

Abstract
The genetic demyelinating mouse “twitcher” is a model of the human globoid cell leukodystrophy, caused by galactosylceramidase (GALC) deficiency. Demyelination in the twitcher brain is secondary to apoptotic death of oligodendrocytes (OLs). Lipocalin-type prostaglandin (PG) D synthase (L-PGDS), a protein expressed in mature OLs, was progressively upregulated in twitcher OLs; whereas expression of OL-associated proteins such as carbonic anhydrase II, myelin basic protein, and myelin-associated glycoprotein was downregulated during demyelination in twitcher brains. The upregulation of L-PGDS was more remarkable in perineuronal OLs than in interfascicular OLs. A larger number of L-PGDS-positive OLs was found in selected fiber tracts of twitcher brains where fewer apoptotic cells were detected. The distribution of L-PGDS-positive OLs was inversely related to the severity of demyelination, as assessed by accumulation of scavenger macrophages. Mice doubly deficient for L-PGDS and GALC disclosed a large number of apoptotic neurons, which were never seen in twitcher brains, in addition to an increased number of apoptotic OLs. A linear positive correlation was observed between the population of L-PGDS-positive OLs in the twitcher brain and the ratio of apoptotic nuclei in the double mutant versus those in the twitcher, suggesting a dose-dependent effect of L-PGDS against apoptosis. These lines of evidence suggest that L-PGDS is an anti-apoptotic molecule protecting neurons and OLs from apoptosis in the twitcher mouse. This is a novel example of OL–neuronal interaction.
- prostaglandin D2
- lipocalin-type prostaglandin D synthase
- twitcher
- oligodendrocyte
- demyelination
- galactosylceramidase
- apoptosis
Prostaglandin (PG) D2 is a major PG in the CNS of rats, mice, and humans, and functions as a neuromodulator of several central actions, such as regulation of the sleep–wake cycle (Urade and Hayaishi, 1999,2000b; Hayaishi and Urade, 2002), body temperature, hormone release, and pain responses (Eguchi et al., 1999). In the CNS, PGD2 is mainly synthesized by lipocalin-type PGD synthase (L-PGDS; Urade et al., 1985a; Ujihara et al., 1988). Although L-PGDS is mainly expressed in the leptomeningeal cells (Urade et al., 1993; Beuckmann et al., 2000), immunohistochemistry and in situ hybridization revealed that L-PGDS is also produced in oligodendrocytes (OLs) after commencement of myelination in rats (Urade et al., 1987, 1993; Garcia-Fernandez et al., 1997; Beuckmann et al., 2000), mice (Eguchi et al., 1999), and humans (Blodorn et al., 1996). Because L-PGDS is not expressed in Schwann cells of the peripheral nerve system, this protein is a specific marker for mature OLs (Urade et al., 1985b).
The twitcher mouse (C57BL/6J-GALCtwi/twi ), a homozygote for a nonsense point mutation of the galactosylceramidase (GALC) gene, is a model of the human genetic demyelinating disease called globoid cell leukodystrophy (Duchen et al., 1980; Wenger et al., 2000). The cause of demyelination in twitcher mice has been suggested to be the result of an accumulation of the toxic metabolite psychosine (Igisu and Suzuki, 1984b; Suzuki and Taniike, 1995), leading to the dysfunction of myelin-forming cells, i.e., OLs and Schwann cells. Because demyelination in the twitcher brain progresses in an orderly manner (Taniike and Suzuki, 1994), this mutant is a valuable model for investigating the molecular events occurring in OLs during the demyelinating process. The expression of OL markers, such as myelin basic protein, myelin-associated glycoprotein, and proteolipid protein, all decreased after the commencement of demyelination in the twitcher brain (Taniike et al., 1998). We previously reported that the depletion of OLs in the twitcher brain is caused by apoptosis (Taniike et al., 1999).
Because L-PGDS is expressed in normal mature OLs, we investigated L-PGDS expression in the twitcher brain during the demyelination process. Unexpectedly, we found that expression of L-PGDS was upregulated in twitcher brains. Furthermore, we found that the distribution of L-PGDS+ OLs and the severity of demyelination were in inverse relationship. Thus, we hypothesized that upregulation of L-PGDS may suppress apoptosis of OLs, since demyelination is secondary to the apoptosis of OLs in the twitcher brains. We finally confirmed the anti-apoptotic role of L-PGDS by generating double-mutant, L-PGDS-deficient (L-PGDS−/− ) twitcher (GALCtwi/twi ) mice.
MATERIALS AND METHODS
Animals. All animal experiments were performed according to the Japanese Law for the Protection of Experimental Animals and also conformed to the regulations issued by the National Institutes of Health and the Society for Neuroscience. Twitcher heterozygous pairs (GALCtwi/+ , the inbred C57BL/6J-twi strain) were originally purchased from Jackson Laboratory (Bar Harbor, ME), and the mutation was maintained by interbreeding of known heterozygous mice.L-PGDS−/− mice were generated through gene targeting technology (Eguchi et al., 1999) and backcrossed to the inbred C57BL/6J strain. Southern blot and/or PCR analysis of DNA prepared from clipped tails was conducted as previously described (Sakai et al., 1996; Eguchi et al., 1999). For the detection of wild-type or mutant alleles of L-PGDS, 5′ primer (P1: 5′-TGTCAGGAATGTGGTATGCTC-3′) was used together with either P2 (5′-AATACAGCTTTCTTCTCCCGGAAC-3′) or P3 (5′-GTAGCCGGATCAAGCGTATGC-3′) primers.
For generation of double mutant mice (L-PGDS−/−GALCtwi/twi ),L-PGDS−/− C57BL/6J offspring (L-PGDS−/−GALC+/+ ) were mated withL-PGDS+/+GALCtwi/+ to create F1 (L-PGDS+/−GALC+/+ andL-PGDS+/−GALCtwi/+ ). Of the F2 litters produced by mating of F1,L-PGDS−/−GALCtwi/+ were mated with each other to produceL-PGDS−/−GALCtwi/twi F3. ControlL-PGDS+/+GALCtwi/twi F3 were produced by matingL-PGDS+/+GALCtwi/+ F2 mice. These two types of F3 mice thus produced were subjected to the analysis. The frequency of the each genotype in all generations conformed to Mendelian inheritance patterns.
