

Abstract
Conflicting data have been collected so far on the action of nitric oxide (NO) on cholinergic interneurons of the striatum. In the presentin vitro electrophysiological study, we reported that intracellularly recorded striatal cholinergic interneurons are excited by both hydroxylamine andS-nitroso-N-acetylpenicillamine, two NO donors. This excitation persisted unchanged in the presence of glutamate, dopamine, and substance P receptor antagonists as well as after blockade of tetrodotoxin (TTX)- and calcium channel-sensitive transmitter release, suggesting that NO produces its effects by modulating directly resting ion conductances in the somatodendritic region of striatal cholinergic cells. The depolarizing effect of hydroxylamine was greatly reduced by lowering external concentrations of sodium ions (from 126 to 38 mm) and did not reverse polarity in the voltage range from −120 to −40 mV. The sodium transporter blockers bepridil and 3′,4′-dichlorobenzamil were conversely ineffective in preventing NO-induced membrane depolarization. Intracellular cGMP elevation is required for the action of hydroxylamine on striatal cholinergic cells, as demonstrated by the findings that the membrane depolarization produced by this pharmacological agent was prevented by bath and intracellular application of two inhibitors of soluble guanylyl cyclase and was mimicked and occluded by zaprinast, a cGMP phosphodiesterase inhibitor. Finally, intracellular Rp-8-Br-cGMPS, a protein kinase G (PKG) inhibitor, blocked the hydroxylamine-induced membrane depolarization of cholinergic interneurons, whereas both okadaic acid and calyculin A, two protein phosphatase inhibitors, enhanced it, indicating that intracellular PKG and phosphatases oppositely regulate the sensitivity of striatal cholinergic interneurons to NO. The characterization of the cellular mechanisms involved in the regulation of striatal interneuron activity is a key step for the understanding of the role of these cells in striatal microcircuitry.
- acetylcholine
- basal ganglia
- brain slices
- intracellular recordings
- NOS-positive interneurons
- protein phosphatases
In recent years, electrophysiological, histochemical, and morphological criteria allowed the identification of distinct classes of striatal interneurons. They comprise ∼2–4% of the neuronal population of the striatum, the remaining being represented by medium spiny GABAergic projection cells. According to their physiological characteristics, striatal interneurons have been divided into fast-spiking (FS), low-threshold spikes (LTS), and long-lasting afterhyperpolarization (LA) cells (Kawaguchi, 1993;Kawaguchi et al., 1995). FS and LTS cells are GABAergic interneurons and are immunoreactive for parvalbumin and nitric oxide synthase (NOS), respectively. LA cells, conversely, the most studied group of these interneurons, are the main source of acetylcholine (Ach) in the striatum and provide this brain area with one of the highest contents of this transmitter in the brain (Weiner et al., 1990; Graybiel et al., 1994; Hersch et al., 1994). In spite of their numerical disadvantage, striatal interneurons posses very large axonal and dendritic fields and densely innervate virtually all neuronal species within this nucleus (Kawaguchi, 1992, 1993; Kawaguchi et al., 1995).
Experimental evidence obtained during microdialysis studies in vivo suggested that NO might be involved in the control of Ach release in the striatum. Few and conflicting data, however, have been collected so far on this interesting issue. In particular, some authors reported that NO donors cause a pronounced increase in striatal Ach release (Prast and Philippu, 1992; Guevara-Guzman et al., 1994; Prast et al., 1995, 1998), which has been attributed, at least in part, to a concomitant increased release of glutamate. Various NO donors, in fact, have been found to be ineffective in triggering Ach release from the striatum in the presence of antagonists of glutamate receptors (Prast et al., 1998). Other studies, however, reported that the release of striatal Ach was unaffected by NO (Sandor et al., 1995) and also that endogenous NO attenuated rather than favored NMDA-induced Ach release (Ikarashi et al., 1998).
Indeed, these results do not allow conclusions to be drawn on the possible interaction between NO and cholinergic cells within the striatum. In the attempt to clarify this issue, in the present study we used an electrophysiological approach in vitro to study the effects of NO on striatal cholinergic interneurons recorded intracellularly.
MATERIALS AND METHODS
Preparation and maintenance of the corticostriatal slices. Male Wistar rats (2–3 months) were used for the electrophysiological experiments. Preparation and maintenance of the slices have been described in detail previously (Calabresi et al., 1992, 1999a). In brief, animals were killed under ether anesthesia by cervical dislocation, the brain was quickly removed, and corticostriatal coronal slices (200- to 300-μm-thick) were cut, from tissue blocks, with the use of a vibratome. Slices were maintained at 31°C in an oxygenated solution for ∼30 min. A single slice was then transferred into a recording chamber and fully submerged in a continuously flowing Krebs' solution (30°C, 3 ml/min) gassed with 95% O2 and 5% CO2. The composition of the solution was (in mm): 126 NaCl, 2.5 KCl, 1.3 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 glucose, and 18 NaHCO3. In some experiments choline chloride was used to replace Na+ chloride. In these experiments Na+ chloride was reduced to 30% (38 mm).
Electrophysiological recordings. In all the electrophysiological experiments the intracellular recording electrodes were filled with 2 m KCl (30–60 MΩ). An Axoclamp 2A (Axon Instruments, Foster City, CA) amplifier was used for both current- and voltage-clamp recordings. In single-electrode voltage-clamp mode the switching frequency was 3 kHz. The headstage signal was continuously monitored on a separate oscilloscope. Traces were displayed on an oscilloscope and stored in a digital system.
