wild-type mouse (inset F2) and, in p35−/− mice (F1 andinset F3), dispersed granule cells (dGC) in the IML surrounded by the CRP. G, H, Photomicrographs show parvalbumin immunoreactivity in dentate interneurons (arrows) and the axonal plexus (arrowheads) in the wild-type (G1,inset G2) and p35−/− mice (H1,inset H2). There is dramatically less parvalbumin immunoreactivity in the axonal plexus surrounding the granule cells in p35−/− mice (H2). Scale bars: A,D, E, F1, 100 μm;B, C, G1,H1, 200 μm; F2–3, G2, 50 μm.
To further characterize the dentate region of p35−/− mice, Timm staining was used to label zinc in synaptic vesicles of MF boutons. In the wild-type (Fig. 4C), Timm-stained MF axons normally project from the granule cell layer throughout the hilus toward the CA3 pyramidal cell layer, forming a compact projection in s. lucidum; in addition, MF axon bundles are also localized infrapyramidally and in the pyramidal cell layer. Timm staining in p35−/− mice shows a similar pattern of MF projection in hilus and CA3 subfield (Fig.4D). However, in contrast to the wild-type, numerous Timm-stained MF boutons/axons are also present in the granule cell layer and particularly within the inner molecular layer (Fig.4D, arrows). Although there was significant animal-to-animal variability in the degree of granule cell dispersion and MF distribution in granule cell and molecular layers, these abnormal patterns were typical of p35−/− mice and not seen in wild-type animals.
Analysis of granule cell dispersion
Quantitative cell counts
To assess the extent of granule cell dispersion in the dentate gyrus of p35−/− mice, we analyzed the following histological parameters from 2-month-old p35−/− mice and age-matched wild-type mice: (1) numbers of granule cells and (2) volumes of granule cell and molecular layers; from these measures, we calculated the numerical density of granule cells. Each measure was estimated separately for the granule cell and molecular layers. Because in p35−/− mice the border between granule cell layer (GCL) and inner molecular layer is irregular, the distinction between granule cell and molecular layers was artificially (but consistently) defined in each section. The data (Table 1, Fig.5A,C) revealed small but nonsignificant differences between p35−/− and wild-type mice for the total number of granule cells per dentate (Fig.5C) and the number of granule cells in the granule cell layer (Fig. 5A). However, p35−/− mice do show significantly more granule cells in the molecular layer than wild-type mice (Fig. 5B). Approximately 29.8% of all granule cells in p35−/− mice are localized within the molecular layer (compared with 14.4% in the wild-type). Statistical comparison of the mean volume of the dentate layers revealed a difference between p35−/− mice and wild-type mice: molecular layer and the total value of the granule cell plus molecular layers of p35−/− animals are significant smaller (p < 0.016 andp < 0.03, respectively), but the difference in the granule cell layer is not significant (p < 0.44). However, the numerical density of granule cells in the molecular layer, the dispersed granule cells, is significantly higher in p35−/− mice compared with the wild-type. The intra-animal CE for the number of cells counted and points counted (see Materials and Methods), as well as the between-subjects CV and CE2/CV2ratio, were in the range that indicated adequate sampling (West et al., 1991).
Total number and numerical density of granule cells (GCs), and volume of granule cell layer (GCL) and molecular layer (ML), in the dentate gyrus of 2-month-old wild-type and p35−/− mice

Graphs show the numbers of granule cells counted in the dentate granule cell and molecular layers per hippocampus of the wild-type and p35−/− mice at 2 months of age. A, The number of granule cells within the granule cell layer per entire hippocampus. The granule cell number is slightly greater (nonsignificantly) in wild-type mice than in p35−/− animals.B, The number of heterotopic granule cells within the molecular layer. In p35−/− mice, ∼30% of the total number of granule cells is heterotopically localized within the molecular layer, compared with ∼14% in wild-type mice (*p < 0.001). C, The total number of granule cells per hippocampus. There is no significant difference in the total number of granule cells between wild-type and p35−/− mice. Although the number of dispersed granule cells is significantly greater in p35−/− mice (+125% in the molecular layer), the absolute number of displaced neurons is relatively small compared with the number of cells in the granule cell layer. Thus, there is no statistical difference when total numbers are compared.