Materials. Rabbit polyclonal and rat monoclonal anti-mouse L-PGDS antibodies were raised and purified as described previously (Eguchi et al., 1999). Preabsorbed antibody was prepared by incubation of polyclonal antibody with an excess amount of purified recombinant L-PGDS. These antibodies were used at a dilution of 1:1000. The other antibodies used were as follows: rabbit polyclonal anti-mouse pi-form of glutathione S-transferase (pi-GST) antibody (1:1000; Biotrin International, Dublin, Ireland), rabbit polyclonal anti-mouse carbonic anhydrase II (CA II) antibody (1:1000; a generous gift from Dr. W. Cammer, Department of Neuroscience, Albert Einstein College of Medicine), rabbit polyclonal anti-cow glial fibrillary acidic protein (GFAP) antibody (prediluted; Dako, Glostrup, Denmark), rabbit polyclonal anti-S-100 antibody (1;1000; DAKO), rat monoclonal anti-mouse F4/80 antibody (1:400; Serotec, Oxford, UK), and mouse monoclonal anti-rat microtubule-associated protein 2 (MAP2) antibody (1:200; Sigma, St. Louis, MO). Biotinylated Ricinus communis-agglutinin-1 (RCA-1; 50 μg/ml) was purchased from Vector Laboratories (Burlingame, CA).
RNA isolation, electrophoresis, and hybridization.Under deep ether anesthesia, mice were killed at postnatal day 15 (P15), P20, P30, and P45. Entire brains or brains divided into cerebellum and cerebrum were used. When indicated, leptomeninges and choroid plexus were removed under a dissecting microscope. The brains were quickly frozen in liquid nitrogen, with subsequent total RNA isolation by the guanidinium thiocyanate–phenol–chloroform method (Chomczynski and Sacchi, 1987). Total RNA (10 μg) was electrophoresed in an agarose gel, transferred to Zeta Probe nylon membranes (Bio-Rad Laboratories, Hercules, CA), and hybridized with32P-labeled cDNA probes specific for mouse L-PGDS (Eguchi et al., 1999), mouse CA II, or mouse glyceraldehyde-3-phosphate dehydrogenase (G3PDH). The blots were visualized by autoradiography with Kodak XAR-5 film and an intensifying screen. The relative amount of each transcript was estimated by quantifying the associated radioactivity with the BAS-2000 system (Fuji film, Tokyo, Japan).
In situ hybridization. Paraffin sections of mouse brains were hybridized with 35S- or digoxigenin-labeled antisense riboprobe for mouse L-PGDS as previously described (Urade et al., 1993; Gerashchenko et al., 1998). Signal specificity was assessed by incubation of adjacent sections with the labeled sense riboprobe.
Measurement of L-PGDS activity and psychosine content in brain. L-PGDS activity was determined in mouse brains with 40 μm [1-14C] PGH2 in the presence of 1 mm dithiothreitol (Urade et al., 1985a). Protein concentrations were determined by use of bicinchoninic acid reagent (Pierce, Rockford, IL) with bovine serum albumin used as the standard by following the manufacturer's protocol. The level of psychosine in mouse brains was measured by the method previously described (Igisu and Suzuki, 1984a).
Immunohistochemistry, lectin histochemistry, and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining
We used four mice per genotype for each age for immunohistochemical analysis for L-PGDS. Under deep ether anesthesia, mice were briefly perfused through the left cardiac ventricle with physiological saline followed by 4% paraformaldehyde in 0.1m phosphate buffer (PB; pH 7.4). Their brains were immediately removed, postfixed overnight in the same solution, and processed for paraffin embedding.
The immunohistochemical results with a polyclonal rabbit antibody and a monoclonal rat antibody against mouse L-PGDS (Eguchi et al., 1999) were basically the same. Therefore, monoclonal antibody was used in most cases because of lower background staining. Deparaffinized and hydrated sections (5-μm-thick) were preincubated with 0.3% H2O2 in methanol followed by PBS containing 0.2% Triton X-100. After pretreatment with trypsin for 15 min, the sections were sequentially incubated with L-PGDS antibody, appropriate biotinylated secondary antibody, and avidin–biotin complex (ABC; Vector Laboratories) according to the manufacturer's protocol. Immunoreactivity was visualized with 0.03% H2O2 solution in 50 mm Tris-HCl, pH 7.6, containing 0.05% diaminobenzidine (Dotite, Kumamoto, Japan).
For double immunostaining, anti-pi-GST/CA II, GFAP/S-100, and F4/80 antibodies were used to identify OLs, astrocytes, and microglia–macrophages, respectively (Tansey and Cammer, 1991; Toyooka et al., 1993; Taniike and Suzuki, 1995). Deparaffinized sections were incubated at 4°C overnight with one of these primary antibodies together with anti-mouse L-PGDS antibody. Rat anti-L-PGDS antibody was used in all cases except for the staining combined with F4/80, in which case rabbit L-PGDS antibody was used. Sections were then reacted with Texas Red-conjugated anti-rabbit IgG antibody (2 μg/ml; ICN Biomedicals, Aurora, OH) and biotin-conjugated anti-mouse IgG antibody (Vector Laboratories) for 2 hr, followed by FITC-conjugated avidin D (2 μg/ml; Vector Laboratories) for 2 hr. Absence of cross-reactivity between secondary antibodies was confirmed by omission of either primary antibody. All sections were observed under an Axiovert 100M microscope connected to a Zeiss laser-scanning microscope 510 (Carl Zeiss, Oberkochen, Germany).
For RCA-1 lectin histochemistry, after pretreatment with 3% H2O2 and 0.1% bovine serum albumin, deparaffinized sections were incubated with biotinylated RCA-1 in PBS for 30 min. The incubation with ABC and the visualization of horseradish peroxidase activity were performed as described (Toyooka et al., 1993).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) histochemistry combined with immunohistochemistry for pi-GST or MAP2 was performed as described (Taniike et al., 1999). In the case of MAP2 staining, pretreatment with a mouse-on-mouse immunodetection kit (Vector Laboratories) was necessary.
Electron microscopy. After fixation with 3% glutaraldehyde and 1% paraformaldehyde in 0.1 m PB, pH 7.2, followed by postfixation with 1% osmium tetrahydroxide, coronally cut brains were routinely processed and examined. After observation of 1-μm-thick semithin sections stained with Toluidine blue, areas for ultrathin sections were selected and cut. Ultrathin sections of the cerebellar hemisphere were stained with uranyl acetate and lead citrate and examined with a JEM-100CX electron microscope (JEOL, Tokyo, Japan) at 80 kV.