Morphological and histochemical characterization of cholinergic interneurons. In some experiments, for simultaneous optical and electrical recordings, the tip of the recording electrode was filled with a solution of 2 mm fura-2 (pentapotassium salt; Molecular Probes, Leiden, The Netherlands) and 100 mm KCl, whereas the shank was filled with a 2m KCl solution. After cell impalement, cells (n = 64) were loaded with fura-2 by injecting, through the recording electrode, 0.1–0.5 nA negative current for 10–15 min. In these cases, the recording chamber was mounted on the stage of an upright microscope (Axioscop FS, Zeiss), equipped with a 60× water immersion objective (Zeiss). Excitation light passed through a shutter and was filtered at 340 and 380 nm. Emission light was filtered by a long-pass barrier filter (470 nm) and detected by a CCD camera (Photonic Science, East Sussex, UK). Images were stored and analyzed with a software (IonVision; ImproVision, Birmingham, UK) running on PowerMac 8100. Ratio images were calculated from pairs of 340 and 380 nm images after background fluorescence was subtracted (backgrounds were acquired from regions free of dye fluorescence). Ratiometric measurements were converted into intracellular calcium concentration values (Grynkiewicz et al., 1985; Pisani et al., 1999).
In other experiments (n = 46) biocytin was used in the intracellular electrode to stain the neurons. In these cases, biocytin at concentration of 2–4% was added to a 2 m KCl pipette solution. Slices containing neurons stained with biocytin were fixed in 4% paraformaldehyde in 0.1 m phosphate buffer (PB) overnight at 4°C. After incubation in PB containing sucrose 30% in 0.1 m PB for 3 hr, the slices were frozen and further resectioned in a cryostat at 40 μm thickness. Free-floating sections were incubated with fluorescein isothiocyanate (FITC) conjugated to avidin (Sigma, St. Louis, MO; diluted 1:200 in PBS containing 0.1% Triton X-100) overnight at 4°C. The sections were then washed in PB several times and mounted on slides with glycerol in PB (1:3). The sections were observed and photographed in a fluorescence microscope (Leitz, Wetzlar, Germany) using epifluorescence B-2E (barrier filter, 520–560) for FITC to examine biocytin-positive cells. Selected sections, in which a large aspiny neuron had been identified, were further processed for double staining of biocytin and choline acetyltransferase (ChAT) immunoreactivity. The sections were removed from the slides, and after washing in PB, incubated with a rat monoclonal antibody against ChAT (Boehringer Mannheim, Mannheim, Germany; 1:250) in PB containing 10% normal goat serum and 2% bovine serum albumin, for 3 hr at room temperature. After washing in PB, the sections were incubated in a mixture containing goat anti-rabbit IgG (Sigma; 1:50) conjugated to tetramethylrhodamine isothiocyanate (TRITC) and avidin-conjugated FITC (1:200) for 2 hr at room temperature. After washing, the sections were mounted on slides with glycerol in PB (1:3). In this case, the slices were observed and photographed in the fluorescence microscope using epifluorescence G-2A (barrier filter, >590 nm) for TRITC, and epifluorescence B-2E (barrier filter, 520–560 nm) for FITC, so that ChAT-immunoreactive neurons were seen in red, and biocytin positive cells in yellow–green. In several cases, sections were further processed to make permanent staining of biocytin-loaded cells.
Data analysis and drug application. Values given in the text and in the figures are mean ± SEM of changes in the respective cell populations. Wilcoxon's test or Student's t test (for paired and unpaired observations) were used to compare the means and ANOVA was used when multiple comparisons were made against a single control group. Drugs were applied by dissolving them to the final concentration in the saline and by switching the perfusion from control saline to drug-containing saline. Drug solutions entered the recording chamber within 40 sec after a three-way tap had been turned on. In some experiments, however, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), 4-H-Bromo-1,2,4-oxadiazolo(3,4-d)benz(b)oxazin-1-one (NS 2028), or guanosine 3′,5′-cyclic monophosphorothioate, 8-bromo, Rp-isomer (Rp-8-Br-cGMPS) were applied intracellularly through the recording electrode, as previously reported (Calabresi et al., 1999a). Hydroxylamine was from Merck (Darmstadt, Germany). 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), (+)-MK 801 maleate (MK-801), ODQ, SCH 23390, (RS)-α-methyl-4-carboxyphenylglycine (MCPG), andS-nitroso-N-acetylpenicillamine (SNAP) were from Tocris Cookson (Bristol, UK). Rp-8-Br-cGMPS was from Calbiochem (La Jolla, CA). Zaprinast (M&B 22948, 2-O-propoxyphenyl-8-azapurin-6-one) was from Rhône-Poulenc Rorer (Dagenham, UK). Okadaic acid, 4-H-Bromo-1,2,4-oxadiazolo(3,4-d)benz(b)oxazin-1-one (NS 2028), and Calyculin A were from Alexis (Läufelfingen, Switzerland). Bepridil, biocytin, [d--Arg1,d--Pro2,d--Trp7,9,Leu11]-SP, nifedipine, 2-Phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO), and TTX were from Sigma (Rome, Italy). 3′,4′-dichlorobenzamil (DCB) was from E. J. Cragoe, Jr (Nacogdoches, TX). ω-agatoxin TK and ω-conotoxin GVIA were from Alomone Labs (Jerusalem, Israel).
