

Abstract
Dopamine receptor subtypes D1 and D2, and many other seven-transmembrane receptors including adenosine receptor A2A, are colocalized in striatum of brain. These receptors stimulate or inhibit adenylyl cyclases (ACs) to produce distinct physiological and pharmacological responses and interact with each other synergistically or antagonistically at various levels. The identity of the AC isoform that is coupled to each of these receptors, however, remains unknown. To investigate the in vivo role of the type 5 adenylyl cyclase (AC5), which is preferentially expressed in striatum, mice deficient for the AC5 gene were generated. The genetic ablation of the AC5 gene eliminated >80% of forskolin-induced AC activity and 85–90% of AC activity stimulated by either D1 or A2A receptor agonists in striatum. However, D1- or A2A-specific pharmaco-behaviors were basically preserved, whereas the signal cascade from D2 to AC was completely abolished inAC5−/−, and motor activity of AC5−/− was not suppressed by treatment of cataleptic doses of the antipsychotic drugs haloperidol and sulpiride. Interestingly, both haloperidol and clozapine at low doses remarkably increased the locomotion ofAC5−/− in the open field test that was produced in part by a common mechanism that involved the increased activation of D1 dopamine receptors. Together, these results suggest that AC5 is the principal AC integrating signals from multiple receptors including D1, D2, and A2A in striatum and the cascade involving AC5 among diverse D2 signaling pathways is essential for neuroleptic effects of antipsychotic drugs.
Dopaminergic systems play central roles not only in movement behaviors and motivated behaviors but also in pathological states, including Parkinson's disease, drug addictions, and schizophrenia (Ebadi and Srinivasan, 1995; Missale et al., 1998). Neuroanatomical substrates underlying these physiological and pathological responses include the striatum, where dopamine receptors are highly enriched (Levey et al., 1993; Surmeier et al., 1996). Historically, all five dopamine receptor isoforms cloned in mammals have been envisioned to stimulate or inhibit adenylyl cyclase (AC) to generate their physiological and pharmacological responses. After activation, D1-type dopamine receptors (D1 and D5) stimulate AC, whereas D2-type dopamine receptors (D2, D3, and D4) inhibit AC (Creese et al., 1983; Missale et al., 1998; Sidhu and Niznik, 2000). Despite the long history of dopamine receptor biology, the identity of the AC subtype that is coupled to specific dopamine receptor isoforms in the brain has not been verified.
To date, more than 10 different types of ACs, including cytosolic AC, have been cloned and characterized in mammals (Buck et al., 1999;Hanoune and Defer, 2001). Studies on the expression of ACs showed that many AC isoforms were expressed at various levels in striatum (Glatt and Snyder, 1993; Cali et al., 1994; Mons and Cooper, 1994; Lane-Ladd et al., 1997; Matsuoka et al., 1997; Antoni et al., 1998; Liu et al., 1998; Mons et al., 1998). The type 5 adenylyl cyclase (AC5) has been thought to be a component of dopamine receptor signaling, primarily because it was highly concentrated in striatum where D1 and D2 were expressed abundantly (Glatt and Snyder, 1993; Matsuoka et al., 1997). Unfortunately, further progress in understanding the in vivorole of ACs, including AC5, in dopamine receptor biology has been hampered by the lack of AC subtype-specific inhibitors.
The A2A receptors are highly concentrated in striatum, particularly in GABAergic striatopallidal neurons where A2A and D2receptors are colocalized (Fink et al., 1992; Augood and Emson, 1994). Antagonistic interactions between A2A and D2 receptors in the basal ganglia system have been proposed to play a key role in the motor depressant effects of adenosine receptor agonists and the motor stimulant effects of adenosine receptor antagonists, such as caffeine (Ferre et al., 1997). In addition, many other seven-transmembrane receptors, including opioid receptors and muscarinic acetylcholine receptors, are also highly concentrated in striatum. These receptors use AC as their effector and interact with dopaminergic systems synergistically or antagonistically at various levels (Gomeza et al., 1999; Jang et al., 2000). However, the integrity of the effector system of these receptors in vivo is basically undetermined.
One strategy to dissect the functional receptor–effector system is to create mice lacking an effector candidate and to examine the signaling pathway of receptors of interests with the mutant animals. Therefore, the current study was undertaken to unravel the interaction of AC5 with dopamine receptors and other receptors in striatum using AC5-deficient mice. We demonstrated that AC5 interacted with many receptors including dopamine receptors; consequently it acted as an effector of integrating signals from multiple receptors.
MATERIALS AND METHODS
AC5 knock-out cassette, homologous recombination of the AC5 gene, and genetics. The N-terminal 369 bp cDNA fragment of the rat AC5 (GenBank accession no. M96159) (Premont et al., 1992) was amplified by PCR using two primers, 5′-GTCGAGGAAAAGGCCGAGCGGCCGAGG-3′ and 5′-CAGCCATGATAAGGATCACGCCCACAG-3′. The fragment was used to screen for mouse AC5 genomic DNA clones from a mouse 129SVJ genomic DNA library (Stratagene). Two overlapping phage clones were isolated and characterized to contain the 1.3 kb ApaI fragment that covered the first half of predicted mouse AC5. The genomic DNA sequences around the 1.3 kb ApaI fragment were determined, and an exon identified in the fragment was designated as exon 2. A part of the AC5 sequences was deposited into GenBank (AF417936). The targeting cassette was constructed by subcloning the 2.2 kb short-arm (BamHI–exon 2) and the 5.3 kb long-arm (ApaI–XhoI) at each side of pGK-neo and TK in front of the short-arm as depicted in Figure 1A. The pGK-neo clones were provided by P. Soriano (Fred Hutchison Cancer Research Center, Seattle, WA) and D. S. Lim (Korea University, Seoul, Korea). ES cell culture and embryo handlings were conducted by following a standard procedure (Joyner, 1993; Kim et al., 1997). Briefly, J1 embryonic stem (ES) cells were used for homologous recombination, and seven independent chimeric mice were generated. They were bred to C57BL/6J mice to obtain heterozygote F1 mice. Intercrossing between heterozygote F1 produced F2 hybrids of homozygote (AC5−/− ), heterozygote (AC5+/− ), and wild-type (AC5+/+ ) littermates, which were used in this study.
Northern, Southern, and Western blot analyses. Northern blot analysis was performed as described (Kim et al., 1999). Briefly, membrane blot carrying 30 μg of total RNA in each lane was prepared and hybridized with 32P-labeled probes prepared from the PCR-amplified 529 bp fragment (4232–4760) of the 3′-end of rat AC5 cDNA (Premont et al., 1992). For genomic Southern blots, a blot carrying XbaI-digested genomic DNA was hybridized with the 32P-labeled 0.5 kbSalI–HindIII fragment as indicated (see Fig.1A). A PCR method was also used in genotyping. The PCR primers were AC5-(A), 5′-ACCGTCGAGGATGGAGACGG-3′ (971–990); AC5-(A+), 5′-GTGGCTGTGGCAGC AACAGG-3′ (1383–1402); and AC5-(pGK2r), 5′-CAGCGCGGCAGACGTGCGCT-3′. The PCR using the (A) and (A+) or (A) and (pGK2r) combination generates 432 bp of wild-type allele and 665 bp of mutant allele, respectively. For Western blot analyses, primary antibodies for PKA Cα (Santa Cruz Biotechnology), PKA RI (Transduction Laboratories), PKA RIIα (Santa Cruz Biotechnology), Gsα, Giα, Goα, and Gzα (Santa Cruz Biotechnology) were purchased.