Cell counts and statistical analysis.L-PGDS+ cells and TUNEL+ cells were counted in paraffin sections prepared from mice at P35 and P45, six mice at each time. L-PGDS+ cells were counted in 45-d-old L-PGDS+/+ GALCtwi/twi brain parenchyma.
Statistical comparisons were made by Student's t test. Values of p < 0.05 were considered to be significant.
RESULTS
Upregulation of L-PGDS in OLs ofGALCtwi/twi mice
Figure 1A shows that L-PGDS mRNA in GALCtwi/twi brains increased progressively after P30 when demyelination was already obvious (Taniike and Suzuki, 1994), whereas its level in wild-type (GALC+/+ ) brains had already reached a plateau by P30. It reached a level 1.7 times higher inGALCtwi/twi than inGALC+/+ by P45. In contrast, mRNA for CA II, which is a myelin/OL-associated protein, did not increase after P20 in GALCtwi/twi brains and was 61% of theGALC+/+ level at P45. L-PGDS activities in mouse brains at P39 were as follows (mean ± SE, n = 4):GALC+/+ cerebrum, 0.27 ± 0.05 nmol · min−1 · mg−1protein; GALCtwi/twi cerebrum, 0.72 ± 0.06 nmol · min−1 · mg−1protein; GALC+/+ cerebellum, 0.28 ± 0.04 nmol · min−1 · mg−1protein; GALCtwi/twi cerebellum, 0.74 ± 0.11 nmol · min−1 · mg−1protein. Thus, L-PGDS activity was significantly increased in both cerebrum (p < 0.005) and cerebellum (p < 0.01) inGALCtwi/twi as compared with that inGALC+/+ .

Upregulation of mRNA for L-PGDS inGALCtwi/twi OLs. A,Northern blot analysis for L-PGDS, CA II, and G3PDH. Wand T represent GALC+/+ and GALCtwi/twi,respectively. Postnatal days are indicated at the top of the columns. The corrected values (/G3PDH) are shown in the lower graph. B, Northern blot analysis for L-PGDS and G3PDH in brain parenchyma without leptomeninges and choroid plexus (P), the isolated leptomeninges and choroid plexus (L), and the whole cerebellum (C) from two mouse brains at P30. W-1 and W-2 areGALC+/+ , and T-1 and T-2 areGALCtwi/twi. C–H, In situ hybridization for L-PGDS mRNA in coronal mouse brain sections at P40 with 32P-labeled probe (C–E) or digoxigenin-labeled probe (F–H). C–E, Cerebellum and medulla. DCN is indicated by asterisk inD. F–H, GALCtwi/twi. F, The ventral part of the forebrain. The leptomeninges (arrows) as well as OLs (arrowheads) possess mRNA for L-PGDS. G, Cerebellum. Interfascicular OLs displaying strong signals are indicated byarrowheads. The astrocytes, having characteristically large pale nuclei, are negative for L-PGDS mRNA (small arrows). IG and CWM represent the internal granular layer and the cerebellar white matter, respectively.H, Facial nuclei. Perineuronal OLs (neurons are shown byn) show strong signals for L-PGDS. Scale bars:C–E, 2 mm; F, 100 μm;G, H, 20 μm.
To exclude the possibility that the increase in L-PGDS inGALCtwi/twi brains was attributable to its increased production in leptomeninges and choroid plexus, we compared the expression level of L-PGDS mRNA between brains with and without leptomeninges (Fig. 1B). The level of L-PGDS mRNA inGALCtwi/twi cerebral parenchyma without leptomeninges (designated as “P” in Fig. 1B) increased fivefold to eightfold as compared with that ofGALC+/+ mice, whereas the levels in isolated leptomeninges and choroid plexus (designated as “L” in Fig. 1B) and in whole cerebellum (“C” in Fig. 1B) increased only 10–40% inGALCtwi/twi mice. The upregulation of L-PGDS mRNA in the GALCtwi/twi brain parenchyma was also confirmed by in situ hybridization (Fig.1C–E). Except for its presence in leptomeninges, L-PGDS mRNA was undetectable in either GALC+/+ orGALCtwi/twi brains before the onset of demyelination at P15 (data not shown). The obvious leptomeningeal signal was recognized in both GALC+/+ andGALCtwi/twi (Fig. 1C,D, F, arrows). In addition, at P40 when demyelination had already advanced, a strong signal was recognized throughout the cerebellar white matter and medulla, particularly in the deep cerebellar nuclei (DCN) (Fig. 1D, asterisk), inGALCtwi/twi . Higher magnification disclosed that the intense signal was observed only in OLs in theGALCtwi/twi (Fig.1F–H). L-PGDS mRNA was recognized in both interfascicular OLs of the white matter (Fig. 1G) and perineuronal OLs of the gray matter (Fig. 1H).
