

Abstract
The amyloid precursor protein (APP) of Alzheimer's disease (AD) has a copper binding domain (CuBD) located in the N-terminal cysteine-rich region that can strongly bind copper(II) and reduce it to Cu(I) in vitro. The CuBD sequence is similar among the APP family paralogs [amyloid precursor-like proteins (APLP1 and APLP2)] and its orthologs (including Drosophila melanogaster, Xenopuslaevis, andCaenorhabditis elegans), suggesting an overall conservation in its function or activity. The APP CuBD is involved in modulating Cu homeostasis and amyloid β peptide production. In this paper, we demonstrate for the first time that Cu-metallated full-length APP ectodomain induces neuronal cell death in vitro. APP Cu neurotoxicity can be induced directly or potentiated through Cu(I)-mediated oxidation of low-density lipoprotein, a finding that may have important implications for the role of lipoproteins and membrane cholesterol composition in AD. Cu toxicity induced by human APP,Xenopus APP, and APLP2 CuBDs is dependent on conservation of histidine residues at positions corresponding to 147 and 151 of human APP. Intriguingly, APP orthologs with different amino acid residues at these positions had dramatically altered Cu phenotypes. The corresponding C. elegans APL-1 CuBD, which has tyrosine and lysine residues at positions 147 and 151, respectively, strongly protected against Cu-mediated lipid peroxidation and neurotoxicity in vitro. Replacement of histidines 147 and 151 with tyrosine and lysine residues conferred this neuroprotective Cu phenotype to human APP, APLP2, andXenopus APP CuBD peptides. Moreover, we show that the toxic and protective CuBD phenotypes are associated with differences in Cu binding and reduction. These studies identify a significant evolutionary change in the function of the CuBD in modulating Cu metabolism. Our findings also suggest that targeting of inhibitors to histidine residues at positions 147 and 151 of APP could significantly alter the oxidative potential of APP.
Alzheimer's disease (AD) is characterized by progressive neuronal dysfunction, reactive gliosis, and the formation of amyloid plaques in the brain. The major constituent of AD plaques is the amyloid β peptide (Aβ) that is cleaved from the membrane-bound amyloid precursor protein (APP) (Glenner and Wong, 1984; Masters et al., 1985; Kang et al., 1987; Koh et al., 1990; Yankner et al., 1990; Hardy et al., 1998). The cause of the neuronal cell loss in AD is unclear but may be related to increased oxidative stress from excessive free radical generation (Martins et al., 1986; Smith et al., 1997; Bush, 2000; Sayre et al., 2000). One of the major potential sources of free radical production in the brain is from the transition metals, copper (Cu) and iron (Fe) (Bush, 2000;Sayre et al., 2000). These metals are vital for normal cellular function because of their high redox activity. This redox potential has been successfully harnessed by a number of enzymatic pathways, including cellular respiration. However, if the redox reactivity of Cu and Fe is not strictly regulated, this can result in the generation of toxic reactive oxygen intermediates (ROIs) such as the hydroxy radical (•OH) (Smith et al., 1997). The potential for oxidative damage from ROI in the aging brain is further enhanced by the high oxygen consumption and relatively low antioxidant levels in brain tissue. To prevent transition metal-mediated oxidative stress, cells have evolved complex metal transport systems that deliver Cu and Fe to metalloenzymes and proteins. These include mammalian Cu chaperones that are involved in intracellular Cu trafficking to Cu/Zn superoxide dismutase and the Wilson's disease Cu ATPase (Waggoner et al., 1999). The chaperones target the Cu atoms to specific intracellular proteins, which results in unbound Cu being essentially absent in the intracellular environment (Rae et al., 1999). Therefore, cuproproteins have an important role in maintaining cellular Cu metabolism (Andrews, 2001).