RESULTS
Electrophysiological, morphological, and histochemical properties of striatal cholinergic interneurons
All striatal neurons included in this study were identified as cholinergic interneurons for both their electrophysiological and morphological characteristics. They comprised 46 of 260 cells when electrodes were placed into the striatum without visual control, the remaining neurons having electrophysiological characteristics of spiny neurons. With visual placement of the recording electrode, a further 64 cholinergic interneurons were obtained. The high proportion of cholinergic cells recorded is also attributable to the increased probability to obtain stable recordings (lasting at least 30 min) from these large cells compared with the smaller spiny neurons. Membrane properties of these cells closely resembled the membrane properties reported previously for rat cholinergic interneurons (Kawaguchi, 1992;Bennett and Wilson, 1998; Calabresi et al., 1998). The distinguishing features of these neurons were: low resting membrane potential (RMP) (−58 ± 4 mV) and high input resistance (180 ± 60 MΩ) compared with other striatal neurons, accommodation of action potential discharge and marked afterhyperpolarization, prominent cesium-sensitive decline in hyperpolarizing electrotonic potential (Ih cation current), and large somata (30–60 μm) with three to five primary dendrites bearing no spines. Thirty cells were further processed for double staining of biocytin and ChAT immunoreactivity. All these neurons, which showed the electrophysiological and morphological characteristics typical of cholinergic interneurons, were found to be positive for ChAT immunoreactivity.
Effects of the NO donors hydroxylamine and SNAP on striatal cholinergic interneurons
In current-clamp experiments, bath application of the NO donor hydroxylamine (100 μm, 2–7 min) invariably induced a small but significant membrane depolarization of the recorded striatal cholinergic interneurons. This membrane depolarization averaged 5 ± 1.4 mV in amplitude and triggered action potentials when the RMP of the cell was sufficiently depolarized. Higher doses of hydroxylamine (300 μm) failed to produce more pronounced effects, whereas lower concentrations (3, 10, and 30 μm) revealed a dose-dependent action of this agent on membrane potential of the recorded striatal cholinergic interneurons (Fig.1). The effect of 100 μmhydroxylamine was mimicked by another NO donor, SNAP (100 μm; 3–7 min; p < 0.05) (n = 5), and was fully reversible after 8–15 min washout of both drugs. Neither hydroxylamine- nor SNAP-induced membrane depolarization of cholinergic cells was coupled to significant changes in intracellular calcium concentration (n = 52;p > 0.05; data not shown). In some experiments, hyperpolarizing current pulses (100–200 pA; 2–3 sec duration; 10 sec interval) were applied to monitor input resistance of the cells. To measure the effects of NO on this electrophysiological parameter, during the hydroxylamine- or SNAP-induced membrane depolarization, membrane potential value was returned to the control level through the injection of continuous negative current. In the recorded cells, the input resistance was not significantly affected by these NO donors (n = 6 for each experimental condition;p > 0.05; Fig.2A).
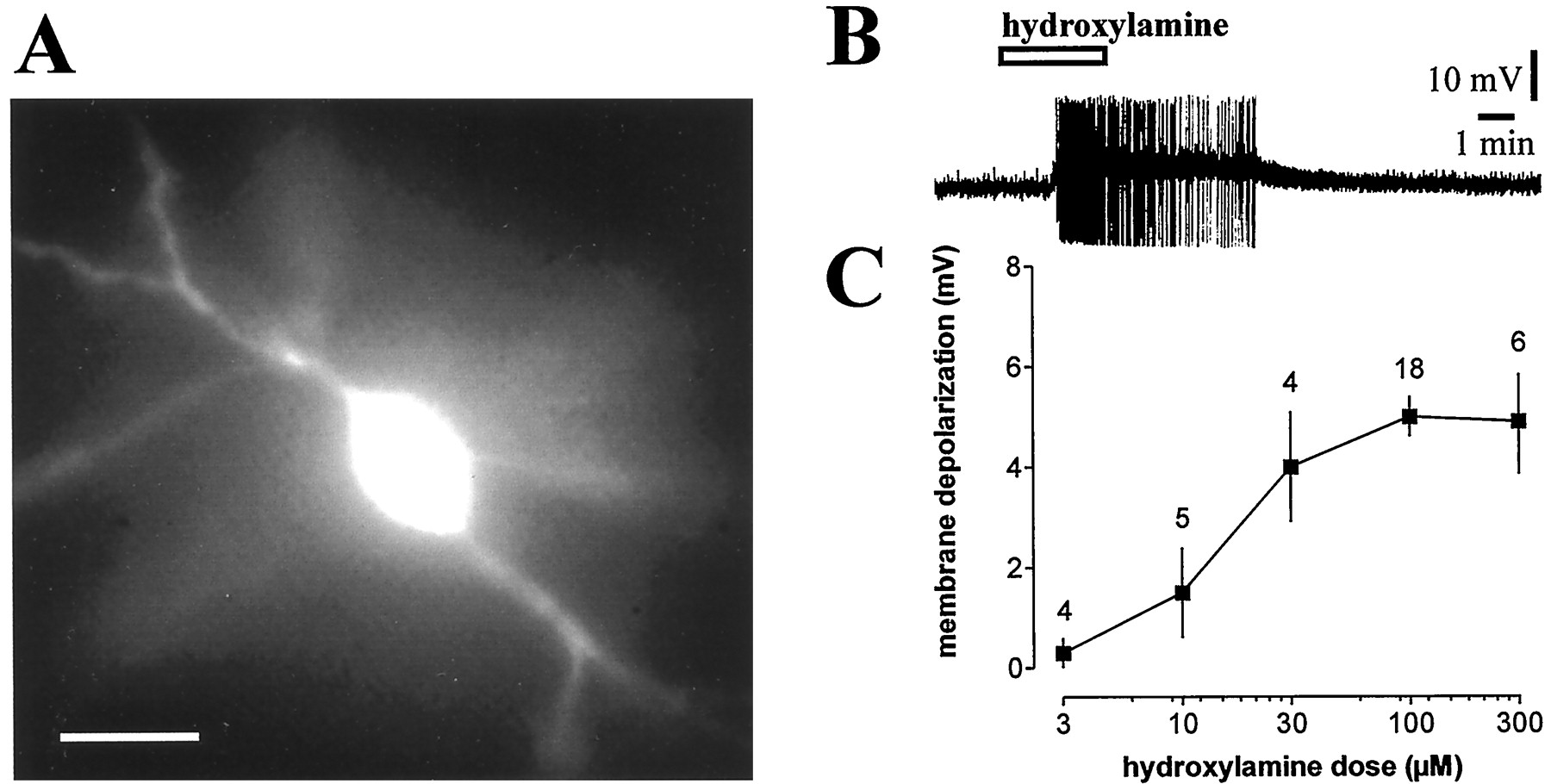
Hydroxylamine depolarizes striatal cholinergic interneurons in a dose-dependent manner. A,Morphological identification of a fura-2-filled striatal cholinergic interneuron. Scale bar, 50 μm. B, Hydroxylamine (100 μm) depolarized a striatal cholinergic interneuron and caused action potential firing. Resting level is −55 mV; full action potential height not captured by pen recorder. C, The graph shows the dose–response curve for the hydroxylamine-induced membrane depolarization of striatal cholinergic interneurons. In this graph and in the following ones, the number of observations is indicated for each experimental condition.