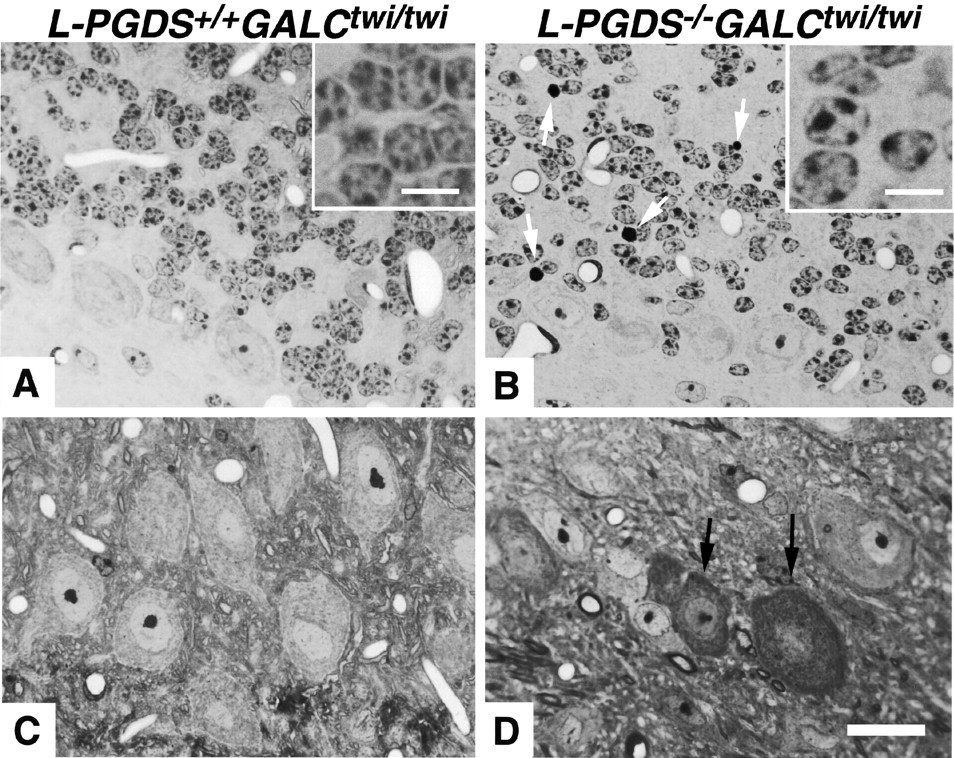
In GALC+/+ , only a few L-PGDS+ perineuronal OLs were detected throughout all ages examined (Fig.2A,C, insets). In contrast, in GALCtwi/twi brains, L-PGDS+ OLs were increased in number in both the gray and white matter after P25, and many cells with morphological features of degenerating OLs with multiple varicose processes (Taniike et al., 1999) showed intense L-PGDS immunoreactivity at P40 (Fig. 2B,D, insets). The use of the double immunofluorescence technique showed that L-PGDS+ cells were positive for neither GFAP nor S-100, markers for astrocytes, nor for F4/80, a marker for microglia/macrophages (Taniike and Suzuki, 1995; data not shown). Confocal images of GALCtwi/twi brains disclosed that all L-PGDS+ cells were immunoreactive for CAII (Fig. 2E) or pi-GST (Fig.2F,G) and were thus identified as OLs (Taniike et al., 1999). In addition, all perineuronal OLs were also immunoreactive for L-PGDS (Fig. 2F, arrows), whereas interfascicular OLs were occasionally L-PGDS− (Fig.2G, arrowheads). L-PGDS+ OLs were abundant in the DCN (Fig. 2D), cerebellar cortex, anterior commissure, and optic nerve; but they were rare in the corpus callosum (Fig. 2G, arrows) and internal capsule, and L-PGDS was not detected at any age examined in interfascicular OLs of the intermedullary portion of the eighth nerve root (8n) and spinal trigeminal tract (sp5) (Fig.3A,E), which are sites of extensive demyelination in GALCtwi/twi (Taniike and Suzuki, 1994).

Increase in number of L-PGDS-immunoreactive OLs inGALCtwi/twi brains at P40.A, B, The primary motor area in the forebrain. Scale bars: A, B, 200 μm;insets, 20 μm. C,D, DCN facing the fourth ventricle. Scale bars:C, D, 100 μm; insets, 5 μm. Arrows and arrowheadsindicate L-PGDS+ cell somas and their varicose processes, respectively, in aGALCtwi/twi brain.E–G, Confocal images for the double immunostaining for L-PGDS (green) and CAII (E, red) or pi-GST (F, G, red) of the brainstem (E), cerebral cortex (F), and corpus callosum (G) ofGALCtwi/twi at P40.Arrows in E–G andarrowheads in G indicate L-PGDS+ OLs and L-PGDS− OLs, respectively. Scale bars, 10 μm.

Inverse relationship between the distribution patterns of L-PGDS+ cells and microglia/macrophages in GALCtwi/twibrains at P40. A–D, Cerebellopontine angle.Asterisks indicate 8n in A andB. Scale bars, 1 mm. C andD are higher magnification views of the boxed areas in A and B, respectively.Asterisks and daggers indicate CWM and DCN, respectively. Scale bars, 50 μm. E, F, Medulla.Asterisks indicate sp5. Scale bars, 0.5 mm.
These results clearly show that L-PGDS was upregulated inGALCtwi/twi OLs in a region-specific manner.
Inverse relationship between the distribution of L-PGDS+ OLs and the severity of demyelination inGALCtwi/twi brains
Next we investigated the relationship between L-PGDS immunoreactivity and the severity of demyelination inGALCtwi/twi brains. Figure 3 shows serial sections of the cerebellopontine angle region (Fig.3A–D) and the medulla (Fig. 3E,F) of theGALCtwi/twi at P40. These regions were already described to be the most severely demyelinated inGALCtwi/twi brains (Taniike and Suzuki, 1994). Accumulation of RCA-1+ scavenger macrophages, a pathological hallmark of demyelination, was obvious in 8n (Fig. 3A,B, asterisks) and in sp5 (Fig. 3E,F, asterisks), in which no L-PGDS+ OLs were detected (Fig. 3A,E). On the contrary, a close-up view of the interface between cerebellar white matter (CWM) (Fig.3C,D, asterisks) and DCN (Fig. 3C,D, daggers) revealed that very few macrophages had infiltrated into the DCN where L-PGDS+ OLs were abundant (Fig.3C). These data clearly show that the regional distribution profiles of L-PGDS+ OLs and macrophages were in inverse relationship.
Apoptotic cells significantly increased in L-PGDS−/− GALCtwi/twibrains
Because OL depletion and subsequent demyelination in GALCtwi/twi are attributable to apoptosis of OLs (Taniike et al., 1999), we predicted that L-PGDS may be upregulated in OLs to provide resistance against the apoptosis and subsequent demyelination in GALCtwi/twi . To verify this hypothesis, we established a mouse line doubly deficient for GALC and L-PGDS (Fig.4).

Enhanced apoptosis and macrophage aggregation inL-PGDS−/−GALCtwi/twi. A, The restriction map for the wild-type allele and the mutated allele for L-PGDS. Restriction sites: P,PvuII; N, NcoI;A, AcyI.Neor , neomycin resistance gene. The combination of P1/P2 primers or P1/P3 primers was used to detect wild-type or null allele for L-PGDS, respectively.GALC genotype was identified by the different length of the EcoRV-digested fragments of the PCR fragments; i.e., the twitcher allele has a shorter fragment length. B–G,Cerebellar cortex at P45. H–M, DCN at P45. Scale bars, 100 μm.