Both APP and Aβ can strongly bind Cu(II) and reduce it to Cu(I)in vitro (Hesse et al., 1994; Multhaup et al., 1996; Atwood et al., 1998; Cherny et al., 1999; Huang et al., 1999a,b). The APP Cu binding domain (CuBD) is located in the N-terminal cysteine-rich region next to the growth factor-like domain (Hesse et al., 1994; Rossjohn et al., 1999). APP is a member of a multigene family that contains the paralog amyloid precursor-like proteins (APLP1 and APLP2). Orthologs have been identified in a diverse range of species, includingDrosophila melanogaster, Xenopus laevis, Caenorhabditis elegans, puffer fish (Fugu rubripes and Tetraodon fluviatilis), and electric ray (Narke japonica) (Wasco et al., 1992; Daigle and Li, 1993; Slunt et al., 1994; Okado and Okamoto, 1995; Torroja et al., 1996; Iijima et al., 1998; Villard et al., 1998). The CuBD sequence is similar among the different APP family paralogs and orthologs, suggesting an overall conservation in its function or activity.
APP expression modulates Cu homeostasis because APP−/− mice have elevated Cu levels in the liver and cerebral cortex when compared with APP+/+ mice (White et al., 1999b). In addition, elevated Cu concentrations reduce Aβ production and increase secretion of APP in a cell line transfected with human APP cDNA (Borchardt et al., 1999). This effect could be influenced by Zn or with Zn and Cu chelators (Borchardt et al., 2000). These studies provide strong evidence that APP has an important role in modulating cellular Cu metabolism in certain tissues, including the brain. Moreover, wild-type APP-expressing neurons (APP+/+) are significantly more sensitive to Cu toxicity than APP-deficient neurons (APP−/−) (White et al., 1999a), and interaction between APP Cu(I) species with hydrogen peroxide can result in Cu(I) oxidation to Cu(II) and APP fragmentation (Multhaup et al., 1998). Therefore, alterations to APP and/or Cu metabolism, as found in AD, could potentially result in increased APP Cu(I)-mediated ROI generation and increased oxidative stress as well as altered APP processing to Aβ (Lovell et al., 1998; Borchardt et al., 1999; Cherny et al., 1999; Sayre et al., 2000). However, full-length APP-mediated Cu neurotoxicity has not been directly demonstrated in vivo orin vitro.
In this paper, we used cell culture and cell-free lipid peroxidation assays to define the role of the APP CuBD in Cu toxicity. We demonstrate for the first time that Cu-metallated human brain-derived and recombinant full-length APP ectodomain oxidizes low-density lipoprotein (LDL) and induces neuronal cell death in vitro. Recombinant and synthetic proteins corresponding to the APP metal-binding ectodomains were used to demonstrate that APP Cu toxicity was specifically mediated by the Cu-binding ectodomain between residues 135 and 166 of human APP. Toxicity was generated by this sequence in the presence of Cu but not other metals and involved reduction of Cu(II) by APP. Mutagenesis of the APP CuBD revealed that APP-mediated Cu toxicity was dependent on the central histidine residues H147, H149, and H151. The importance of the central histidine region in APP Cu toxicity was further supported by the fact that APLP2 and nonmammalian APP orthologs, which have a highly conserved central histidine region, could also potentiate Cu-mediated toxicity. Importantly, APP orthologs with different amino acid residues at the histidine positions have dramatically altered phenotypes. The C. elegans APL-1 peptide (APL-1CuBD), which has tyrosine 147 and lysine 151, strongly protected against Cu toxicity in vitro. Substitution of histidine residues 147 and 151 for tyrosine and lysine, respectively, in human and other toxic APP CuBDs conferred a protective phenotype to these peptides. These findings identify a significant evolutionary change in the function of the CuBD. The data also highlight the important role of the APP CuBD in both APP metabolism and neurotoxicity, possibly through a gain of function activity of APP resulting in perturbed Cu homeostasis.