Histological examinations. Tissue sections for immunohistochemical staining, receptor binding study, or hematoxylin-eosin staining were prepared as described (Kim et al., 1999; Lee et al., 1999). Briefly, brain, heart, or kidney sections cut at 40 μm by vibratome, at 14 μm by cryostat, or at 4 μm by microtome after embedding with paraffin were fixed in 4% paraformaldehyde for 15 min and placed for histological study. Animals were handled in accordance with the guideline of animal care at Ewha Womans University School of Medicine. The polyclonal anti-AC5 antibody was purchased from Santa Cruz Biotechnology. Primary antibodies for substance P (Chemicon), dynorphin (Oncogene), neuropeptide Y (Chemicon), enkephalin (Chemicon), tyrosine hydroxylase (Chemicon), GAD (Chemicon), and parvalbumin (Santa Cruz Biotechnology) were purchased.
AC assay. Striatum, frontal cortex of cerebrum, and cerebellum of adult brain were excised and subjected to AC assays. In preparations of the striatum, the dorsal striatum (caudate and putamen) and ventral striatum (the nucleus accumbens and its surrounding regions) were included unless indicated otherwise. In preparations of the frontal cortex, we included most cortical regions of the frontal lobe but excluded olfactory bulbs, striatum, thalamus, and the cortical regions posterior to the motor and sensory cortices. In preparations of the cerebellum, anterior and posterior lobes were included.
AC assay was performed as described (Onali et al., 1985; Olianas et al., 1997) with a modification using125I-cAMP and anti-cAMP antibody (Amersham Biosciences). Prepared brain tissues were homogenized separately using a Teflon/glass homogenizer in 10 mm imidazole, pH 7.3, 2 mm EDTA, and 10% sucrose. After tissue debris was spun down at 1000 rpm (23 × g), crude membrane fractions were prepared by centrifugation at 25,000 × g at 4°C for 30 min and suspended in 10 mm imidazole or 10 mm Tris-HCl (for D2 assay) to a final concentration of 2–3 μg/μl. The resulting membrane samples were aliquoted in 25–30 μl volume and stored at −70°C until use. For each AC reaction, 20 μg of total protein was used. All reactions below were prepared in 0.1 ml volume, and membrane samples were added last.
The forskolin-induced AC activation was produced in 100 mmHEPES, pH 7.4, 100 mm NaCl, 4 mmMgCl2, 2 mm EDTA, 0.5 mm3-isobutyl-1-methylxantine (IBMX), 2 mm ATP, 20 mm phosphocreatine, and 5 U of creatine phosphokinase with 10 μm forskolin at 30°C for 15 min in vitro.
For D1 and A2A activation assays, reactions were performed in 10 mm imidazole, pH 7.3, 0.5 mm MgCl2, 0.5 mmIBMX, 0.2 mm EGTA, 0.5 mm DTT, 0.01 mm pargyline, 1 μm GTP, 2 mm ATP, 20 mm phosphocreatine, and 5 U of creatine phosphokinase at 30°C for 15 min in vitro.
For the D2 activation assay, reactions were incubated in 80 mm Tris/HCl, pH 7.4, 2 mmMgSO4, 1 mm EGTA, 150 mmNaCl, 0.5 mm IBMX, 0.5 mm DTT, 0.05 mm GTP, and 0.2 mm ATP at 30°C for 5 minin vitro. For the muscarinic acetylcholine receptor assay, reactions were made in 80 mm Tris/HCl, pH 7.4, 2 mm MgCl2, 0.3 mm EGTA, 1 mm DTT, 0.5 mm IBMX, 0.1 mm GTP, and 0.2 mm ATP at 30°C for 5 min in vitro.
Reactions were terminated by adding 0.5 ml of 0.1N HCl and centrifuged at 22,000 × g for 5 min, and supernatant was taken. The amount of cAMP formed was determined by the125I-cAMP assay system (Amersham Biosciences). The assay was based on the competition between unlabeled cAMP and a fixed quantity of 125I-cAMP and anti-cAMP antibody. According to the manufacturer's instructions, higher assay sensitivity was obtained by acetylation of protein samples. Three microliters of the reaction sample were mixed with 100 μl of 0.05 m acetate buffer, pH 5.8. After adding 8 μl of the mix of 1 vol of acetic anhydride and 2 vol of triethylamine, reactions were vortexed vigorously for 7–8 min. Twenty microliters of acetylated sample were mixed with 80 μl of assay buffer (0.05 m acetate buffer, pH 5.8), followed by adding 100 μl of anti-cAMP antibody. After incubation at 4°C for 3 hr, they were added with 100 μl of125I-cAMP (30,000 cpm/ml) and incubated further at 4°C overnight. Then they were mixed with 500 μl of Amerlex-M secondary antibody conjugated with magnetic beads and incubated at room temperature for 15 min. After centrifugation at 3000 × g for 10 min, pellet was used to determine the bound radioactivity by Packard gamma counter. The radioactivity was converted to picomoles of cAMP by comparison to the reference curve that was constructed with standards. AC activities were presented by averaging three to six independent measurements with duplicates, for which protein samples were taken from more than three animals for each genotype.
Molecular cloning ACs present in the striatum ofAC5−/−. To identify AC responsible for the AC activity in the striatum ofAC5−/− , RT-PCR analyses were performed using a battery of primer sets: 5′-GACATTGTGGGCTTCAC-3′ and 5′-CTTCAGTAGCCTCAGCC-3′ for AC1, 5′-CCTCGACACACTCTGGACGG-3′ and 5′-GCTGGCAGTGCAGTAGCTC for AC2, 5′-GATGCAGCTGCTGAGGGAG-3′ and 5′-CAGTCTTGGTCTTCTCCCGC for AC3, 5′-GGATTGCTGTCTTCTCTGG-3′ and 5′-GTAGGTGATGATCAGAGCTG-3′ for AC4, 5′-ACCGTCGAGGATGGAGACGG-3′ and 5′-GTGGCTGTGGCAGCAACAGGC-3′ for AC5, 5′-CCTGATACTCGGGATTTATG-3′ and 5′-CCACAGCTGGGCAGTCCAG-3′ for AC6, 5′-CAACATTGAATCACCTGGAC-3′ and 5′-GATGGCCTGGAGTGTACTTC-3′ for AC7, and 5′-CCGGCCTGGGCACATCTTTG-3′ and 5′-CGGCGGGGCTCAGGCAGTC-3′ for AC8. Undesired amplification of genomic DNA in RT-PCR was monitored by using total RNA as template. The partial sequences of mouse AC3 cDNA clone was deposited in GenBank (AF253540).
D1 and D2dopamine receptor binding assays. Striatum was homogenized using a Teflon/glass homogenizer in 10 mmTris·HCl, pH 7.4, and crude membrane fractions were prepared. Receptor-ligand binding experiments were performed using3H-SCH23390 or3H-spiperone for D1and D2 receptors, respectively. Cold SCH23390 or butaclamol was used for blocking of nonspecific binding. Binding reactions were performed for 1 hr at room temperature. For the125I-sulpiride binding study, brain sections mounted on a glass slide were overlaid with ligand binding buffer (50 mm HEPES, pH 7.5, 1 mm EDTA, and 0.1% BSA) containing 0.5 nm125I-sulpiride (2000 Ci/mmol). Nonspecific binding was blocked with cold sulpiride. After incubation at room temperature for 60 min, the slides were washed with ice-cold ligand binding buffer and ice-cold water, air dried, and autoradiographed.