There was no significant difference betweenL-PGDS−/−GALCtwi/twi andL-PGDS+/+GALCtwi/twi in terms of the life span (50.3 ± 0.3 d inL-PGDS+/+GALCtwi/twi vs 50.1 ± 0.6 d inL-PGDS−/−GALCtwi/twi ;n = 9). The neuropathology of both genotypes was similar until P35. Psychosine, which is a potent neurotoxin that accumulates in the GALCtwi/twi brain as a consequence of the genetic defect, has been suggested to induce OL dysfunction and apoptosis. The content of psychosine at P45 was the same in theL-PGDS−/−GALCtwi/twi (cerebrum, 6.67 ± 1.07 ng/mg protein; cerebellum, 22.74 ± 6.87 ng/mg protein) and in theL-PGDS+/+GALCtwi/twi (cerebrum, 7.68 ± 1.92 ng/mg protein; cerebellum, 22.40 ± 6.41 ng/mg protein) brains.
However, at P45,L-PGDS−/−GALCtwi/twi brains showed more severe demyelination than theL-PGDS+/+GALCtwi/twi ones. At P35, the number of TUNEL+ nuclei was already moderately increased in the caudate–putamen and 8n ofL-PGDS−/− GALCtwi/twi brains; however, by P45, this number was drastically increased in theL-PGDS−/−GALCtwi/twi brains (Fig. 4F,L) as compared with the number in theL-PGDS+/+GALCtwi/twi ones (Fig. 4C,I). InL-PGDS−/−GALCtwi/twi brains, TUNEL+ cells were especially increased in number in the gray matter such as the internal granular layer of the cerebellum (Fig. 4C,F, compare IG) or DCN (Fig.4, compare I, L). The number of apoptotic nuclei was also increased in the white matter such as CWM (Fig. 4, compareC, F) and corpus callosum. In both the DCN and IG ofL-PGDS+/+GALCtwi/twi brains, a large number of L-PGDS+perineuronal OLs was observed (Fig.4B,H). InL-PGDS−/− GALCtwi/twi brains, however, apoptotic cell death was accompanied by many F4/80+ activated microglia and/or infiltrating macrophages in the IG (Fig. 4G) and DCN (Fig.4M), where F4/80+ cells were quite few (Fig. 4D,J) with very mild demyelination, as previously described (Fig. 3, daggers), in theL-PGDS+/+ GALCtwi/twi brains.
Figure 5A summarizes the spatiotemporal distributional change in RCA-1+ microglia/macrophages, a pathological hallmark of demyelination (Taniike et al., 1994), and L-PGDS+ OLs in theL-PGDS+/+GALCtwi/twi brains. In the most severely affected areas such as the 8n or sp5, very few L-PGDS+ OLs were observed throughout the ages examined. On the other hand, in the least affected areas such as the lateral cerebellar nucleus (Fig. 5A, Lat) and dorsal cochlear nucleus (Fig. 5A, DC), L-PGDS+ cells were found in abundance from the early stage of demyelination. Figure 5B summarizes the population of L-PGDS+ OLs and apoptotic nuclei counted in seven brain regions. Because perineuronal OLs tended to express more intense L-PGDS immunoreactivity than interfascicular OLs as described above, we chose to analyze four regions from gray matter (cerebral cortex, caudate–putamen, cerebellar cortex of anterior vermis, and DCN) and two regions from white matter (corpus callosum and 8n). In addition, because the increase in number of TUNEL+ nuclei was the most remarkable in the paraflocculus/flocculus of cerebellum, the gray and white matter of this region was analyzed as a whole. Whereas TUNEL+ nuclei density (nuclei/mm2) was already significantly increased at P35 (Fig. 5B, top panel) in the caudate–putamen (from 5.6 ± 0.5 inL-PGDS+/+GALCtwi/twi to 18.3 ± 5.0 inL-PGDS−/−GALCtwi/twi ;p < 0.05), and 8n (from 14.4 ± 6.1 to 32.1 ± 7.7; p < 0.005), it was remarkably increased at P45 (Fig. 5B, middle panel) in the DCN (from 36 ± 5 inL-PGDS+/+GALCtwi/twi to 183 ± 32 inL-PGDS−/−GALCtwi/twi ;p < 0.01), cerebellar cortex (from 47 ± 3 to 251 ± 32; p < 0.05), paraflocculus/flocculus (from 79 ± 36 to 307 ± 34; p < 0.005), caudate–putamen (from 333 ± 34 to 603 ± 54;p < 0.01), and corpus callosum (from 47 ± 3 to 80 ± 10; p < 0.05). No significant difference in the number of TUNEL+ nuclei was observed in the cerebral cortex (31 ± 5 and 40 ± 2) and 8n (60 ± 9 and 86 ± 17) between the respectiveL-PGDS+/+GALCtwi/twi andL-PGDS−/− GALCtwi/twi brains at P45. The density of L-PGDS+ OLs (in cells per square millimeter) in 45-d-oldPGDS+/+GALCtwi/twi brains (Fig. 5B, bottom panel) was high in DCN (153.4 ± 6.2) and cerebellar cortex (140.9 ± 6.3), moderate in the paraflocculus/flocculus (99.4 ± 16.7) and corpus callosum (95.8 ± 10.2), and low in the cerebral cortex (35.3 ± 1.3), caudate–putamen (15.0 ± 1.5), and 8n (1.8 ± 1.8). When the ratio of apoptotic nuclei inL-PGDS−/−GALCtwi/twi versus those inL-PGDS+/+GALCtwi/twi was plotted against the population of L-PGDS+ OLs in theL-PGDS+/+GALCtwi/twi (Fig. 5C), a linear positive correlation between these two parameters was observed, suggesting a dose-dependent effect of L-PGDS against apoptosis. In addition, the finding that apoptotic nuclei were more increased in number in the gray matter (DCN, cerebellar cortex, caudate–putamen) than in the white matter (corpus callosum and 8n) implies not only a larger anti-apoptotic effect by perineuronal OLs than by interfascicular OLs but also the possibility of neuronal apoptosis inL-PGDS−/−GALCtwi/twi brains.