MATERIALS AND METHODS
Materials. Poly-l-lysine, bathocuproine disulfonate (BC), bovine serum albumin (BSA), LDL, and trypsin were purchased from Sigma (St. Louis, MO). Metal salts were obtained from Ajax Chemicals or BDH Chemicals. Minimal Essential Medium (MEM) was obtained from Life Technologies. Fetal calf serum (FCS) and horse serum (HS) were from the Commonwealth Serum Laboratories.
Recombinant APP18–611, APP18–146, APP124–189, APLP2, and APLP1. Recombinant secreted APP (APP18–611), APLP2, APLP1, APP18–146, and APP124–189 were produced in the methylotrophic yeast,Pichia pastoris. The expression of APP18–611, APLP2, and APLP1 has been described elsewhere (Henry et al., 1997, 1998; White et al., 1998). APP18–146 was generated by PCR using the primers CCC CGG GAT GCT GGA GGT ACC CAC TGA TGG and CCC CCG GGC TAA GTT TCG CAA ACA TCC ATC CTC. The PCR product was cloned as an XmaI fragment into the P. pastoris vector pHIL-S1 (Invitrogen, San Diego, CA). APP124–189 was generated by PCR using the primers GCT CGA GAA AA GAG AGG CTA GTG ATG CCC TTC TCG and GAA TTC TTA CAG TGG GCA ACA CAC AAA CTC. The PCR product was cloned as a XhoI–EcoRI fragment into the P. pastoris vector pIC9 (Invitrogen). The constructs were transformed into P. pastoris strain GS115 as described previously (Henry et al., 1997). Expressing clones were identified by silver stain SDS-PAGE analysis of the culture supernatants.
APP124–189 was purified to homogeneity in two steps. First, supernatant from a P. pastoris culture expressing the domain was concentrated, and buffer was exchanged into 20 mm TRIS buffer, pH 8.5, containing 5 mm EDTA and applied to a QHyperD 1.6 × 13 cm column (Biosepra) equilibrated in the same buffer. APP124–189, which eluted in the column flow-through, was concentrated and further purified on a Superdex 75 HR 10/30 gel filtration column (Amersham Biosciences) in 20 mm sodium phosphate buffer, pH 6.8, containing 1 mm EDTA. Pure APP124–189 eluted as a single peak. N-terminal amino acid sequencing and mass spectrometry confirmed the N terminus was intact, and the mass correlated with that of the predicted sequence. The protein was concentrated by ultra-filtration to a final concentration of 5 mg/ml in 20 mm phosphate buffer, pH 6.8.
Purification of human brain-derived APP. Human brain-derived secreted APP was prepared as described previously (Moir et al., 1992). The proteins were concentrated, and buffer was exchanged into 20 mm HEPES, 138 mm NaCl, pH 7.4.
Chemical synthesis and purification of APP CuBD peptides. For solid-phase synthesis of APP CuBD (see Tables 1-3), we used the Fmoc strategy (Merrifield, 1963; Carpino and Han, 1972) in a fully automated synthesizer (ABI 433). Peptide chain assembly was performed using in situ activation of amino acid building blocks by 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate. The purified material was analyzed by HPLC and laser desorption mass spectrometry (Vision 2000, Finnigan MAT). Purified peptides were dissolved in double-distilled water (dH2O) at a concentration of 700 μm and stored at −70°C until use.
Metallation of proteins and peptides. Brain purified APP, recombinant proteins, or synthetic peptides (500 μl samples) were mixed with metal–glycine solutions [Cu(II), Fe(II), or Zn(II) at a metal to glycine ratio of 1:6] at an equimolar or a two-fold metal to protein concentration unless stated otherwise. Metal–protein mixtures were incubated overnight at 37°C and then extensively dialyzed [24 hr against two changes of dH2O (3 liters per change) at room temperature] using mini-dialysis cups with a 3500 kDa cutoff (Pierce, Rockford, IL). Dialysis of proteins was also performed against PBS, pH 7.4, which resulted in metallated proteins with activity identical to dH2O dialysis.