Field potential analysis
A laminar profile of synaptic responses, recorded throughout the granule cell and molecular layers of the superior blade of the dentate gyrus, revealed clear differences between p35−/− and wild-type mice (Fig. 6). The amplitude of the evoked fEPSPs was measured at a fixed latency (Fig. 6A) and plotted as a function of location along a model granule soma–dendrite axis (Fig. 6C). Consistent with the morphological findings of a dispersed granule cell layer in p35−/− animals, the fEPSP positivity of p35−/− dentate evoked by perforant path stimulation was much broader in slices from p35−/− mice (Fig. 6B). This positivity is thought to reflect a “somal” current source [see Sutula et al. (1998) for similar analysis of “sprouted” dentate], suggesting that the heterotopic granule cells are activated by incoming perforant path fibers.

A, Field EPSPs evoked by stimulation in the dentate molecular layer were recorded from sites in the superior blade of the dentate gyrus in wild-type and p35−/− mice.B, The amplitude of these EPSPs was measured at a fixed latency [peak of EPSP recorded in the granule cell layer (GCL)] and plotted as a function of recording location along an axis perpendicular to the GCL (wild-type, ●; p35−/−, ▾).C, Schematic drawing showing location of normally placed granule cells (solid ovals) and dispersed granule cells (dotted ovals). IML, Inner molecular layer; MML, middle molecular layer; OML, outer molecular layer.
Immunocytochemistry of astrocytes and interneurons reveals cell-specific differences between p35−/− and wild-type mice
ICC techniques were used to examine the expression of several markers specific for astrocytes and inhibitory interneurons (Fig.4E–H). Using an antibody against GFAP (Fig. 4E), we found no obvious differences in distribution and density of GFAP-positive cells within the hippocampus proper containing heterotopic neurons. However, in the dentate gyrus, a characteristic population of astrocytes that is normally localized along the hilar–granule cell border was absent in p35−/− mice; furthermore, occasional aberrant astrocyte somata were seen between the granule cells.
The distribution of hippocampal inhibitory interneurons in p35−/− hippocampus was examined using an antibody against GAD67 (synthesizing enzyme of the inhibitory neurotransmitter GABA) to stain all GABAergic neurons. In addition, we used a set of antibodies—calretinin, parvalbumin, and somatostatin—that were previously associated with various GABAergic subpopulations of interneurons. The pattern of GAD67-positive neuron distribution in p35−/− and wild-type mice was similar to that seen in other ICC and in in situhybridization studies on rodents, including mice (data not shown) (for review, see Houser and Esclapez, 1994; Freund and Buszaki, 1996; Fukuda et al., 1997). In both p35−/− and wild-type mice, GAD immunoreactive neurons were found in the hippocampal pyramidal cell layer, s. oriens and s. radiatum/moleculare. In the dentate, GAD-positive cells were seen in the dentate hilus, in the granule cell layer, and scattered in the molecular layer. GAD67 reveals consistent “punctate” (presumed axon terminals) immunostaining surrounding somata of principal cells, including the dispersed granule cells in the dentate molecular layer (data not shown). This expanded region of GABA-containing terminals surrounding granule cells constitutes the major difference in the pattern of GAD67 immunoreactivity distribution between p35−/− and wild-type animals.
Immunostaining for SOM revealed a large number of neurons in all subfields of the hippocampus and dentate gyrus, with laminar distribution consistent with previous studies (data not shown) (for review, see Buckmaster et al., 1994; Freund and Buszaki, 1996). In agreement with these previous studies on mouse, we found only light labeling of SOM-positive axons in the outer two-thirds of the dentate molecular layer (compared with a dense axon and terminal plexus in rats). There were no apparent differences in the patterns of distribution and intensity of SOM staining between p35−/− and wild-type mice.