Electrophysiological characteristics of hydroxylamine- and SNAP-induced excitation of striatal cholinergic interneurons. A, In a current-clamp experiment, hydroxylamine (100 μm) (a) and SNAP (100 μm) (b) depolarized a striatal cholinergic interneuron without affecting the apparent input resistance of the cell. To compare the effects of both compounds on this electrophysiological parameter, membrane potential value was returned to the control level by continuous injection of 100 pA negative current. Resting level was −61 mV. Downward deflections are hyperpolarizing electrotonic potentials evoked by rectangular current pulses (200 pA, 2 sec); their decline after the initial peak reflects the prominent Ih in these cells.B, In another striatal interneuron recorded in the voltage-clamp mode, 100 μm hydroxylamine produced an inward current (a). This effect persisted unchanged in the presence of 1 μm TTX, to block voltage-dependent sodium channels (b). Holding potential was −60 mV. C, Current–voltage relationship of a cholinergic interneuron before (open circles) and during 100 μm hydroxylamine (filled circles). The values were calculated by measuring the steady-state current generated by 3 sec voltage steps of progressively increasing and decreasing amplitude. Holding potential was −60 mV.
When the cholinergic interneurons were voltage-clamped at or near the RMP (−60 mV), bath application of hydroxylamine produced an inward shift of the holding current in all the tested neurons (−140 ± 25 pA; p < 0.01; n = 12). This effect persisted unchanged after the application of 1 μm TTX, a voltage-dependent sodium channel blocker, and recovered within 15 min of washout (Fig.2B). TTX was also found to be ineffective on hydroxylamine- (p > 0.05; n = 6; Fig. 3C) and SNAP-mediated electrophysiological effects in current-clamp experiments (p > 0.05; n = 4; data not shown). In an attempt to uncover the ionic mechanisms for hydroxylamine-induced membrane depolarization/inward current, voltage steps (3 sec) of progressively increasing amplitude were applied in the presence of 1 μm TTX to voltage-clamped interneurons before and during the application of this NO donor. The resulting current–voltage relationships were calculated by measuring the steady-state current during each voltage step. As shown in Figure2C, a net inward current was present at all membrane potentials with no apparent reversal potential within the tested voltage range. Although poor space clamp is assumable given the size of these neurons and the limitation of the recording technique, this evidence might indicate that this current does not result, at least in the totality, from the blockade of potassium channels, and might suggests that either an increased sodium and/or calcium conductance with a reversal potential positive to the explored membrane potentials, or the modulation of a membrane ion transporter might be involved.

Various receptor antagonists and calcium channel blockers fail to prevent hydroxylamine-induced membrane depolarization of striatal cholinergic interneurons. A, The membrane depolarization produced by 100 μm hydroxylamine (a) was not altered in the presence of 30 μm MK-801 and 10 μm CNQX (7 min) to block both NMDA and non-NMDA glutamate receptors (b). Resting membrane potential was −58 mV. B, Application of the DA D1 receptor antagonist SCH 23390 (10 μm, 7 min) (a) failed to affect the membrane response produced in control medium by 100 μm hydroxylamine (b). Resting membrane potential was −58 mV.C, Summary of pharmacological experiments on 100 μm hydroxylamine-induced membrane depolarization of striatal cholinergic interneurons. Cocktail solution contained: ω-conotoxin GVIA, nifedipine, ω-agatoxin TK, MCPG, and [d--Arg1,d--Pro2,d--Trp7,9,Leu11]-SP. Concentrations were: MK-801 30 μm, CNQX 10 μm, SCH 23390 10 μm, TTX 1 μm, ω-conotoxin GVIA 1 μm, nifedipine 20 μm, ω-agatoxin TK 20 nm, MCPG 300 μm, and [d--Arg1,d--Pro2,d--Trp7,9,Leu11]-SP 10 μm.