Lipoprotein oxidation. Two different assays of metal-mediated lipid peroxidation were used. The first assay involved measuring the oxidative activity of metallated proteins. This was determined by mixing dialyzed metallated or native protein (at designated concentrations) with 0.5 mg/ml LDL for 24 hr (37°C). Lipid peroxidation (LPO) was measured using a lipid peroxidation assay kit (LPO 486, Oxis International Inc., Portland, OR) as per kit instructions. The level of LPO was determined by comparing absorbance (486 nm) with LDL alone (100% LPO). The second assay was used to measure the LPO activity of native proteins in the presence of free, nonprotein-bound Cu. This involved adding nonmetallated peptides (140 μm) to 0.5 mg/ml LDL together with 20 μm Cu-Gly and assaying for LPO as for the metallated proteins. The level of LPO was determined by comparing the absorbance (486 nm) with LDL + Cu-Gly (100% LPO). As a negative control, LDL was also exposed to dialyzed Cu-Gly solutions comparable with those used to Cu metallate the proteins.
Primary neuronal cultures. Cortical cultures were prepared as described previously (White et al., 1999a). Briefly, embryonic day 14 BL6Jx129sv wild-type or APP−/− mouse cortices were removed, dissected free of meninges, and dissociated in 0.025% (w/v) trypsin. Dissociated cells were plated in 24-well culture plates (Greiner GmbH) at a density of 2 × 106 cells/ml in MEM with 10% (v/v) FCS and 10% (v/v) HS. Cultures were maintained at 37°C in 5% CO2. This method produced cultures that were 95% pure for neurons (White et al., 1999a). Before experiments, the culture medium was replaced with MEM plus N2 supplements.
Cytotoxicity induced by the Cu-metallated APP. To determine the neurocytotoxic effects of the APP CuBD, metallated and native proteins and peptides were added to 2-d-old primary neuronal cultures. Where indicated, cultures were also exposed to Cu-Gly (5 or 10 μm) or LDL. Positive control cultures were treated with Cu-Gly + LDL or the LPO product, 4-hydroxy-nonenol (HNE; Sigma Chemicals). Cultures were assayed for cell death using the lactate dehydrogenase (LDH) assay kit (Roche Molecular Biochemicals, Nunawading, Australia) as per kit instructions (White et al., 1999a).
Cu(I) detection in Cu-metallated human-derived and recombinant proteins. Cu(I) generated by human brain-derived and recombinant proteins was measured using a modification of the BC Cu(I) detection assay (Huang et al., 1999a,b). APP was metallated and dialyzed as described above. Metallated or native recombinant protein (25 μm) or human-derived APP (2 μm) was mixed with BC (250 μm) in PBS, pH 7.4. Protein–BC mixtures were incubated at 37°C for 4 hr, and absorbance was measured with a Bio-Rad Model 550 plate reader. Absorbance readings for BC in PBS alone were subtracted from protein–BC readings to give Cu(I) levels as absorbance units per milliliter.
Real-time surface plasmon resonance analysis. Real-time binding experiments were performed on a BIACORE system equipped with the Upgrade kit (BIACORE). All experiments were performed at 37°C. To prepare a metal chelating sensor surface, a nitrilotriacetic acid (NTA) immobilized sensor chip (sensor chip NTA, BIACORE) was exposed to copper solution (100 μmCuCl2 in Milli-Q water) for 4 min at a flow rate of 5 μl/min. For control experiments the sensor surface was treated as above, but EDTA (1 μm) was injected for 4 min.