Immunostaining for calretinin identifies another subpopulation of interneurons in all hippocampal subfields and strata (for review, seeFreund and Buszaki, 1996). In addition, in the hilus of the ventral dentate gyrus of wild-type and p35−/− animals, calretinin immunoreactivity labeled the somata and processes of mossy cells (data not shown) as reported previously by Liu et al. (1996). Axon terminals of these mossy cells form a dense supragranular-IR band in the inner molecular layer in both dorsal (Fig. 4F) and ventral dentate gyrus. In p35−/− mice, this supragranular calretinin-positive band appeared more prominent than in wild-type mice and overlapped with the dispersed granule cells in this layer.
In the hippocampus and dentate gyrus, parvalbumin ICC shows parvalbumin positivity in interneuron somata, dendrites, and axon terminals. Parvalbumin-containing cell bodies are mainly localized in the pyramidal cell layer and s. oriens of the hippocampus, in the granule cell layer and hilus of the dentate gyrus of wild-type mice (Fig.4G), and only occasionally in other layers (e.g., s. lucidum of the CA3 region). The dendrites of parvalbumin-positive neurons arise parallel to the granule cell dendrites and exhibit prominent varicosities spanning all layers of the molecular layer. In both p35−/− and wild-type mice, the axons and terminals immunoreactive for parvalbumin form a dense axon plexus within the cell layers, surrounding pyramidal and granule cells, including the dispersed granule cells within the dentate molecular layer of p35−/− mice. Although no systematic cell counting was performed, the number of parvalbumin-containing neurons appears to be reduced in p35−/− mice as compared with the wild-type; furthermore, the intensity of the parvalbumin staining of the axon plexus and terminals in the dentate granule cell layer was weaker in p35−/− mice (Fig.4G,H).
Electron microscopy of mossy fiber connections
Electron microscopic examination of the granule cells in p35−/− mice localized in granule cell and molecular layers showed ultrastructural characteristics similar to the wild-type and those described previously for dentate granule cells (Laatsch and Cowan, 1966; Ribak and Anderson, 1980; Wenzel et al., 1981; Seress, 1992). Despite the variable granule cell localization (Fig.7A) and dendritic orientation in p35−/− mice (see next section), dendrites from both genotypes are covered with spines with a broad range of shape, size, and length. Dendrites and dendritic spines form simple and multiple asymmetric synaptic contacts with axon terminals. In p35−/− dentate, MFs form a widely distributed axon plexus around granule cell somata and proximal dendrites, in both the granule cell and molecular layers; these axons are associated with numerous terminals (i.e., MF boutons) that vary considerably in size and shape (Fig.5B–E). The identity of terminals as MF boutons is confirmed by their ultrastructural features (packed with clear round synaptic vesicles, occasional dense-core vesicles, a few coated vesicles, and numerous mitochondria) and EM localization of ZnT3 IR of synaptic vesicle membranes in these terminals (identical to those seen in hilus and CA3 s. lucidum). The MF boutons, identified by ZnT3 immunoreactivity in both granule cell (Figs.7D,E) and molecular layers (Figs.7B,C), form asymmetric synapses predominantly with multiple simple dendritic spines (Fig.7B); some MF boutons also form synapses with complex spines and with dendrites of presumed granule cells (Figs.7B,E). Occasionally, MF boutons were found making synapses with the soma of heterotopic granule cells and aspiny dendrites of interneurons (data not shown). The EM observation on ZnT3-positive MF boutons abnormally localized in the molecular layer (Fig. 7C) is consistent with the Timm staining pattern of p35−/− dentate and was not observed in wild-type mice. However, there is some Timm labeling (seen at the light microscopic level) within the granule cell layer of wild-type mice, suggesting that even in normal dentate there may be some MF synapses in this region.

Electron microscopy of mossy fiber boutons (MFB) heterotopically localized within the granule cell layer (GCL) and inner molecular layer (IML) of p35−/− mice. MFBs are identified with ZnT3 immunocytochemistry (black DAB-nickel reaction product inC and E). A, Low-power magnification of the dentate gyrus (superior blade) with heterotopic GCs within the IML. B, In the IML (indicated bytop box and arrow inA), an MFB forms asymmetric synapses with spines (S) of granule cells. C, A ZnT3-positive MFB makes an asymmetric synapse with a simple spine.D, E, In the granule cell layer (indicated by bottom box and arrow), a nonlabeled (D) and ZnT3-positive (E) MFB make asymmetric synapses with spines (S) and dendrites (D) of presumed granule cells. Scale bars: B–E, 0.25 μm.