NO-mediated membrane depolarization of striatal cholinergic interneurons does not depend on the release of endogenous neurotransmitters
The effect of hydroxylamine on striatal cholinergic interneurons was not inhibited preincubating the slices (5–10 min) with MK-801 (30 μm) plus CNQX (10 μm), antagonists of NMDA and non-NMDA glutamate receptors, respectively, suggesting that this effect was not mediated by an increased release of glutamate (p > 0.05; n = 5; Fig.3A,C). Because it has been reported that NO also increases dopamine release in the striatum both in vitro (Zhu and Luo, 1992; Black et al., 1994; Stewart et al., 1996) and in vivo(Spatz et al., 1995; West and Galloway, 1996, 1997) and that D1-like dopamine receptor activation depolarizes striatal cholinergic interneurons with a time course similar to that one produced by hydroxylamine (Aosaki et al., 1998; Pisani et al., 2000), we also tested the possibility that hydroxylamine caused its effects through the release of endogenous dopamine. In five experiments, therefore, we bath-applied the NO donor after pretreatment of the slices with 10 μm SCH 23390 (5–10 min), a D1-like dopamine receptor antagonist able to fully block membrane depolarization of striatal cholinergic neurons induced by dopamine (Aosaki et al., 1998). We found that this pharmacological agent failed to prevent the membrane depolarization of cholinergic interneurons induced by hydroxylamine (p > 0.05), ruling out the contribution of endogenous dopamine in this effect (Fig. 3B,C).
NO might favor transmitter release in the striatum independently of the activation of TTX-sensitive sodium channels and might depolarize cholinergic cells indirectly via glutamate acting on metabotropic glutamate receptors (mGluRs) or via the release of other transmitters. We measured, therefore, the effects of 100 μmhydroxylamine after 10 min preincubation of the slices in a solution containing 1 μm ω-conotoxin GVIA, 20 μmnifedipine, and 20 nm ω-agatoxin TK (to block, respectively, N-type, L-type, and P-type high voltage-activated calcium channels) and 300 μm MCPG plus 10 μm[d--Arg1,d--Pro2,d--Trp7,9,Leu11]-SP (to block mGluRs and substance P receptors, respectively). Noticeably, ω-conotoxin GVIA has already been found to block synaptic inputs to striatal cholinergic interneurons (Pisani et al., 2000), and MCPG and [d--Arg1,d--Pro2,d--Trp7,9,Leu11]-SP prevented in previous studies the depolarizing effects mediated by mGluR and substance P receptor activation on these cells (Aosaki and Kawaguchi, 1996; Takeshita et al., 1996). This pharmacological treatment, however, failed to affect the amplitude of the NO-induced membrane depolarization of striatal interneurons (p > 0.05; n = 5; Fig.3C). These data, coupled to the observation that TTX (1 μm) did not affect the membrane responses to 100 μm hydroxylamine (see above) strengthen the conclusion that NO depolarizes striatal cholinergic interneurons through a direct postsynaptic action.
Effects of low sodium-containing solution and of sodium transporter blockers on hydroxylamine-induced membrane depolarization of striatal cholinergic interneurons
To investigate whether the membrane depolarization produced by NO donors was mediated by sodium influx, in some experiments we reduced this extracellular ion by substituting it with choline chloride. As shown in Figure 4, the 100 μm hydroxylamine-evoked membrane depolarization recorded in a solution containing 126 mm NaCl showed a remarkable reduction when the perfusing solution was switched to a solution containing 38 mm NaCl (n = 6), indicating that sodium ions are the major carrier of this depolarization.

Hydroxylamine-induced membrane depolarization is significantly attenuated by low sodium-containing external solution but not by sodium exchanger blockers. A, In this current-clamp experiment, the membrane depolarization produced by 100 μm hydroxylamine (a) was blocked by 7 min perfusion of the slices with a solution containing 38 mm sodium ions (b). Resting membrane potential was −61 mV. B, Summary of experiments on hydroxylamine (100 μm)-induced membrane depolarization of striatal cholinergic interneurons. Concentration of both DCB and bepridil was 100 μm (**p < 0.01).
Sodium transport and some sodium channels are sensitive to the amiloride derivative DCB (Wacholtz et al., 1993; Calabresi et al., 1999b) and to bepridil (Stys et al., 1992; Kiedrowski et al., 1994). We tested, therefore, these two pharmacological agents on the membrane responses induced by bath application of 100 μmhydroxylamine on striatal cholinergic interneurons. Preincubation of the slices with either DCB (5–10 min; 100 μm;n = 7) or bepridil (5–10 min; 100 μm;n = 5) produced per se no change in the RMP, and apparent input resistance of the recorded neurons and failed to affect the membrane responses evoked by the application of hydroxylamine (100 μm; 2–7 min) (p > 0.05 for both experimental conditions) (Fig. 4B). Noticeably, at the concentrations used in this study, both DCB and bepridil neither affected RMP of striatal spiny neurons recorded in vitro. During in vitroischemia, however, the ability of both agents in inhibiting sodium–calcium exchanger was unmasked (Calabresi et al., 1999b). This observation confirms that although DCB- and bepridil-sensitive transporters do not seem involved in the excitatory action of NO in striatal cholinergic cells, 100 μm DCB and 100 μm bepridil are able to target ion transporters in our slice preparation.