Behavioral assessments. For the open field test, mice (10–20 weeks) were placed individually in a test chamber and monitored on a TV screen after video recording. A test chamber consisted of a 60 × 60 cm2 floor with 40-cm-high walls in which the floor was marked by lines to have 25 equal squares. Animals were allowed to spend 15 min in the chamber for acclimation. At the end of acclimation, each drug dissolved in 120 μl of 0.9% saline was administered intraperitoneally. SKF38393, SCH23390, CGS21680, quinpirole, oxotremorine, haloperidol, clozapine, sulpiride, and dihydrexidine were purchased from Tocris. Horizontal locomotor activity was judged by cumulative counts of line crossovers of animals for a 60 min period (15–75 min). Later, we established a computerized video tracking system that was used to obtain the data for the open field system in Figures 3D, 4D, and8A,B. For this system, we used a test chamber consisting of a 45 × 45 cm2 floor with 40-cm-high walls. Horizontal locomotor activity was judged by the distance of the animal's movement for 60 min after drug administration. In all experiments, mice that were administered drugs were not used further. For behavioral assessments, n = 4–9 for each genotype, unless indicated otherwise.
Catalepsy was measured by a bar test with a cutoff time of 3 min. The forepaws of the mice were placed on a 1-cm-diameter bar held 5 cm above the floor and 5 cm from the front wall, and the time spent in the given posture was regarded as an indication of catalepsy.
Statistical analysis. Two-sample comparison was performed using Student's t test, and multiple comparisons were made using one-way ANOVA followed by the Newman–Keuls multiple range test. All data were presented as the mean ± SEM, and a statistical difference was accepted at 5% level unless indicated otherwise.
RESULTS
Generation of AC5 knock-out mice
To elucidate the in vivo role of AC5, we generated mice deficient for the AC5 by means of homologous recombination (Fig.1A,B). Homozygous males and females were viable and healthy. Northern analysis revealed that the AC5 expression was totally abolished in the striatum of AC5−/− mice (Fig. 1C). In agreement with these data, anti-AC5 immunoreactivity was absent in the brain ofAC5−/− mice (Fig.1D).

Targeted disruption of the AC5 gene locus. A, Restriction map of 129SVJ genomic DNA clone (wild type), targeting cassette (pAC5-neo-TK), and homologous recombinant (mutant). The 1.3 kb ApaI fragment in wild type carried an exon covering most of the first half of AC5. Predicted mouse AC5 cDNA at the top was drawn on the basis of the published rat cDNA sequence, in which TMindicates the transmembrane domains and C1 and C2represent the catalytic domains located in the cytoplasmic side. Drawing is not to scale. X, XbaI;Xh, XhoI; S,SalI; H, HindIII;B, BamHI; A,ApaI. B, Genomic Southern blot of F2 littermates. Arrows indicate wild-type (13 kb) and mutant (9 kb) bands on the XbaI-digested genomic DNA blot. C, Northern blot analysis shows noAC5 messages in the striatum ofAC5−/−. The sizes of AC5 message were ∼6 and 7.4 kb. D, Immunohistochemical detection of AC5 expression in brain sections. Anti-AC5 antibody detected a background level of immunoreactivity in the brain ofAC5−/− .
Viability and development ofAC5−/−mutant animals
The numbers of homozygotes, heterozygotes, and wild-type littermates were 97 (23.7%), 210 (51.2%), and 103 (25.1%), respectively, among the first 410 progenies generated from F1 intercrosses. There was no sex bias in F2 offspring. Both homozygous males and females were fertile and bred normally. TheAC5−/− mice were active in their home cage and morphologically indistinguishable from their wild-type littermates. Body weight was slightly reduced to 90–95% of that of AC5+/+ during the first 20 weeks after birth (data not shown). AC5 has been thought to be an important component in the function of heart and kidney during development and in adult life (Chabardes et al., 1999; Hanoune and Defer, 2001). Histological examination, however, revealed no obvious difference between AC5−/− and wild-type littermates in most respects, including the sizes and microscopic structures of heart and kidney (data not shown). Together, these results indicate that although the absence of the AC5 gene results in slightly delayed growth after birth, the AC5 is not essential for survival in mice.
Impairment of adenylyl cyclase activity in the brain ofAC5−/− mice
To assess the AC activity abolished by the mutation of theAC5, we compared AC activity in the brain of mutant animals with that of wild-type littermates. In the striatum, the baseline AC activity of AC5−/− in the absence of exogenous activators was slightly reduced when compared with that of AC5+/+ . However, forskolin (10 μm) treatment enhanced AC activity >20-fold inAC5−/− , which reached 18% of that in AC5+/+ (Fig.2A). Other AC subtypes present in the striatum ofAC5−/− , for which we identified AC1 through AC8, the expression of which was not altered as determined by semiquantitative RT-PCR analyses (data not shown), may account for the forskolin-induced AC activity inAC5−/− . In the frontal cortex of cerebrum and in the cerebellum, the forskolin (10 μm)-enhanced AC activity inAC5−/− was diminished to 73 and 60%, respectively, of that in wild-type littermates (Fig.2B,C). Overall, these results suggest that AC5 is the major AC in the striatum, whereas it constitutes a minor AC in the frontal cortex and cerebellum of normal mice.

Impairment of AC activity inAC5−/− mice. The forskolin (10 μm)-stimulated AC activity inAC5−/− was reduced to 18% in the striatum (A), 73% in the cerebral cortex (B), and 60% in the cerebellum (C) of that inAC5+/+ . The baseline AC activity inAC5−/− was slightly reduced when compared with that ofAC5+/+ (p< 0.05). ** indicates a difference between two groups (p < 0.01). Base, Baseline;FSK, forskolin.
General motor behaviors of naı̈ve mutant animals
We examined whether the genetic ablation of the AC5gene with the concomitant abolishment of the AC5 activity in the brain could produce any abnormality in the expression of motor behaviors. Regarding spontaneous motor activities including walking, running, climbing, grasping, writhing, and motor skills related to eating,AC5−/− mice were indistinguishable from their wild-type littermates.AC5−/− animals displayed no overt sign of ataxia or tremor, despite the fact that the forskolin-stimulated AC activity in the cerebellum was reduced to 60% of that in AC5+/+ (Fig. 2C). Thus, spontaneous general motor activities ofAC5−/− appeared to be normal.