Intracellular labeling with biocytin reveals abnormal patterns of granule cell localization, axonal and dendritic morphology, and mossy fiber reorganization
Light microscopic examination of individual granule cells obtained after in vitro injection of biocytin into the s. lucidum of hippocampal slices, or intracellular labeling with biocytin, revealed a high degree of granule cell abnormality in p35−/− dentate with respect to the localization of granule cell somata and in the orientation of their dendritic fields and axon arbors (Figs.8, 9). Granule cell somata of wild-type mice have oval or round-shaped cell bodies, typically giving rise to a spiny apical dendritic tree that arborizes within the molecular layer; the mossy fiber axon arises from the hilar side of the soma and runs toward the CA3 subfield. As shown with Golgi and recent intracellular labeling studies (Spigelman et al., 1998; Ribak et al., 2000), our examination of granule cells of wild-type mice revealed only exceptional abnormalities. In contrast, as shown in Figure 8, granule cells of p35−/− mice are often displaced, with cell bodies located either in the inner molecular zone (close to the GCL) or in clusters within the hilus. Normal-appearing granule cells in p35−/− dentate show the expected monopolar-oriented dendritic branching pattern within the molecular layer (Fig.8A1,B, cell 1). However, many granule cells exhibit a bipolar dendritic arborization (Fig. 8, cells2, 3, 5; Fig.9B1–2,C1–2). The spiny dendrites of some p35−/− granule cells arose from the hilar side of the cell body and then turned 180° to form “recurrent” basal dendrites ascending through the molecular layer (Fig. 8, cell 2; Fig.9C); some dendrites originating from the hilar pole of the cell body also extend into the subgranular hilar region (Fig.8B, cell 5; Fig.9D1-2). Granule cells heterotopically localized within the hilus gave rise to one or two primary dendrites that ran through the hilus toward the granule cell layer to branch within both granule cell and molecular layers (Fig. 8, cell 4; Fig.9B).

Photomicrographs (A) and camera lucida drawings (B) of biocytin-filled granule cells from a p35−/− mouse. Granule cells and mossy fibers were visualized after terminal uptake and retrograde transport of biocytin, after in vitro injection of biocytin into the s. lucidum of a hippocampal slice. A1, Biocytin-labeled granule cell bodies (1-5) are present in the granule cell layer (GCL), inner molecular layer (IML), and hilus (H).A2, Photomicrograph of a biocytin-labeled granule cell (5) with basal dendrites (arrows) in an adjacent section of the same slice. A3, Higher magnification of the same granule cell showing a spiny dendrite (arrows) and the mossy fiber axon (arrowhead). B, Drawings of the biocytin-labeled granule cells (GCs) shown in A, demonstrating abnormal morphology with respect to location and orientation of the cell body, and dendrites and axon arborization: neuron 1, normal-appearing GC; neuron 2, inverted GC with recurrent basal dendrites and an axon (MF, arrowhead) originating from the basal dendrite; neuron 3, GC with dendrites emerging from the apical and hilar pole of the soma, and axon (MF, arrowhead) arising from a dendrite; neurons 4, heterotopic GCs within the hilus (H), with dendrites extending into the molecular layer; neuron 5, GC with basal dendrites branching and extending into the hilus. Dashed lineindicates border between “normal” GCL and IML.

Photomicrographs (A1–D1) and camera lucida drawings (A2–D2) of single biocytin-filled granule cells in wild-type (A1, A2) and p35−/− mice (B1–D2). Hippocampal subregions CA3, dentate hilus (H), granule cell layer (GCL), and inner (IML), middle (MML), and outer (OML) molecular layer are indicated. A1, A2, Photomicrograph and drawing show the regular dendritic pattern and axonal arborization of a dentate granule cell (GC) within the GCL from a wild-type mouse; an arrowhead indicates mossy fiber axon (MF). B1, B2, Heterotopically localized GC in the dentate hilus that gives rise to two apical dendrites that extend into the GCL/IML and branch within the ML. Note the extensive axon arborization (MF) within the hilus.C1, C2, GC with abnormal dendritic and axonal morphology. Dendrites emerge from the apical and hilar pole of the GC body and extend into the ML. A recurrent hilar axon collateral (arrows) arborizes within the GCL, indicating MF sprouting (reorganization). D1, D2, Biocytin-filled GC with abnormal apical dendritic arborization and formation of basal dendrites (D, arrows) extending into the hilus.