Requirement of postsynaptic cGMP elevation in the hydroxylamine-induced membrane depolarization of striatal cholinergic interneurons
NO elevates intracellular cGMP levels by stimulating soluble guanylyl cyclase (sGC) activity. Thus, to address the involvement of cGMP in the electrophysiological effects produced by hydroxylamine, we first investigated whether the pharmacological inhibition of sGC was able to prevent the membrane depolarization caused by this NO donor. Preincubation of the slices with 10 μm ODQ (5–10 min), a selective inhibitor of sGC, did not cause any change in the membrane potential of the recorded striatal interneurons (n = 12) but fully blocked the membrane depolarization produced in these cells by 100 μm hydroxylamine (p < 0.01). This pharmacological inhibition was reversible after a period of wash-out of 10–15 min. Interestingly, ODQ (100 μm) strongly reduced hydroxylamine-induced membrane depolarization also when this sGC inhibitor was applied intracellularly through the recording pipette (15 min), demonstrating that NO produces its effects by stimulating sGC activity in cholinergic interneurons themselves (n = 7; p < 0.01; Fig. 5C). Similar results were obtained by applying intracellularly (15 min) NS 2028 (50 μm), another specific inhibitor of sGC (n = 4; p < 0.01; Fig. 5C). Although neither ODQ nor NS 2028 are membrane-impermeable compounds, these data, together with those obtained with TTX, calcium channel blockers and receptor antagonists, are all supportive of the idea that the enzymes mediating the response to NO are located in cholinergic interneurons themselves.
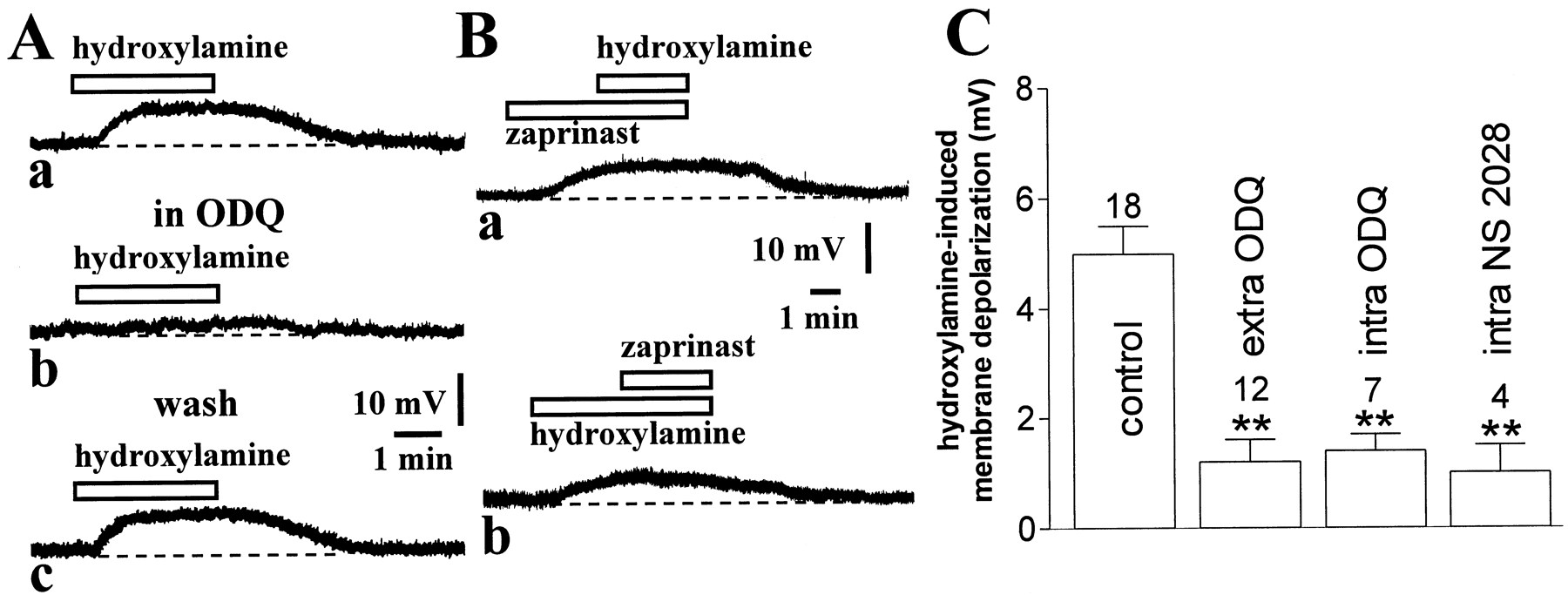
Hydroxylamine-induced membrane depolarization of striatal cholinergic interneurons requires cGMP elevation. A, The pharmacological blockade of sCG by ODQ (5 min, 10 μm) (b) fully prevented the membrane depolarization of a cholinergic interneuron produced by 100 μm hydroxylamine (a). This inhibition was reversible after 15 min wash of this compound (c). Resting membrane potential was −60 mV.B, The cGMP phosphodiesterase inhibitor zaprinast (30 μm) produced a membrane depolarization of another neuron and prevented further depolarization when 100 μmhydroxylamine was added (a). After 10 min wash of both pharmacological agents, the ability of 100 μmhydroxylamine to depolarize the recorded cell was restored and 30 μm zaprinast failed to produce significant depolarization when applied in the presence of this NO donor (b). Resting membrane potential was −62 mV.C, Summary of experiments on hydroxylamine (100 μm)-induced membrane depolarization of striatal cholinergic interneurons. Concentrations were (in μm): extracellular ODQ 10, intraelectrode ODQ 100, and intraelectrode NS 2028 50 (**p < 0.01).