Coupling of D1 dopamine receptors and AC5
The genetic disruption of the AC5 and the presence of the forskolin-activated residual AC activity in the striatum ofAC5−/− prompted us to examine the receptor–effector interactions in the context of AC5. To delineate the interactions, we relied on biochemical and pharmaco-behavioral assay methods. First of all, we explored whether AC5 is the essential component downstream of D1dopamine receptor activation. In the striatum ofAC5+/+ , the D1agonist SKF38393 (100 μm)-stimulated AC activity was increased markedly (29.04 ± 2.04 → 53.16 ± 4.24 pmol of cAMP per milligram per minute; p < 0.01; 183% of the baseline). Similarly, the AC activity stimulated by dihydrexidine (DHX; 100 μm), a full agonist for D1, was enhanced more than twofold inAC5+/+ (29.04 ± 2.04 → 60.84 ± 5.69 pmol of cAMP per milligram per minute; p < 0.01; 210% of the baseline). In the striatum ofAC5−/− , the SKF38393 (100 μm)-stimulated AC activity was increased at a low level, but it was consistently higher than the baseline control in repeated experiments (5.05 ± 0.16 → 5.73 ± 0.15 pmol of cAMP per milligram per minute; p < 0.01; 113% of the baseline). Consistently, the treatment with DHX (100 μm) produced a comparable level of increase inAC5−/− (5.05 ± 0.16 → 5.80 ± 0.21 pmol of cAMP per milligram per minute;p < 0.01; 115% of the baseline), implying that a functional D1–Gαs–AC system was present in the striatum ofAC5−/− (Fig.3A). Overall, however, the increase of SKF38393- or DHX-stimulated AC activity inAC5−/− was reduced to 10–11% of that in AC5+/+ . We assessed separately the DHX-stimulated AC activity in the dorsal (caudate and putamen) and ventral (nucleus accumbens and its surrounding areas) striatum and found no difference in their activation folds in these subdivisions of striatum (data not shown). Together, these results suggest that D1 receptors use AC5 as their primary AC and other ACs as their alternative pathway. In the frontal cortex of cerebrum in AC5+/+ , where D1 receptor density is not as high as in striatum, we observed that the DHX (100 μm)-stimulated AC activity was relatively low but substantially increased when compared with the baseline control (10.75 ± 0.26 → 14.27 ± 0.73 pmol of cAMP per milligram per minute; p < 0.01; 133% of the baseline). InAC5−/− , the DHX-stimulated AC activity was also enhanced in a similar fold (7.91 ± 0.24 → 10.87 ± 0.29 pmol of cAMP per milligram per minute;p < 0.01; 137% of the baseline), despite the fact that overall the DHX-stimulated AC activity inAC5−/− was decreased to 73% of that in AC5+/+ . Thus, other ACs available in the frontal cortex appear to interact effectively with D1.

D1 and AC coupling. A, AC assays with D1 agonists, SKF38393 and DHX. The fold increment of AC activity by SKF38393 (100 μm) or DHX (100 μm) in the striatum (A) ofAC5−/− was 11 and 10%, respectively, of that in AC5+/+ (p < 0.01 for both genotypes). GTP addition itself increases the baseline AC activity inAC5+/+ as well as inAC5−/−. B, E, The D1 dopamine agonist, SKF38393 (50 mg/kg, i.p.), increased the locomotor activity in both AC5+/+ andAC5−/− (B). The locomotor activity ofAC5−/− was dramatically increased 20 min after administration of SKF38393 (E); however, the mechanism underlying the behavioral response remains unknown. D, The D1-specific dopamine agonist, DHX (30 mg/kg, i.p.), increased the locomotor activity in bothAC5+/+ andAC5−/− . Note that D1 agonist-induced locomotion ofAC5−/− was markedly increased, despite the fact that D1agonist-induced AC activation was severely impaired. C, F, The D1 receptor antagonist, SCH23390 (0.3 mg/kg, i.p.), produced the suppression of locomotion in both genotypes (C). The time-dependent change of the locomotion induced by SCH23390 is shown in F. * and ** indicate a difference between two groups at p < 0.05 andp < 0.01, respectively. Base, Baseline; Veh, vehicle.
We questioned whether the severe but incomplete loss of the D1 agonist-stimulated AC activity in the striatum of AC5−/− would result in any impaired pharmaco-behaviors in response to D1agonists or antagonists. To address this, we relied on a behavioral assay paradigm using the open field test. The open field test has been used widely for the measurement of neuronal outputs of the basal ganglia system responding to agonists and antagonists of dopamine receptors, psychomotor stimulants, or antipsychotic drugs (Xu et al., 1994; Dulawa et al., 1999). Administration of the D1 agonist, SKF38393 (50 mg/kg, i.p.), induced an increase of the horizontal locomotor activity in wild-type littermates as reported previously (Gomeza et al., 1999). InAC5−/− , the same dose of SKF38393 produced a robust enhancement of the locomotion. Interestingly, the increase in the locomotor effects of the D1 agonist inAC5−/−animals was higher than that observed inAC5+/+ (Fig.3B,E). Similar results were obtained by the administration of the D1-specific agonist, DHX (30 mg/kg, i.p.) (Fig. 3D). In addition, the locomotor activity in the open field test after administration of the D1 antagonist, SCH23390 (0.3 mg/kg, i.p.), was fully suppressed in AC5−/− as seen in AC5+/+ (Fig.3C,F). Therefore, the D1 system inAC5−/− appeared to be functional, at least in part, at the behavioral level, despite the fact that the AC activity induced by D1 stimulation was diminished severely. The identity of the non-AC5 effector(s) responsible for the observed D1-dependent pharmaco-behavioral responses was not known in the present study.
Coupling of A2A adenosine receptor and AC5
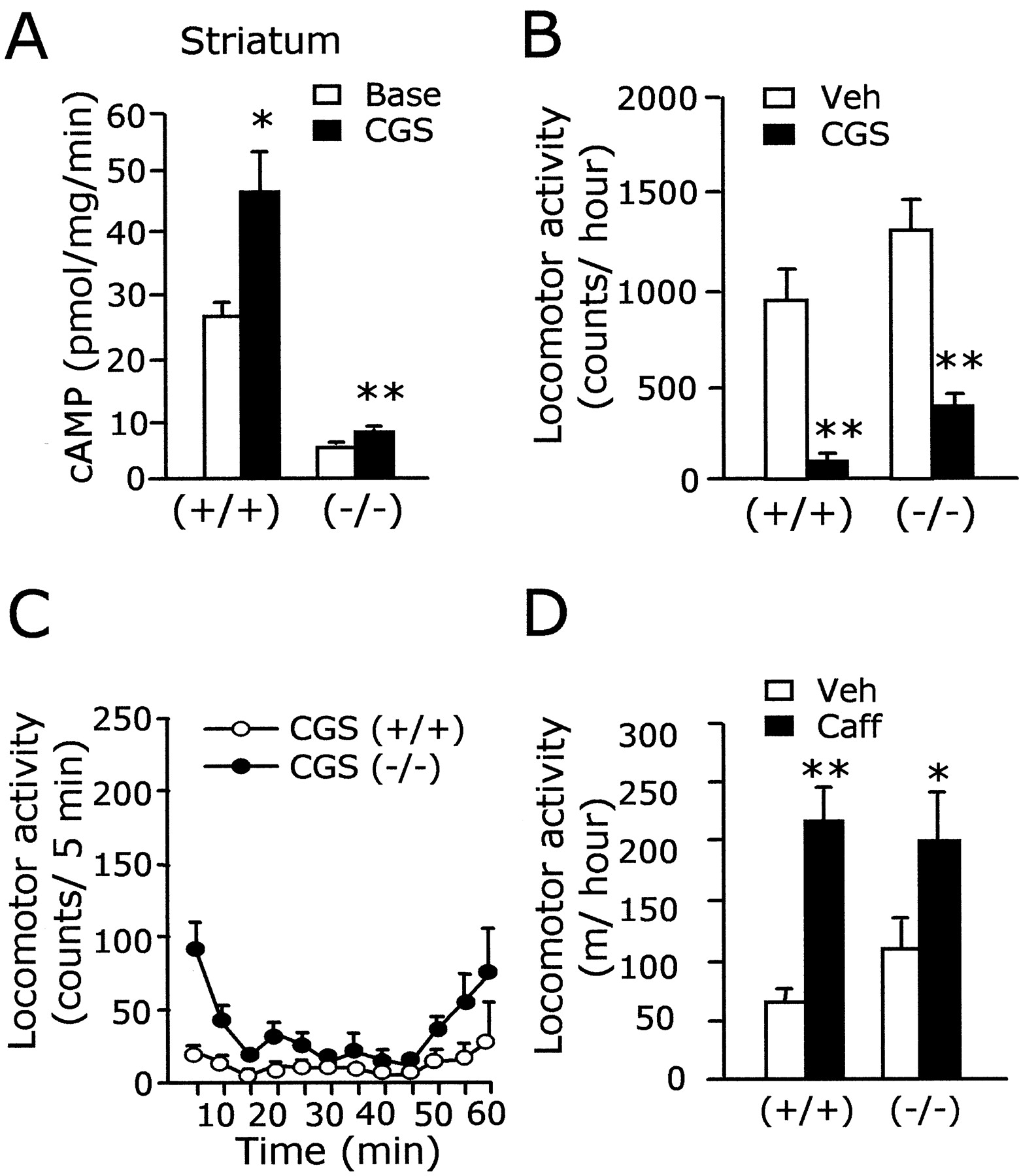
Similar to those of D1 dopamine receptors, A2A receptors are preferentially expressed in the striatum and positively coupled to AC (Ferre et al., 1997; Moreau and Huber, 1999). We tested whether the anatomical and biochemical analogy between A2A and D1receptors can be extended to the receptor–effector coupling mode. Biochemical assessment indicated that the A2Aagonist CGS21680 (10 μm)-stimulated AC activity in the striatum of AC5+/+ was highly increased (26.14 ± 2.51 → 46.51 ± 6.36 pmol of cAMP per milligram per minute; p < 0.05; 178% of the baseline). In the striatum of AC5−/− , the AC activity stimulated by CGS21680 was significantly increased (4.94 ± 0.17 → 7.34 ± 0.52 pmol of cAMP per milligram per minute;p < 0.01; 149% of the baseline), suggesting the existence of a functional A2A/AC system in the striatum of AC5−/− (Fig.4A). However, the AC activity stimulated by CGS21680 (10 μm) inAC5−/− was reduced to 16% of that in AC5+/+ , indicating that AC5 was the major AC for A2A receptors.