Abnormally localized granule cells, as well as morphologically abnormal granule cells localized within the granule cell layer, usually displayed an axon that arose from the cell body and ran toward the CA3 s. lucidum, forming typical small “en passant” boutons and large MF boutons with filipodial extensions (Fig. 9B2,C2). These observations suggest that both heterotopic and normally positioned granule cells contribute to the normal MF projection in p35 −/− mice. However, many of these displaced cells also showed an unusually extensive MF arborization within the hilus (Fig.9B2). The axons of some granule cells arose from anomalous locations, e.g., the lateral side of the cell body, the apical dendrite, or the initial portion of a recurrent basal dendrite. Figures8 (A1 and B, cells 2 and 3) and 9 (C2) indicate some examples of abnormal axon origin. Finally, granule cells of p35−/− mice, either localized within the granule cell layer or displaced into the molecular layer, show recurrent MF sprouting into the granule cell layer and/or inner molecular layer, indicative of MF reorganization. Figure9C1–2 shows an example of a “bipolar” granule cell forming not only recurrent basal dendrites but also an aberrant MF axon collateral that forms a plexus within the granule cell layer.
DISCUSSION
These results expand the previous description by Tsai and her colleagues (Chae et al., 1997; Kwon and Tsai, 1998) showing that normal development of hippocampus and dentate gyrus involves p35-mediated processes. We have, in addition, found that (1) hippocampal pyramidal neurons of the CA1 and CA3 subfields and granule cells of the dentate gyrus are heterotopically localized, (2) a significant proportion of p35−/− mice exhibit spontaneous behavioral and electrographic seizure, and (3) heterotopic granule cells exhibit features (e.g., basal dendrites, abnormal axon origin, and aberrant mossy fiber collaterals and synapses) often observed in epileptic tissue.
The findings of Kwon and Tsai (1998) in p35 knock-out mice, together with similar cortical migration defects in cdk5-deficient mice (Ohshima et al., 1996; Gilmore et al., 1998) suggest a critical signal transmission function of p35/cdk5 kinase for the migration of cortical neurons (or glia) to their proper destinations. In contrast, Reelin, a glycoprotein secreted by the Cajal-Retzius cells in the marginal zone, is thought to act as a “stop signal” for migrating cortical plate neurons at the boundary of the marginal zone (Sheppard and Pearlman, 1997; Kwon and Tsai, 1998), thus explaining the abnormal presence of neurons in this zone in the reeler mutant (Frotscher, 1998). Developmental processes in the hippocampus are similar to those in the neocortex, except for the long-lasting neurogenesis of granule cells in the dentate gyrus and their “outside-in” gradient of positioning (Cowan et al., 1980; Soriano et al., 1994). The abnormal positioning of dentate granule cells within the hilus and molecular layer and of hippocampal pyramidal neurons in adjacent layers (and absence of a specific astrocyte population) indicates a defect in the migration of later generated cells. It is yet not clear, however, whether the heterotopic granule cell positioning in the hilus results from a migration defect or whether these granule cells might be generated at a later time point, from progenitor cells within the hilus (Altman and Bayer, 1990; Parent et al., 1997), and do not migrate into the granule cell layer (Schlessinger et al., 1975; Soriano et al., 1994).