To further confirm that an increase in cGMP levels mediated the physiological effects of hydroxylamine, we tested whether the inhibition of cGMP breakdown by zaprinast could mimic and occlude the physiological responses of this NO donor. Zaprinast is a rather selective inhibitor of cGMP phosphodiesterase, which has been already demonstrated to cause both significant elevation of cGMP levels in extracts of striatal slices and long-term depression of the efficacy of excitatory synaptic transmission in striatal projection cells (Calabresi et al., 1999a). In current-clamp recordings, bath application of this pharmacological agent (30 μm, 3–5 min) caused a membrane depolarization in five of eight striatal cholinergic interneurons. This membrane depolarization had similar amplitude than that one recorded in the presence of 100 μm hydroxylamine (4.8 ± 1 mV; p < 0.05; Fig. 5Ba) and was coupled with no change in the apparent input resistance of the recorded neurons (data not shown). Moreover, in a different manner than the effects on excitatory transmission in striatal projection neurons, the electrophysiological effects of zaprinast on cholinergic cells were quickly reversible at the wash-out of the drug. The ability of zaprinast in producing per se the reported electrophysiological effects might suggest the existence of a resting NO tone in striatal slices. To address this issue, therefore, we studied the effects of the NO scavenger PTIO (30 μm; 5–7 min; n = 4) on the resting membrane potential of striatal cholinergic cells. This agent produced no effect on these cells, suggesting that endogenous NO is unable to depolarize cholinergic cells in the absence of a concomitant inhibition of cGMP breakdown (data not shown). In six experiments, 30 μm zaprinast was added to the bathing solution during the steady state of the membrane depolarization produced by 100 μm hydroxylamine, and in other four experiments the application of zaprinast preceded that one of hydroxylamine. When hydroxylamine was applied first, the subsequent application of zaprinast did not enhance the membrane depolarization caused by hydroxylamine (6 ± 0.8 vs 6.5 ± 0.5 mV; p> 0.05). Similarly, when zaprinast preceded the application of hydroxylamine, this latter compound produced only a small potentiation of the depolarizing effect of zaprinast alone (5 ± 0.8 vs 5.8 ± 1 mV; p > 0.05) (Fig. 5B). These data show that hydroxylamine- and zaprinast-induced membrane depolarizations of striatal cholinergic cells were mutually occlusive, that is, during the hydroxylamine- or zaprinast-induced membrane depolarization, further depolarizations were not inducible by, respectively, zaprinast or hydroxylamine.
Role of protein kinase G and protein phosphatases in the hydroxylamine-induced membrane depolarization of striatal cholinergic interneurons
Intracellular elevation of cGMP levels results in the stimulation of the cGMP-dependent protein kinase (PKG). This effector, in turn, modulates the function of a series of cellular substrates by increasing their phosphorylation state. Therefore, to test whether PKG stimulation was involved in the action of NO on cholinergic cells of the striatum, we intracellularly applied 1 μm Rp-8-Br-cGMPS, a selective inhibitor of this kinase. This agent did not affect resting membrane potential and input resistance of the recorded cells, but prevented their membrane depolarization after the application of 100 μm hydroxylamine. These observations suggest that an increased phosphorylation of critical PKG-sensitive substrates mediates the effects of NO on these striatal cells. Because the phosphorylation state of cellular substrates also depends on the action of specific phosphatases (Greengard et al., 1999), we also studied the effects of either okadaic acid (30–100 nm) or calyculin A (100 nm) on hydroxylamine-induced membrane depolarization. These two pharmacological agents are rather selective inhibitors of protein phosphatase-1 (PP-1) and protein phosphatase-2A (PP-2A), enzymes that counteract the action of various intracellular kinases. In the presence of 1 μm TTX, both these compounds, bath-applied 5–7 min before the further application of hydroxylamine, did not produce per se any change in the RMP and input resistance of the recorded cell, but slightly enhanced the amplitude of hydroxylamine-induced membrane depolarization in striatal cholinergic interneurons, suggesting that in physiological conditions PP-1 and/or PP-2A limit the NO-stimulated PKG activity (Fig. 6).

Role of intracellular PKG and protein phosphatases in the hydroxylamine-induced membrane depolarization of striatal cholinergic cells. The histogram shows the effects of the pharmacological blockade of postsynaptic PKG and protein phosphatases on the membrane depolarization produced by 100 μmhydroxylamine (see Results for details). Concentrations were: Rp-8-Br-cGMPS 1 μm, okadaic acid 30 (n = 3) 100 nm (n = 4), and calyculin A 100 nm (* p < 0.05; **p < 0.01).