A2A and AC5 coupling.A, The A2A adenosine receptor agonist, CGS21680 (10 μm), induced an increase of AC activity in the striatum of AC5+/+ andAC5−/− . The CGS21680-induced increment of AC activity inAC5−/− was 16% of that in wild-type littermates. B, C, The A2A agonist CGS21680 (0.5 mg/kg, i.p.) produced the suppression of locomotion in bothAC5+/+ andAC5−/− (B). The time-dependent change of locomotion induced by CSG21680 is shown in C. D, Administration of caffeine (15 mg/kg, i.p.) increased the locomotion of both AC5+/+ andAC5−/− mice. * and ** indicate a difference between two groups at p < 0.05 andp < 0.01, respectively. Base, Baseline;CGS, CGS21680; Veh, vehicle;Caff, caffeine.
We examined whether the severe loss of the A2Aagonist-stimulated AC activity in the striatum ofAC5−/− could result in impaired behavior in response to A2A agonist treatment. Administration of the A2A agonist CGS21680 (0.5 mg/kg, i.p.) induced akinesis broadly inAC5+/+ . Similarly, administration of the same dose of CGS21680 produced a strong suppression of motor behaviors inAC5−/− (Fig. 4B,C). The locomotor activity in the open field after administration of the nonselective adenosine receptor antagonist, caffeine (15 mg/kg, i.p.), was increased inAC5−/− as seen inAC5+/+ (Fig. 4D). These results indicate that the typical A2A receptor-specific pharmacology was retained, at least in part, at behavioral levels inAC5−/− .
Coupling of D2 dopamine receptors and AC5
A possibility of coupling between AC5 and D2was investigated. Because D2 dopamine receptors are negatively coupled to AC via Gαi protein, the effect of D2 activation on AC activity can be measured clearly when D2 is coactivated together with D1 or other Gαs-coupled receptors. Indeed, in the striatum ofAC5+/+ , D2activation by quinpirole, a D2 agonist, suppressed the D1 agonist DHX (0.5 μm)-stimulated AC activity in a dose-dependent manner, as was reported previously (Mottola et al., 1992). However, D2 activation by quinpirole even at high doses failed to inhibit the DHX-stimulated AC activity inAC5−/− (Fig.5A). Similarly, the D2 agonist quinpirole did not produce any inhibitory effects on the AC activation induced by the neuropeptide VIP (1 μm) or the A2AR agonist, CGS21680 (0.1 μm) inAC5−/− (Fig.5B,C). More decisively, the inhibitory effect of D2 activation on the forskolin (0.1 μm)-stimulated AC activity, unlike that in AC5+/+ , was totally abolished in AC5−/− (Fig.5D). Together, these data consistently indicate that the D2–Gαi–AC system is completely impaired in the striatum ofAC5−/− .

D2 and AC5 coupling. A, The D1 agonist DHX (0.5 μm)-induced AC activation in AC5+/+ was suppressed in a dose-dependent manner by quinpirole, but not inAC5−/−. Q(1) and Q(10) denote, respectively, 1 and 10 μm of quinpirole. Base, Baseline. B–D, The AC activity stimulated by A2A agonist CGS21680 (0.1 μm) (B), neuropeptide VIP (1 μm) (C), or forskolin (0.1 μm) (D) was suppressed in a dose-dependent manner by quinpirole in AC5+/+ but not inAC5−/− . The concentrations of DHX (0.5 μm), CGS21680 (0.1 μm), VIP (1 μm), and forskolin (0.1 μm) for B–D were chosen on the basis of preliminary experiments, because they produced ∼110–130% of AC activation of the baseline control, which was also applied for the assay of muscarinic acetylcholine receptor activation in Figure 6. * and ** indicate a difference between two groups atp < 0.05 and p < 0.01, respectively.
Coupling of other Gαi-coupled receptors and AC5
We explored whether other Gαi-coupled receptors also strictly require AC5. The receptors that are expressed abundantly in the striatum and negatively coupled to AC through Gαi protein include muscarinic acetylcholine receptors M4 and M2 (Weiner et al., 1990; Gomeza et al., 1999). Biochemical assessment indicated that in the striatum ofAC5+/+ , the forskolin-stimulated AC activity was inhibited in a dose-dependent manner by oxotremorine, an agonist for muscarinic acetylcholine receptors. The forskolin-induced AC activity in the striatum ofAC5−/− was notably suppressed by oxotremorine, although the oxotremorine-induced inhibition appeared to be partially defective (Fig.6A). In the frontal cortex of cerebrum inAC5−/− , the forskolin-induced AC activation was also suppressed by oxotremorine (Fig. 6B). Although this study did not aim at differentiating the identity of the oxotremorine-activated receptors, the oxotremorine-responsive receptors in the striatum might be M4 or M2 (Gomeza et al., 1999). Therefore, despite the fact that >80% of forskolin-stimulated AC activity was abolished and the D2–Gαi–AC system was completely defective in the striatum ofAC5−/− , at least a part of the machinery of the M4–Gαi–AC and M2–Gαi–AC system in the striatum ofAC5−/− appeared to be functional.

A functional muscarinic acetylcholine receptor system is present in the striatum ofAC5−/−.A, B, Oxotremorine (Oxo), an agonist for muscarinic acetylcholine receptors, suppressed the forskolin (0.1 μm)-stimulated AC activity, in a dose-dependent manner, in the striatum (A) and frontal cortex (B) of bothAC5+/+ andAC5−/− . The muscarinic acetylcholine receptors that are negatively coupled to AC and expressed abundantly in the striatum include M4 and M2 receptors. The amount of AC activity suppressed strongly inAC5+/+ by a high dose of oxo (1,000 μm) appears to include the contribution of the GTP-stimulated AC activity as well as the forskolin-stimulated AC activity. * and ** indicate a difference between two groups atp < 0.05 and p < 0.01, respectively.Base, Baseline; FSK, forskolin.Oxo 10, Oxo 100, and Oxo 1000 were, respectively, 10, 100, and 1,000 μm of oxotremorine.