Surface plasmon resonance analysis (SPR) buffers and solutions were filtered and degassed: eluent buffer (PBS, 0.005%n-octylglycopyranoside, 1 μmEDTA, pH 7.4), dispensor buffer (PBS, 0.005%n-octylglycopyranoside, 3 mmEDTA), and regeneration solutions I (50 mm EDTA) and II (45 mm EDTA, 1 mmBC). After extensive washing to reset the surface with regeneration buffer I followed by eluent buffer, individual flow cells were loaded with copper solution to saturate the surface with Cu(II). The signal for binding of Cu(II) to NTA was 40 response units (RU) (see Fig. 4A). Peptide stock solutions were prepared in 1 μm EDTA (1 mg/ml), diluted in PBS (30 μg/ml), and injected onto the surface for 2 min (10 μl) by using the KINJECT command. The sensorgram was allowed to run for an additional 20 min after the end of injection to determine the dissociation kinetics. The following treatment with regeneration solution II for 6 min resulted in return to the baseline signal, indicating that the surface had been cleaned completely. Two observations are central to demonstrating the reliability of the present approach. First, peptides did not show binding to the NTA surface when it had not been loaded previously with Cu(II). Second, the injection of 1 mm BC for 2 min onto the Cu(II)-charged NTA surface did not affect surface-bound RU, showing that peptide binding was specific and exclusively mediated by Cu(II) but not Cu(I).
Sensorgrams were analyzed using the BIAevaluation 3.0 program (BIACORE), and kinetic constants were obtained by fitting curves to a single-site binding model (A + B = AB).
Statistical analysis. Data represent the mean and SE of at least three experiments performed in triplicate. ANOVA and Newman–Keuls tests were used to analyze data.
RESULTS
Human brain-derived and recombinant APP Cu oxidize lipoproteins
Although previous studies have shown that a peptide to the APP CuBD peptide (APP142–166) can induce Cu-mediated neurotoxicity (White et al., 1999a), the ability of a physiologically relevant form of APP to be toxic in the presence of Cu is unknown. To examine this, we tested human brain-derived and recombinant APP for their ability to induce Cu-mediated lipid peroxidation. Purified APP was first metallated by incubation with twofold molar excess of Cu-Gly and then dialyzed extensively against distilled water to remove unbound Cu. Human brain APP Cu (60 nm) was incubated with LDL (0.5 mg/ml) for 24 hr, and LPO levels were measured. Soluble and membrane-associated APP Cu significantly elevated LPO levels (136 ± 7 and 132 ± 6%, respectively) when compared with nonmetallated APP (**p < 0.05) (Fig.1A). Recombinant α-secretase-cleaved APP695 ectodomain (APP18–611-Cu) (60 nm) also induced a significant increase (214 ± 20%) in LDL oxidation compared with either LDL alone or nonmetallated APP18–611 (*p < 0.01) (Fig.1A). Cu-metallated recombinant APLP2 ectodomain (APLP2-Cu, 60 nm) induced elevated LPO levels (168 ± 21%) (*p < 0.01) (Fig.1A), but neither APLP1-Cu nor BSA-Cu (60 nm) had any effect on LPO, demonstrating that the LPO induced by APP18–611-Cu and APLP2-Cu is protein specific and not caused by a nonspecific protein–Cu–LDL interaction. Similarly, LPO was not induced by a dialyzed Cu-Gly solution of the same concentration used to metallate APP, indicating that LPO is not caused by residual unbound Cu in the APP Cu solution.

A, LDL oxidation induced by Cu-metallated recombinant and human brain-derived APP. LDL (0.5 mg/ml) was incubated with 60 nm Cu-metallated or nonmetallated recombinant APP, APLP2, APLP1, or human brain-derived soluble (Sol) or membrane-associated (Mem) APP. Cu-metallated APP and APLP2 induced significantly elevated LDL oxidation compared with untreated LDL or nonmetallated protein (*p < 0.01, **p < 0.05). Cu-metallated APLP1 or BSA did not increase LDL oxidation. B, Neuronal cell death induced by APP Cu. Primary cortical neurons were treated with APP18–611 (100–500 nm) or BSA-Cu (500 nm) (4 treatments over 6 d). Cell death was determined with the LDH assay. APP18–611 and BSA-Cu had no effect on cell survival, whereas APP18–611-Cu induced a dose-dependent increase in neuronal death (*p < 0.05, **p < 0.01).C, Neuronal cell death induced by APP Cu and LDL. APP18–611-Cu, APLP2-Cu, APLP1-Cu, or BSA-Cu (100 nm; 2 treatments over 4 d) was incubated with LDL (100 μg/ml). APP18–611-Cu and APLP2-Cu potentiated cell death from LDL exposure (**p < 0.01). APLP1-Cu or BSA-Cu had no effect on LDL-mediated toxicity, and LDL alone had no effect on neurons. The LPO product HNE (10 μg/ml) or 5 μm Cu + LDL (25 μg/ml) induced significant cell death (**p < 0.01).