Positive relationship between the regional distribution of L-PGDS+ OLs and the increase in apoptotic cells caused by L-PGDS deficiency. A, The population of RCA-1+ microglia/macrophages (pink triangles in the left half) and L-PGDS+ cells (blue dots in the right half) inL-PGDS+/+GALCtwi/twi brains at P35 and P45. Abbreviations: 7, facial nucleus;8n, vestibulocochlear nerve root; DC, dorsal cochlear nucleus; Gi, gigantocellular reticular nucleus; GiA, gigantocellular reticular nucleus (α part); IntA, interposed cerebellar nucleus (anterior part); icp, inferior cerebellar peduncle;Lat, lateral cerebellar nucleus; Lve, lateral vestibular nucleus; Mve, medial vestibular nucleus; py, pyramidal tract; RMg, raphe magnus nucleus; scp, superior cerebellar peduncle;sp5, spinal trigeminal tract; Sp5, spinal trigeminal nucleus; VCP, ventral cochlear nucleus (posterior part). B, The population of TUNEL+ apoptotic nuclei (P35 and P45; hatched columns, L-PGDS+/+GALCtwi/twi ;open columns, L-PGDS−/−GALCtwi/twi ) and L-PGDS+ cells ofL-PGDS+/+GALCtwi/twi at P45 (solid columns) in seven regions. *p < 0.05; **p < 0.01; ***p < 0.005. C, The ratio of the number of TUNEL+ cells inL-PGDS−/−GALCtwi/twi versus that inL-PGDS+/+GALCtwi/twi at P45 was plotted against the population of L-PGDS+cells inL-PGDS+/+GALCtwi/twi brains. r = 0.875 (p < 0.01).
Neuronal apoptosis is induced inL-PGDS−/−GALCtwi/twi
We chose the cerebellum for further analysis, because a large number of TUNEL+ nuclei were recognized there. The observation of 1 μm cerebellar sections disclosed many apoptotic nuclei (Fig. 6B, white arrows) and the loss of granular cells in theL-PGDS−/−GALCtwi/twi . Granular cells were present in a cluster or a row inL-PGDS+/+GALCtwi/twi (Fig. 6A), whereas these cells were often solitary inL-PGDS−/−GALCtwi/twi (Fig. 6B). In addition, the nuclear size of granular cells was much more variable and usually smaller inL-PGDS−/−GALCtwi/twi than inL-PGDS+/+ GALCtwi/twi . The chromatin ofL-PGDS−/−GALCtwi/twi granular cells was condensed into fewer and larger nucleoli than that ofL-PGDS+/+GALCtwi/twi ones, making these nuclei lighter and clearer (Fig. 6A,B, insets). These nuclear changes are suggestive of the early phase of apoptosis. In the DCN, degenerating neurons with a dark cytoplasm were frequently observed in L-PGDS−/−GALCtwi/twi brains (Fig. 6D, arrows) but not at all inL-PGDS+/+GALCtwi/twi ones (Fig. 6C).

Degeneration and enhanced apoptosis of cerebellar neurons inL-PGDS−/−GALCtwi/twi brains at P45. Semithin sections (1 μm) stained with Toluidine blue.A, B, The Purkinje cells and IG. Apoptotic nuclei are indicated by white arrows. C, D, DCN neurons. Dark degenerating neurons are indicated by arrows in D. Scale bars: 15 μm; insets, 5 μm.
In the IG ofL-PGDS−/−GALCtwi/twi , most of the TUNEL+ cells were pi-GST− (Fig.7A) and MAP2+neurons (Fig. 7B), whereas a considerable number of TUNEL+cells in the CWM and molecular layer (ML) (Fig. 7A, arrows) were identified as pi-GST+ OLs. We observed no TUNEL+ cells inL-PGDS−/− GALC+/+ and no MAP2+ TUNEL+ cells inL-PGDS+/+ GALCtwi/twi (data not shown).
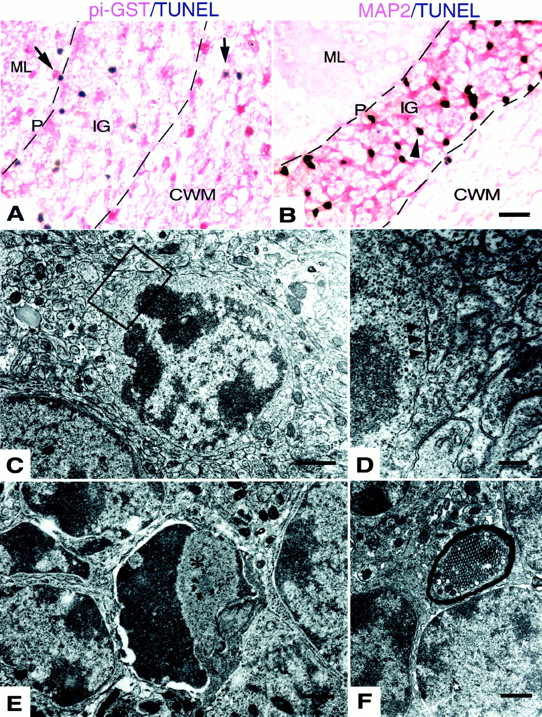
Most apoptotic cells in theL-PGDS−/−GALCtwi/twi cerebellum at P45 were neurons. A, B, TUNEL histochemistry. P and ML represent the Purkinje cell layer and molecular layer, respectively. Pi-GST+TUNEL+ cells representing apoptotic OLs are indicated by arrows inA. A MAP2− TUNEL+ cell is indicated by the arrowhead in B. Scale bar, 20 μm. C–F, Electron micrographs. D is a higher magnification view of the boxed area inC. An axosomatic synaptic contact between the apoptotic cell and the neighboring axon is indicated by thearrowheads in D. An apoptotic cell inE, indicated by an asterisk, is surrounded by apparently intact internal granular layer neurons. A tubulovesicular structure (asterisk) is seen inF. Scale bars: C, E,F, 1 μm; D, 0.2 μm.
Electron microscopic investigation disclosed that many cells in the IG of L-PGDS−/− GALCtwi/twi brains showed nuclear changes characteristic of apoptosis such as ruffled nucleoli or condensed and clumpy chromatin beneath their nuclear membrane (Fig. 7C,E). Some of these apoptotic cells showed synaptic contact (Fig. 7D, arrowheads), indicating them to be cerebellar granular cells. A tubulovesicular structure was frequently visible in axons inL-PGDS−/− GALCtwi/twi (Fig. 7F, asterisk). These structures have been reported to be present in cerebellum under diverse experimental and pathological conditions, and they may represent a stage in the degeneration of axonal collaterals and terminals (for review, see Sotelo and Palay, 1971). Axons with these structures were also recognized in theL-PGDS+/+GALCtwi/twi cerebellum (7.3 ± 4.7/100 myelinated axons), but were significantly increased in number inL-PGDS−/− GALCtwi/twi (33.4 ± 3.6/100 myelinated axons; p < 0.02), suggesting that axonal integrity was perturbed in the L-PGDS−/− GALCtwi/twi because of the loss of axonal contact with apoptotic neurons.