D1 and D2 receptor densities and other gene expression
To test the possibility that the biochemical deficit of the D2–Gαi–AC system is caused indirectly by the severe reduction of D2 expression inAC5−/− , we measured the dopamine receptor density in the striatum. The receptor binding study with D2 antagonist3H-spiperone revealed no significant difference in D2 density betweenAC5−/− andAC5+/+ (Bmax was 1.47 ± 0.10 (+/+) and 1.25 ± 0.09 (−/−) pmol/mg protein; p > 0.05). Consistently, the receptor binding study to brain tissue sections with another D2 ligand,125I-sulpiride, indicated that the D2 receptor density in the brain ofAC5−/− was comparable to that in wild-type littermates (data not shown). We observed that D1 density also was unchanged (Table1). In addition, the expressions of Gαs, Gαi, Gαo, and Gαz proteins and of the PKAα catalytic subunit and PKA regulatory subunits, RI and RIIα, in the striatum ofAC5−/− were unchanged (data not shown). Hence, the impairment of the D2–Gαi–AC system in AC5−/− may be explained not by the reduced expression of D2 or Gαi and Gαo or the increased expression of D1, but by the total deficit of the D2–Gαi–AC5 signaling in the striatal neurons.
D1 and D2 receptor expression in the striatum
We examined potential compensatory changes in the expression of genes for substance P, dynorphin, neuropeptide Y, enkephalin, tyrosine hydroxylase, GAD, and parvalbumin. Light microscopic examination of brain sections stained with antibody for each of those showed no obvious change in the expression of these genes in the brain ofAC5−/− (data not shown). The expression of striatal peptides, for example, substance P, dynorphin, neuropeptide Y, and enkephalin, was known to be influenced by the activity of D1 and D2receptors. In particular, the expression of dynorphin was greatly reduced inD1 −/− , and the expression of GAD and enkephalin was significantly increased in D2 −/− (Xu et al., 1994; Baik et al., 1995). However, the expression of dynorphin and enkephalin in the brain ofAC5−/− mice was not altered. These results may suggest that AC5 is the principal AC but not the sole signaling effector for D1 and D2 receptors.
Responses to the antipsychotic drugs sulpiride, haloperidol, and clozapine
We investigated whether the biochemical deficit of the D2–Gαi–AC system could be manifested with altered responses to D2 antagonists at behavioral levels. Administration of the D2 antagonist, sulpiride (50 mg/kg, i.p.), produced severe immobilization of wild-type littermates over the course of the monitoring period. In contrast, the same dose of the drug did not elicit such suppression inAC5−/− animals (Fig.7A,B). These data are consistent with the notion that AC5 is a crucial component for the D2 receptor signaling in striatal neurons.
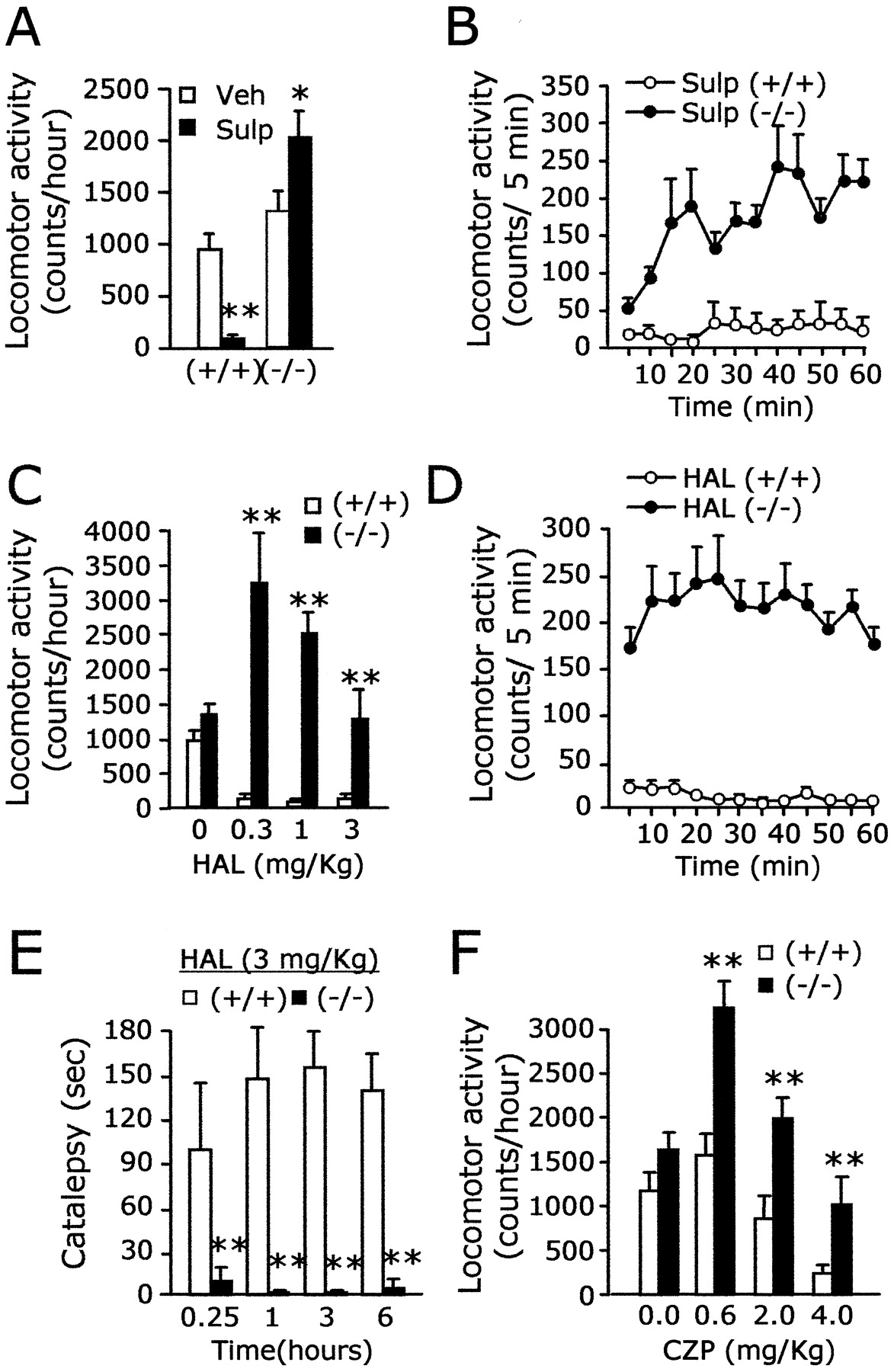
Loss of neuroleptic responsiveness to antipsychotic drugs inAC5−/− mice.A, The D2 antagonist, sulpiride (50 mg/kg, i.p.), induced a decrease of motor activity inAC5+/+ but produced a marked enhancement of locomotion inAC5−/− (p < 0.01 for +/+, andp < 0.05 for −/−). B, The locomotion of AC5−/− was gradually accelerated after administration of sulpiride.C, Haloperidol induced akinesis broadly in wild-type littermates but did not elicit such behavioral suppression in mutant animals. Note the dramatic enhancement of locomotion inAC5−/− at lower doses of haloperidol. D, Time-dependent changes of locomotion induced by 1 mg/kg of haloperidol are presented. E, As opposed to that in AC5+/+ , cataleptic response was induced by haloperidol (3 mg/kg, i.p.) inAC5−/− . For convenience, 180 sec of cutoff time was set for all cases that lasted >180 sec.F, Clozapine produced a substantial decrease of locomotion in both mutant and wild-type littermates in a dose-dependent manner, but its depression effect on the motor activity was markedly diminished in AC5−/− . Note the sharply increased locomotion ofAC5−/− by a low dose (0.6 mg/kg) of clozapine. * and ** indicate a difference between two groups at p < 0.05 and p < 0.01, respectively.Veh, Vehicle; Sulp, sulpiride;HAL, haloperidol; CZP, clozapine.