To establish that the LDL oxidation by brain-derived APP involved reduction of Cu(II) to Cu(I), as described for recombinant APP (Multhaup et al., 1996), we measured Cu(I) generation by the Cu-metallated brain APP using the bathocuproine assay. The absorbance of Cu-metallated brain APP was 0.080 ± 0.009 compared with 0.043 ± 0.004 for nonmetallated APP (p < 0.01) (data not shown), confirming that both recombinant and human brain-derived APP can generate bathocuproine-detectable Cu(I) (Multhaup et al., 1996). Importantly, these results demonstrate that although nonmetallated APP and APLP2 have no effect on Cu-induced LDL oxidation, when loaded with Cu they potentiate high levels of LPO through generation of Cu(I).
APP Cu induces neuronal cell death
To determine whether the peroxidative activity of APP Cu can induce neurotoxicity, we exposed primary mouse cortical cultures to Cu-metallated APP18–611. Cortical neurons were treated with APP18–611 (500 nm, nonmetallated), APP18–611-Cu (100–500 nm), or BSA-Cu (500 nm) (four exposures over 6 d), and cell death was measured with the LDH assay. A dose-dependent increase in cell death was observed in cultures treated with 200 and 500 nm APP18–611-Cu (*p < 0.05, **p < 0.01) (Fig. 1B) but not in cultures exposed to nonmetallated APP18–611 or BSA-Cu. These findings demonstrate for the first time that Cu-metallated full-length APP (ectodomain) is toxic to neurons at physiologically relevant concentrations.
Oxidation of LDL by APP Cu potentiates neuronal cell deathin vitro
Lipoprotein oxidation is a central feature of vascular illness and may have an important role in AD (Pitas et al., 1987; Schippling et al., 2000; Praticò et al., 2001). Our LDL oxidation experiments using Cu-metallated APP18–611 and brain-derived APP demonstrated a potent LPO potential for metallated APP. We therefore investigated the possibility that APP Cu could exacerbate neurotoxicity through oxidation of extracellular lipoproteins. Neuronal cultures treated with subtoxic APP18–611-Cu (100 nm) for 4 d (two exposures over 4 d) in the presence of 100 μg/ml LDL induced significant neuronal cell death (52 ± 3%) compared with LDL alone (2 ± 2%) (**p < 0.01) (Fig. 1C). APP18–611-Cu also induced significant cell death from lower concentrations of LDL (31 ± 5 and 15 ± 2% from 50 and 25 μg/ml LDL, respectively), whereas cell death was not induced by either 100 nm nonmetallated APP18–611 or any concentration of LDL (data not shown). Coincubation of 100 nmAPLP2-Cu with LDL resulted in 23 ± 6% cell death (**p < 0.01) (Fig. 1C). Importantly, cell death was not induced by APLP1-Cu, BSA-Cu, or a dialyzed Cu solution and LDL. The toxicity mediated by 100 nmAPP18–611-Cu with 100 μg/ml LDL was similar to the level induced by 10 μg/ml HNE or 5 μm Cu-Gly + 25 μg/ml LDL (Fig. 1C). These data clearly demonstrate that physiological levels of soluble APP and APLP2 have the potential to induce neuronal cell death through binding and reduction of Cu.