From these lines of evidence, we concluded that the augmentation of apoptosis inL-PGDS−/−GALCtwi/twi was caused by disappearance of the anti-apoptotic function of L-PGDS. L-PGDS produced by perineuronal OLs thus appears to play a protective role against neuronal degeneration.
DISCUSSION
L-PGDS as the first OL-associated protein upregulated during demyelination
In this report, we demonstrate that L-PGDS expression is upregulated in GALCtwi/twi OLs with progressive demyelination (Figs. 1, 2). As noted in other models of demyelination, such as experimental autoimmune encephalomyelitis (Itoyama and Webster, 1982; Yao et al., 1995), virus-induced demyelination (Rodriguez et al., 1994; Barac-Latas et al., 1997), and cuprizone intoxication (Fujita et al., 1990; Tansey et al., 1996;Morell et al., 1998), the expression of myelin/OL-associated proteins, such as CA II (Fig. 1A), myelin basic protein, myelin-associated glycoprotein, proteolipid protein, and UDP–galactose:ceramide galactosyltransferase was downregulated after the commencement of demyelination inGALCtwi/twi (Taniike et al., 1998). The expression of pi-GST, another OL-associated protein, remained constant during demyelination in GALCtwi/twi (Taniike et al., 1999). Besides the possible upregulation of apoptosis-associated proteins such as caspases, L-PGDS is the first non-apoptosis-associated protein demonstrated to be upregulated in OLs during progressive demyelination. In a recent report, L-PGDS was the second most abundant gene upregulated in the brains of patients with multiple sclerosis (Chabas et al., 2001). Moreover, our preliminary data strongly suggested that L-PGDS was upregulated in active lesions in mice with experimental autoimmune encephalomyelitis (data not shown). These findings suggested that the upregulation of L-PGDS was a common phenomenon among the demyelinating diseases, regardless of their primary etiology.
Because of the inverse correlation between L-PGDS+ OLs and the severity of demyelination in GALCtwi/twi brains (Fig.3), we hypothesized an anti-apoptotic function of upregulated L-PGDS in the CNS. However, the evaluation was made complex by the presence of a considerable number of hematogeneous cells, which infiltrate intoGALCtwi/twi brains after P30 (Wu et al., 2000) and secrete apoptotic cytokines such as TNF-α inGALCtwi/twi white matter (K. Shimono, Y. Fujitani, I. Mohri, M. Taniike, and Y. Urade, unpublished observations). The reason why the number of L-PGDS+ cells was not simply inversely related to the number of apoptotic nuclei in eitherL-PGDS−/−GALCtwi/twi orL-PGDS+/+GALCtwi/twi brains might be explained by the presence of a significant number of apoptotic macrophages as well as apoptotic effects of TNF-α.
For example, in the caudate–putamen or the white matter of the paraflocculus/flocculus, where TNF-α expression was high with a lot of infiltrating macrophages, the number of TUNEL+ nuclei was unproportionally high. Even in these regions, the anti-apoptotic effect of L-PGDS was obvious, and the TUNEL+ nuclei increased inL-PGDS−/−GALCtwi/twi . On the other hand, in the regions with minimal cellular infiltration such as DCN or cerebellar cortex, TUNEL+nuclei increased multi-fold inL-PGDS−/−GALCtwi/twi when compared with their number inL-PGDS+/+GALCtwi/twi .
The distribution of L-PGDS+ OLs was region specific. TUNEL+ nuclei-rich areas inL-PGDS+/+ GALCtwi/twi brains at P45 such as 8n or caudate–putamen contained only a small number of L-PGDS+ cells, and vice versa for TUNEL+ nuclei-poor areas such as the VCA or DCN (Fig. 5A,B). As compared with the number inL-PGDS+/+GALCtwi/twi , the number of apoptotic cells inL-PGDS−/−GALCtwi/twi increased in 35-d-old as well as 45-d-old mice (Figs. 4,5B,C). From these lines of evidence, we verified the anti-apoptotic function of upregulated L-PGDS in the CNS. In 8n or caudate–putamen of L-PGDS+/+ GALCtwi/twi brains, the number of L-PGDS+ OLs was constantly small, whereas the number of apoptotic OLs recognized as TUNEL+pi-GST+ cells (Taniike et al., 1999) remarkably increased in an age-dependent manner. Thus, we concluded that the upregulation of L-PGDS does not simply reflect an ongoing apoptosis in OL but possibly reflects heterogeneity of OL subpopulations with regard to L-PGDS inducibility.
The dysfunction of Schwann cells leading to peripheral demyelination is the principal cause of death inGALCtwi/twi mice (Taniike et al., 1994). L-PGDS was neither expressed in Schwann cells ofGALC+/+ nor induced in those ofGALCtwi/twi , and there was no significant difference in the life span between theL-PGDS+/+GALCtwi/twi andL-PGDS−/−GALCtwi/twi mice.
Possible anti-apoptotic mechanisms of L-PGDS
Because L-PGDS is a bifunctional protein acting as a PGD2-producing enzyme and as a lipophilic ligand-carrier protein of the lipocalin family (Urade and Hayaishi, 2000a), there are at least two possibilities for its anti-apoptotic mechanism, as discussed below.
PGD2 elicits its biological actions through binding to the Gs-coupled D-type PG receptor (DPR;Mizoguchi et al., 2001) or to the Gαi-coupled CRTH2 receptor (Hirai et al., 2001). Otherwise, PGD2 is dehydrated to yield the J series of PGs such as 15-deoxy-Δ12,14-PGJ2in vitro, which has been identified as a ligand for peroxisome proliferator-activated receptor-γ (PPAR-γ; Forman et al., 1995;Kliewer et al., 1995). PGD2 increases intracellular cAMP through its binding to DPR (Hirata et al., 1994). The increased cAMP prevents oligodendroglial excitotoxicity and the eventual death of these cells (Yoshioka et al., 1998). Thus, PGD2 produced by upregulated L-PGDS in perineuronal OLs may protect these cells from apoptosis by an increase in intracellular cAMP levels mediated via DPR. CRTH2 is expressed in the mouse brain (Abe et al., 1999), but its function and cellular localization are still unknown. Although the natural occurrence of 15-deoxy-Δ12,14-PGJ2in the brain remains to be clarified, activation of PPAR-γ induces apoptosis in several cell lines (Kawahito et al., 2000; Padilla et al., 2000; Rohn et al., 2001). It is, therefore, worth studying the contribution of CRTH2 and PPARγ pathways to the apoptosis of OLs inGALCtwi/twi .