DISCUSSION
The present study demonstrates that NO enhances membrane excitability of cholinergic interneurons. NO-induced membrane depolarization of striatal cholinergic interneurons is not secondary to the release of excitatory amino acids, dopamine, substance P, or other putative excitatory transmitters, because it persisted unchanged in the presence of glutamate, dopamine, and substance P receptor antagonists, as well as in the presence of TTX and calcium channel blockers. This action requires intracellular cGMP elevation via sGC activity stimulation. Accordingly, the pharmacological inhibition of sGC fully prevented hydroxylamine-induced membrane depolarization of striatal cholinergic cells, whereas the inhibition of cGMP breakdown mimicked and occluded this electrophysiological effect. In addition, hydroxylamine-induced membrane depolarization was prevented by intracellular injection of either sGC or PKG inhibitors, indicating that NO likely acts as an anterograde transmitter on striatal cholinergic interneurons, leading to cGMP-dependent stimulation of PKG. These electrophysiological data are in good agreement with ultrastructural and immunocytochemical studies, demonstrating that NOS-positive terminals do synapse on cholinergic cells in the striatum (Vuillet et al., 1992) and that intracellular cGMP synthesis occurs in cholinergic cells but not in dopaminergic or glutamatergic presynaptic fibers of the rat striatum in response to NO (De Vente et al., 2000). It should be noted, however, that the compounds used in this study to block sGC or PKG are all membrane-permeable and, therefore, it is also possible that the enzymes mediating the response of cholinergic cells to NO are located in other cellular subtypes of our slice preparation.
The evidence that in striatal cholinergic interneurons protein phosphatase inhibitors significantly increased the membrane responses to hydroxylamine suggests the existence of a functional antagonism between PKG and phosphatases in determining the sensibility to NO in these cells. It is therefore conceivable that PKG and protein phosphatases oppositely regulate the phosphorylation state of common intracellular substrates, important for the physiological action of NO.
The experiments showing that the NO-induced membrane depolarization of striatal cholinergic interneurons is significantly reduced in low sodium-containing solution strongly suggest that sodium ions are the main carrier of this depolarization. We failed, however, to identify the channel subtypes involved in this action. The evidence that blockade of N-type, L-type, and P-type voltage-dependent calcium channels had no effect on the hydroxylamine-mediated electrophysiological actions seems to rule out the involvement of these channels. The estimation of the reversal potential of the hydroxylamine-mediated current is probably hampered by space-clamp errors. Nevertheless, our results showing that the hydroxylamine-mediated current does not reverse polarity close to the potassium equilibrium potential (approximately −100 mV) do not support the idea that the modulation of potassium conductances is responsible for this electrophysiological effect. It is however possible that the modulation of potassium conductances might be involved in the electrophysiological action of NO as part of a mixed current. Chloride channels are also potential candidates in the generation of this current. Because of the high concentration of chloride ions in the recording pipette, the chloride equilibrium potential is presumably more positive to the average resting membrane potential of cholinergic interneurons and, therefore, an increased permeability to these ions would contribute to the generation of an inward current. Experiments using patch-clamp recordings from isolated neurons appear more appropriate to clarify all these issues.
This study demonstrates that NO is able to influence the physiological activity of central neurons by causing membrane depolarization. Although it has been found that NO acts as a neurotransmitter in the PNS (Briggs, 1992), in the CNS it revealed so far only “nonconventional” actions. In particular, it has been reported that this agent is involved in the induction of hippocampal long-term potentiation (LTP) (O'Dell et al., 1991; Schuman and Madison, 1991), cerebellar (Crepel and Jaillard, 1990; Böhme et al., 1991;Daniel et al., 1993) and striatal long-term depression (Calabresi et al., 1999a), and increases, presumably through an action on presynaptic nerve terminals, the release of various neurotransmitters in the hippocampus (Nei et al., 1996) and basal ganglia (Kuriyama and Ohkum, 1995; Bogdanov and Wurtman, 1997). The ability to increase neurotransmitter release supports the idea that this agent mainly acts as retrograde transmitter and, although this issue remains controversial, it has been reported that injection of inhibitors of NOS into the postsynaptic cell prevented LTP formation (O'Dell et al., 1991; Schuman and Madison, 1991), as well as presynaptic injection of NO-absorbing agents (Arancio et al., 1996).
Several physiological activities have been postulated for NO in the striatum. NO, in fact, is thought to be released by NOS-positive interneurons to control local blood flow in response to cortical or pallidal inputs (Kawaguchi, 1993; Kawaguchi et al., 1995) and also influences striatal neuron activity by interacting directly with glutamate receptors (Manzoni et al., 1992). Moreover, striatal NO increases gap junction permeability in spiny neurons recorded in vitro (O'Donnell and Grace, 1997) and modulates the activity of striatal feedback pathways involved in regulating dopamine neurons in the substantia nigra (West and Grace, 2000). Together with the report of the present work, these studies indicate that NO exerts a complex control of striatal function in processes which may or may not be involved in modulating excitatory synaptic transmission.
Footnotes
This study was supported by Telethon Grant E. 729 (P.C.), by Telethon Grant E. 0930 (A.P.), and by a Ministero dell' Universitá e della Ricerca Scientifica e Tecnologica/Consiglio Nazionale delle Ricerche Grant (legge 95/95; G.B.). We thank Mr. Massimo Tolu for excellent technical assistance.
Correspondence should be addressed to Dr. Paolo Calabresi, Clinica Neurologica, Dipartimento di Neuroscienze, Università di Roma “Tor Vergata,” Via di Tor Vergata 135, 00133 Rome, Italy. E-mail:calabre{at}uniroma2.it.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}