The occasional appearance of heterotopic granule cells (displaced granule cells) in the dentate hilus and molecular layer has been observed in wild-type mice and rats (Ramon y Cajal, 1968; Amaral, 1978;Seress and Pokorny, 1981; Marti-Subirana et al., 1986). Granule cell dispersion in the dentate gyrus, similar to that seen in human epileptic hippocampi (Houser 1990; Lurton et al., 1997; El Bahh et al., 1999), has also been observed in animal models of epilepsy, e.g., after kainic acid injection into the dorsal hippocampus (Cavalheiro et al., 1982; Suzuki et al., 1995; Bouilleret et al., 1999), and in the pilocarpine model (Mello et al., 1992). Two mechanisms have been proposed to explain the genesis of these heterotopic granule cells: (1) a disorder of the migration, occurring early in development (before any seizure event) and perhaps contributing to epileptogenesis (Scharfman et al., 2000), and (2) a consequence of repetitive seizures (as seen after kainic acid or pilocarpine administration), perhaps reflecting enhanced neurogenesis (Mello et al., 1992; Bouilleret et al., 1999). It remains unclear whether the appearance of heterotopic granule cells is related to granule cell loss in the epileptic hippocampus (Babb et al., 1984;Mello et al., 1992; Mathern et al., 1994; Suzuki et al., 1995). Granule cell dispersion has not been observed when cell loss is minimal (Lurton et al., 1997), but both appear to be directly related to mossy fiber sprouting into the molecular layer (Cavazos and Sutula, 1990;Babb et al., 1991; Houser, 1999). The present study, using Timm staining and intracellular labeling with biocytin to vizualize mossy fiber boutons, demonstrates that heterotopic granule cells generate (and receive) aberrant mossy fibers, in both granule cell and molecular layers. In addition, heterotopic granule cells show basal dendrites (entering the hilus) and axons originating from the apical pole of the soma or the apical dendrite. These findings represent abnormal phenomena associated with epilepsy both in human hippocampus (Franck et al., 1995) and in experimental animal models (Spigelman et al., 1998; Ribak et al., 2000). The behavioral observations of the present study substantiate the enhanced susceptibility of p35−/− mice to seizures. Simultaneous video/EEG recordings confirmed spontaneous epileptiform activity, including both intermittent interictal discharge and spontaneous generalized seizures. Although no fatalities were observed during recorded seizures, the severity of observed seizures, together with occasional sudden (unexplained) death of some animals, suggests that the mortality rate of ∼10% [see also Chae et al. (1997)] might be attributable to fatal seizures. Previous experiments comparing PTZ-induced seizures in p35−/− and wild-type mice showed that generalized convulsions were only fatal in p35−/− mice (Chae et al., 1997).
Early in postnatal development (before postnatal day 10), before any spontaneous seizure, p35−/− mice exhibit heterotopic granule cells, indicating that this abnormality is not a result of seizure activity and suggesting a causal relationship between structural abnormality and occurrence of spontaneous seizures. Other studies investigating animal models of cortical dysplasia with experimentally induced disruption of cortical development also suggest a causal link between cortical malformations and seizure activity (Chevassus-au-Louis et al., 1999;Walsh, 1999; Fleck et al., 2000; Chen et al., 2000). Such animal models include migrational malformations induced by freezing (Dvorak and Feit, 1977; Dvorak et al., 1978; Rosen et al., 1992, 1996; Jacobs et al., 1996, Hablitz and DeFazio, 1998), x-irradiation (Roper et al., 1995,1997; Hicks et al., 1959), and chemical treatment (e.g., methylazoxymethanol: Singh, 1977; Cattaneo et al., 1995; De Feo et al., 1995; Baraban and Schwartzkroin, 1996; Germano and Sperber, 1997; Chevassus-au-Louis et al., 1998, 1999; Baraban et al., 2000). Animals with abnormalities paralleling human genetic neuronal migration disorders also show enhanced seizure-sensitivity: e.g., type 1 lissencephaly (Lis1 mutant) (Reiner et al., 1993; Hirotsune et al., 1998; Fleck et al., 2000), and band heterotopia (tish mutant) (Lee et al., 1997). Although many of these animal models exhibit increased excitability (Baraban and Schwartzkroin, 1995; Jacobs et al., 1996, 1999; Luhmann and Raabe, 1996; Roper et al., 1997; Baraban et al., 2000) (for review, see Chevassus-au-Louis et al., 1999), the presence of a cortical malformation does not necessarily result in a spontaneous seizure phenotype. One might speculate that as in humans, a subtle preexisting focal cortical dysgenesis (e.g., heterotopically displaced neurons) provides a substrate for seizure induction (e.g., from a febrile episode) and may also predispose the animal to a seizure-related pathology (e.g., hippocampal sclerosis in TLE) (Lewis, 1999).