On the other hand, PGD2 inhibits the expression of inducible nitric oxide synthase (iNOS) in vascular smooth muscle cells (Nagoshi et al., 1998). iNOS induces demyelination and/or OL apoptosis (Gilg et al., 2000; Liu et al., 2001; Molina-Holgado et al., 2001) and apoptosis of cerebellar granular neurons (Minc-Golomb et al., 1994, 1996; Sato et al., 1996). Many cerebellar granular neurons showed apoptotic changes inL-PGDS−/− GALCtwi/twi (Figs. 4, 6, 7); however, the upregulation of iNOS inL-PGDS−/−GALCtwi/twi was not clearly recognized (data not shown). Therefore, we do not consider iNOS to play a major role in the neuronal apoptosis inGALCtwi/twi brains.
As a lipocalin, L-PGDS is secreted into the CSF and binds to and transports small hydrophobic molecules such as biliverdin, bilirubin, and retinoic acid (for review, see Urade and Hayaishi, 2000a). Therefore, L-PGDS may work as a scavenger of gliotoxic and neurotoxic molecules to protect OLs and neurons from degeneration. Psychosine is also a small hydrophobic molecule and a potent neurotoxin that accumulates in GALCtwi/twi (Igisu and Suzuki, 1984a,b); however, only weak affinity between L-PGDS and psychosine was detected by surface plasmon resonance spectroscopy (data not shown). In addition, psychosine levels ofL-PGDS+/+GALCtwi/twi andL-PGDS−/−GALCtwi/twi brains were not significantly different, as described in Results. It is, therefore, unlikely that L-PGDS is involved in scavenging psychosine.
It was recently reported that L-PGDS induced apoptosis in PC-12 cells (Ragolia et al., 2001) or pig kidney LLC-PK1 cells (Maesaka et al., 2001). Moreover, the 24p3 protein, which is another lipocalin evolutionally closely related to L-PGDS (Toh et al., 1996), was reported to induce apoptosis of IL-3-deprived hematopoietic cells (Devireddy et al., 2001). These lines of evidence strongly suggested that L-PGDS may function as an apoptotic or anti-apoptotic molecule depending on its concentration, cell types, and so on. It is also possible that infiltrating macrophages produced PGs at a level high enough to induce apoptosis, but that in the areas with little cellular infiltration, the moderate amount of PGD2released by OLs is served an anti-apoptotic role. The anti-apoptotic mechanism of L-PGDS in GALCtwi/twi will be clarified by a gene transfer experiment in which either the intact L-PGDS or mutated L-PGDS gene, the latter encoding the protein that has lost enzymatic activity and functions only as a lipocalin, is injected toL-PGDS−/−GALCtwi/twi brains to see by which construct the augmented apoptosis is restored to theL-PGDS+/+GALCtwi/twi level.
Novel OL–neuronal interaction mediated by L-PGDS
As evident from our previous report that showed L-PGDS immunoreactivity in 8-week-old rat brains and not in 2-week-old ones in active myelination (Urade et al., 1987), the function of L-PGDS in OLs is unlikely to be linked to myelination but to cellular maintenance. It is also noteworthy that the L-PGDS immunoreactivity of perineuronal OLs in the gray matter was more intense than that of interfascicular myelinating OLs in the white matter of 40-d-oldL-PGDS+/+GALCtwi/twi mice (Fig. 2). Only a few reports have proposed functions of perineuronal OLs: Ludwin (1979, 1984) found that perineuronal OLs in the periventricular gray remyelinated axons in cuprizone-induced or trauma-induced demyelination and concluded that these OLs functioned similarly as myelinating OLs. D'Amelio et al. (1990) demonstrated that immunoreactivity for glutamine synthetase was distributed in perineuronal OLs as well as in astrocytes and that perineuronal OLs fulfill a functional role more akin to that of astrocytes. We propose here a novel function of perineuronal OLs, i.e., protection of neurons from apoptosis by upregulation of L-PGDS, which was most dramatically demonstrated in granular cell neurons. This is a novel example of OL–neuronal interaction.
The results of this study not only cast a new light on the function of perineuronal OLs but also have potentially important implications for the treatment of neurodegenerative diseases as well as the demyelinating disorders.
Footnotes
↵* M.T., I.M., and N.E. contributed equally to this work.
This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (09670806, M.T.; 13557016, N.E.; 12558078, Y.U.), Japan Science and Technology Corporation (N.E. and Y.U.), Suntory Institute for Bioorganic Research (N.E.), Takeda Science Foundation (N.E. and Y.U.), Yamanouchi Foundation for Research on Metabolic Disorders (N.E.), the National Institutes of Health United States Public Health Service (NS-24453 and HD-03110, K.S), and Osaka City. We thank Kiyokazu Momose (Oriental Yeast Co., Ltd., Tokyo, Japan) for supplying CMS Sprout diet, Dr. Wendy Cammer (Department of Neuroscience, Albert Einstein College of Medicine) for a generous gift of anti-CA II antibody, Shigeko Matsumoto for immunohistochemistry, Daisuke Irikura for Northern blot analysis, and Kosuke Aritake for measurement of psychosine. We also thank Dr. Takashi Inui (Osaka Bioscience Institute) and Dr. Masahiko Takada (Tokyo Metropolitan Institute for Neuroscience) for their comments during the preparation of this manuscript. We are grateful to Dr. Osamu Hayaishi (Osaka Bioscience Institute) and Dr. Shintaro Okada (Osaka University), for their encouragement of this study.
Correspondence should be addressed to Yoshihiro Urade, Osaka Bioscience Institute, 6-2-4 Furuedai, Suita City, Osaka 565-0874, Japan. E-mail: uradey{at}obi.or.jp.
C. T. Beuckmann's present address: Howard Hughes Medical Institute at University of Texas Southwestern Medical Center, Dallas, TX 75390-9050.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}