We further examined the effect of haloperidol, a typical antipsychotic drug the primary target of which is D2 (Seeman and Van Tol, 1994; Blin, 1999). Haloperidol administration (0.3–3 mg/kg, i.p.) quickly led to the immobilization of wild-type littermates for hours of observation. On the contrary, AC5−/− animals that received the same doses of haloperidol displayed no sign of behavioral suppression but instead showed hyperactive locomotion (Fig.7C,D). Consistently, as opposed to that inAC5+/+ , administration of haloperidol (3 mg/kg, i.p.) did not leadAC5−/− animals to catalepsy (Fig. 7E). Thus, despite the presence of other ACs in the striatum, and potential non-AC effectors such as ion channels (Cai et al., 2000; Sidhu and Niznik, 2000), the neuroleptic effects of haloperidol were totally eliminated inAC5−/− . These results indicate that AC5 acts as an indispensable signaling route for the typical neuroleptic effects of haloperidol.
Similar to that for haloperidol,AC5−/− mice were also hyperactive in response to clozapine, an atypical antipsychotic drug (Fig. 7F). After administration of clozapine, the locomotor activity of wild-type littermates was suppressed in a dose-dependent manner, whereas the locomotion ofAC5−/− marked consistently high scores in the same doses tested (Fig. 7F). These data suggest that AC5 is an important component for the neuroleptic action of clozapine, regardless of the fact that the behavioral response to clozapine in the open field test was slightly different from that of haloperidol.
The increased locomotion by haloperidol or clozapine was antagonized by D1 antagonist
Because a list of reports indicated that haloperidol has a property to induce dopamine release (Di Chiara et al., 1977; Pehek, 1999; Westerink et al., 2001), we tested the possibility that the paradoxical haloperidol-induced increase of locomotion inAC5−/− was related to the increased activation of dopamine receptors. First, we determined that the dose of 0.03 or 0.3 mg/kg of D1 antagonist SCH23390 (i.p.) produced, respectively, intermediate or almost complete suppression of locomotion of normal mice in the open field test (data not shown). In AC5−/− , the enhancement of locomotion by haloperidol (0.3 mg/kg, i.p.) was repressed intermediately by 0.03 mg/kg (i.p.) of SCH23390 and completely by 0.3 mg/kg (i.p.) of SCH23390. The total distance traveled for the given period reached the level of the SCH23390 treatment alone (Fig. 8A). Furthermore, the enhanced locomotion by clozapine (0.6 mg/kg, i.p.) was also antagonized intermediately by 0.03 mg/kg (i.p.) of SCH23390 and completely by 0.3 mg/kg (i.p.) of SCH23390 (Fig. 8B). These results suggest that the enhanced locomotion ofAC5−/− in response to haloperidol and clozapine resulted from the increased activation of SCH23390-sensitive D1 class dopamine receptors.

The increased locomotion by haloperidol or clozapine was antagonized by D1 antagonist.A, The enhancement of locomotion by haloperidol (0.3 mg/kg, i.p.) was repressed in a dose-dependent manner by D1antagonist SCH23390. B, The enhanced locomotion by clozapine (0.6 mg/kg, i.p.) was also antagonized in a dose-dependent manner by SCH23390. ** indicates a difference between two groups atp < 0.01. HAL, Haloperidol;SCH, SCH23390; SCH 0.03, 0.03 mg/kg SCH;SCH 0.3, 0.3 mg/kg SCH.
DISCUSSION
Roles of AC5 in D2 receptor function
Targeted disruption of the AC5 gene produced the complete elimination of AC5 expression in the brain, which made it possible to delineate the indispensable role of AC5 downstream D2 receptors. The typical inhibitory effect of D2 activation on D1agonist-, A2A agonist-, and forskolin-stimulated AC activity was completely abolished inAC5−/− (Fig. 5). It appeared that other types of ACs present in the striatum were not responsive to D2 receptor activation. In accordance with this notion, administration of the D2 antagonists, haloperidol and sulpiride, did not produce the typical neuroleptic effects in AC5−/− animals, unlike those in AC5+/+ (Fig.7A–D). Together, these results are consistent with the conclusion that AC5 is the physiologically relevant AC for D2 receptors and that AC5 is a necessary route for the neuroleptic action of these antipsychotic drugs.
It is unlikely, however, that the absence of the neuroleptic effects and the total impairment of the D2–Gαi–AC system in AC5−/− were produced indirectly by the reduced expression of the Gαi system or the D2 receptor itself. First, it was observed that the forskolin-induced AC activation inAC5−/− , although it was low, was suppressed by oxotremorine, an agonist for muscarinic acetylcholine receptors (Fig. 6). This result indicates the presence of the functional Gαi-coupled receptor system in AC5−/− . Furthermore, the expression level of Gαi in the striatum ofAC5−/− was comparable to that of AC5+/+ (data not shown). Therefore, the complete absence of D2-mediated AC inhibition inAC5−/− cannot be attributed to the general property of receptors that are negatively coupled to AC or to the low levels of forskolin-stimulated AC activity in the striatum ofAC5−/− (Fig.2A). Second, receptor-binding studies with3H-spiperone revealed that the D2 receptor density was slightly reduced inAC5−/− , which was insignificant (Table 1). Given that heterozygous mice deficient for dopamine receptor D2(D2 +/− ) that carry ∼50% of D2 receptors were notably suppressed by D2 antagonist in the open field test (Kelly et al., 1998), the lack of neuroleptic effects inAC5−/− cannot be explained by the loss of D2 receptors in the striatum. It remains unknown at present whether the total impairment of the D2–AC system in the striatum ofAC5−/− , even in the presence of other ACs and the Gαiprotein system, was caused by the failure of colocalization of D2 receptor with other ACs in the same neurons or the inability of D2 to pair with other ACs.
Interestingly, the neuroleptic drugs, sulpiride, haloperidol, and clozapine, produced a markedly enhanced locomotion inAC5−/− , especially at low doses (Fig. 7). Although no direct evidence is available at present, these results are tempting us to speculate that inhibition by these drugs of presynaptic D2, or D2S as implied by Usiello et al. (2000), might result in an increase of dopamine release in the striatum, which in turn produces the enhanced locomotion inAC5−/− . This hypothesis is consistent with our finding that the paradoxical enhancement of locomotion by haloperidol (0.3 mg/kg, i.p.) or clozapine (0.6 mg/kg, i.p.) was antagonized by SCH23390 (0.3 mg/kg, i.p.) as shown in Figure8, but not by the A2A agonist, CGS23390, at the dose that produced the complete immobilization (K.-W. Lee and P.-L. Han, unpublished observations). So, D1 receptor activity in postsynaptic neurons seems to be important for the D2 antagonist-induced increase of locomotion inAC5−/− . This speculation agreed with the fact that D1-dependent pharmaco-behaviors were retained inAC5−/− (Fig. 3). However, given that dopamine receptors are expressed at low levels in many brain regions, including frontal cortex, we do not exclude the possibility that AC5-uncoupled D2 receptors in striatal or extrastriatal regions contribute the neuroleptic-induced enhanced locomotion in AC5−/− . Nonetheless, the remarkable increase of locomotion by haloperidol or clozapine in AC5−/− is consistent with the notion that both haloperidol and clozapine had a property of increasing locomotion in the open field test that was produced in part by a common mechanism that involved the increased release of dopamine, as was suggested previously (Di Chiara et al., 1977; Pehek, 1999; Westerink et al., 2001).