The underlying basis for hyperexcitability (and synchrony) associated with cortical disorganization and reorganization remains a topic of intense investigation. Numerous studies have shown that alterations of GABAergic neurotransmission might be critically involved in underlying epileptic disorders (Schwartzkroin, 1998). Although the present study did not reveal any obvious loss of specific interneuron populations in p35−/− mice, both GAD67- and parvalbumin-containing axon terminals were abnormally distributed within the molecular layer, surrounding somata of heterotopic granule cells, reflecting correct target innervation (Fleck et al., 2000). However, the reduced parvalbumin immunoreactivity within the terminal plexus around the granule cells suggests the possibility of functional abnormality in these inhibitory circuits, either as a direct result of the p35−/− disorganization or as a result of earlier seizures. Loss of parvalbumin immunoreactivity has also been observed after kainic acid-induced seizures (Sloviter 1991, 1994; Best et al., 1993;Buckmaster and Dudek, 1997), although transient parvalbumin loss may not be related to cell death (Scotti et al., 1997). Subtle changes of parvalbumin immunoreactivity in the dentate gyrus of p35−/− mice could be of relevance for seizure sensitivity of these animals (Scotti and Nitsch, 1991). In addition, in both wild-type and p35−/− animals, the calcium binding protein calretinin is expressed in excitatory hilar mossy cells (Liu et al., 1996; Blasco-Ibanez and Freund, 1997; Schurmans et al., 1997), which form a dense axonal plexus within the dentate inner molecular layer (Buckmaster et al., 1996). In p35−/− mice, heterotopic granule cells within the inner molecular layer are surrounded by these calretinin-positive terminals; the abnormal position of excitatory mossy cell synapses onto granule cell bodies (data not shown) could undoubtedly alter the balance of excitation and inhibition in this sensitive circuit.
Physiological studies in epileptic human tissue (Masukawa et al., 1991,1992; Williamson, 1994; Franck et al., 1995) and experimental animal models (Scharfman et al., 1990; Ribak et al., 1992; Sloviter, 1994; Wuarin and Dudek, 1996; Patrylo and Dudek, 1998; Okazaki et al., 1999) have shown that the dentate gyrus can play an important role in epileptic conditions (Masukawa et al., 1999). Dentate granule cells are ideally situated to control the spread of excitation. Developmental malformations of the dentate, whether genetically based or caused by early events such as trauma, fever or infection, may lead to various complex changes (e.g., cell loss, mossy fiber sprouting, receptor alterations, granule cell dispersion) that result in an imbalance between excitation and inhibition. The p35−/− mutant mouse represents one example of an experimental model in which anatomical disorganization is closely linked to seizures. Further studies are necessary to clarify how the p35 mutation and related structural abnormalities lead to epileptiform activities.
Footnotes
This study was supported by National Institutes of Health Grant NS18895. We thank the following people for excellent assistance: Norma L. Anderson, electron microscopy; Andrew F. Turella, histology; Ildiko Hegyvary and Kathryn Blott, quantitative histological analysis; and Paul Schwartz and Janet Schukar, photographic and computer imaging.
Correspondence should be addressed to Dr. Philip A. Schwartzkroin, Department of Neurological Surgery, University of Washington, Box 356470, Seattle, WA 98195-6470. E-mail:pas{at}u.washington.edu.
This study was supported by National Institutes of Health Grant NS18895. We thank the following people for excellent assistance: Norma L. Anderson, electron microscopy; Andrew F. Turella, histology; Ildiko Hegyvary and Kathryn Blott, quantitative histological analysis; and Paul Schwartz and Janet Schukar, photographic and computer imaging.
Correspondence should be addressed to: Dr. Philip A. Schwartzkroin, Department of Neurological Surgery, University of Washington, Box 356470, Seattle, WA 98195-6470. E-mail:pas{at}u.washington.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}