Issues of D2–AC5 coupling
Colocalization of AC5 and D2 receptors in striatal neurons (Mons and Cooper, 1994) and the coupling between D2 and AC5 as demonstrated in this study raised the question of whetherAC5−/− mice were the phenocopy ofD2 −/− (Baik et al., 1995; Jung et al., 1999; Wang et al., 2000). The phenotype of AC5−/− mice has a similarity to that ofD2 −/− . For example,AC5−/− mice were not driven into catalepsy by haloperidol (Fig.7E), as wereD2 −/− andD2L −/− (Kelly et al., 1998; Usiello et al., 2000).
It was somewhat unique toAC5−/− , however, that the locomotion was highly increased by haloperidol (Fig. 7). In addition, despite the complete deficit of D2–Gαi–AC signaling in the striatum (Fig. 6),AC5−/− mice did not show Parkinsonian-like phenotypes or the reduced spontaneous general motor activities displayed byD2 −/− orD2L −/− mice (Baik et al., 1995; Jung et al., 1999; Wang et al., 2000). The preservation of spontaneous motor function inAC5−/− mice raises the possibility that the D2–Gαi–AC5 system, although it is normally required for transmitting the neuroleptic effects of antipsychotic drugs, comprises only a part of D2 signaling pathways. In fact, a list of reports indicates that D2 receptors are coupled to multiple types of Gα-proteins including Gαi, Gαz, and Gαo, which in turn link to various effector systems (Cai et al., 2000; Sidhu and Niznik, 2000;Jiang et al., 2001). Given that the D2–Gαi–AC5 cascade was the major or essential signal route for neuroleptic drugs, the significance of D2 receptor signaling cascades should be evaluated in the physiological context.
Last, it may be worth noting that the loss of haloperidol-induced catalepsy was not limited toAC5−/− orD2 −/− . Mice lacking the RIIβ subunit of protein kinase A (PKA) had a markedly reduced PKA activity in the striatum and showed no catalepsy in response to haloperidol (Adams et al., 1997). The complete absence of cataleptic responses to haloperidol in bothAC5−/− andPKA-RIIβ−/− mice implies that an appropriate control of the cAMP pathway in the striatum is essential for the action of the neuroleptic haloperidol.
Roles of AC5 in D1 receptor function
A well established and long-lasting dogma in dopamine receptor biology is that D1 dopamine receptor activation leads to the stimulation of AC to produce distinct physiological and pharmacological responses (Kebabian and Calne, 1979; Creese et al., 1983; Seeman and Van Tol, 1994; Missale et al., 1998). In fact, the treatment of D1 agonists DHX or SKF38393 produced a marked enhancement of AC activity inAC5+/+ . InAC5−/− , the DHX- or SKF38393-stimulated AC activity was significantly increased, although the maximal AC activity in the given condition reached 10–11% of that in wild-type littermates (Fig. 3A). These data are consistent with the view that AC5 is the primary, long-sought AC for D1 receptors (Kebabian and Calne, 1979; Creese et al., 1983) and that D1 receptors use other ACs as well, thus using dual/multiple AC systems in the striatum.
In AC5−/− , administration of the D1 agonist SKF38393 or DHX produced an increase of locomotion in the open field test (Fig.3B,D,E). In addition, administration of the D1 antagonist SCH23390 induced the depression of motor activity, which was similar to that inAC5+/+ (Fig.3C,F). Thus, the behavioral responses ofAC5−/− mice apparently satisfy the pharmacological criteria of D1receptor functions, despite the fact that D1agonist-induced AC stimulation was severely impaired. Interestingly, the SKF38393- or DHX-induced locomotion inAC5−/− was not intact but was double that of AC5+/+ (Fig.3B,D). This D1 agonist-induced amplified locomotion ofAC5−/− cannot be explained by a compensatory increase of D1 expression in the striatum, because D1 expression was not altered (Table 1). The D1 agonist-induced increase of the locomotion inAC5−/− was produced in the absence of the D2–Gαi–AC5-mediated inhibitory tone. Hence, we are tempted to speculate that the enhanced locomotion by SKF38393 or DHX in normal mice is the outcome of the antagonistic interaction of D1 and D2.
Although the D1 agonist DHX or SKF38393 significantly increased AC activity inAC5−/− , how the D1 class-dependent pharmaco-behaviors (Fig. 3) were produced is not clear at present. It may be possible that the D1-dependent behaviors ofAC5−/− were produced by the residual AC system in the striatum (Fig. 2A). However, we do not exclude the possibility that the D1-dependent behavior ofAC5−/− is related to the non-AC effector system, such as D1-coupled phospholipase C (Clifford et al., 1999), or D1class receptors that do not rely on AC5 and are expressed in extrastriatal motor control regions in the brain. It is also possible that complex types of adaptations including altered expression of uncharacterized AC pathway components or developmental changes at a circuit level could produce the D1-dependent pharmaco-behaviors of AC5−/− mutants. Nonetheless, our results indicate that although the behavioral responses controlled by D2 receptors seem to involve the AC5 pathway, the behavioral effects mediated by D1 receptors do not.
Roles of AC5 in A2A receptor function
In the current study, we demonstrated that similar to that of D1 receptors, AC5 is the major AC for A2A in the striatum, and A2A receptors can function, at least in part, independently of AC5. Thus, A2A receptors use dual–multiple AC systems, for which AC5 is the principal AC for A2A receptors (Fig. 4). Because AC5 is required for both D2 and A2A (Figs.4, 5) and D2 and A2Areceptors are colocalized in the same striatal neurons (Fink et al., 1992; Augood and Emson, 1994), the integrative mechanism of two receptor systems should take place at the level of AC5. This feature of the receptor–effector interaction indicates that the signaling pathways of A2A and D2receptors in the striatal neurons constitute a network of a literally unseparable system. Given that in wild-type mice, D1-stimulated AC activity was inhibited by D2 activation (Fig. 5A), at least some of the D1 and D2 receptors appear to localize in a close proximity to each other in the same striatal neurons (Surmeier et al., 1996; Aizman et al., 2000). So, the unseparable signaling network could be extended to the interaction between A2A and D2 and D1 receptor systems, in which AC5 plays a key and common role.
Footnotes
This work was supported by the Neurobiology Research Program of the Korea Ministry of Science and Technology (KMOST) (J.-K.L., P.-L.H.) and partly by the Creative Research Initiative Program from KMOST (H.-S.S.). We thank K.-H. Lim and A.-L. Youn for their excellent help in behavioral works.
Correspondence should be addressed to Pyung-Lim Han, Ewha Institute of Neuroscience, Ewha Womans University School of Medicine, 70 Jongno-6-Ga, Jongno-Gu, Seoul, 110-783, Korea. E-mail:plhan{at}ewha.ac.kr.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}