Localization of APP Cu-mediated toxicity to the APP metal-binding ectodomain
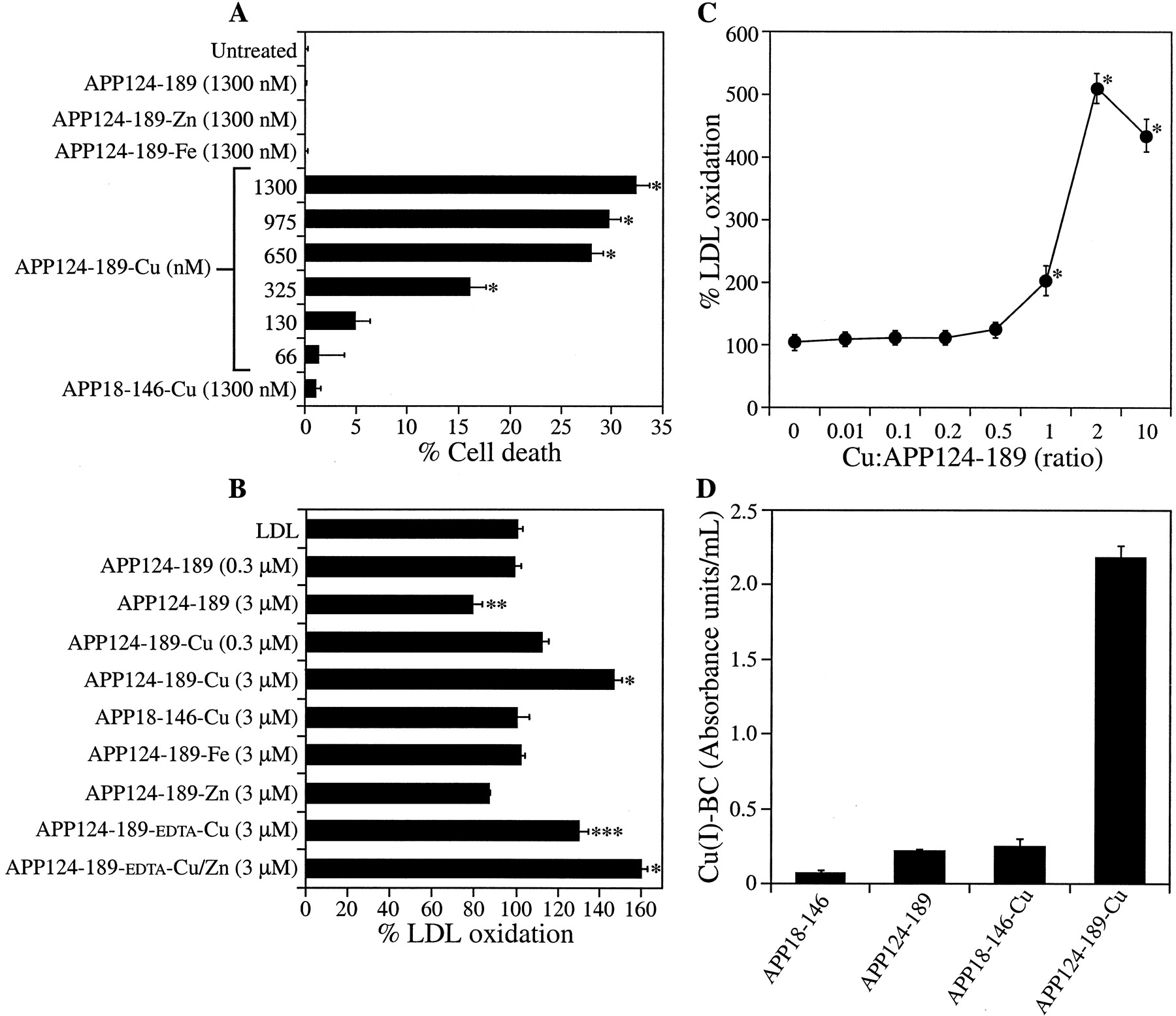
To confirm that the CuBD of APP was responsible for APP18–611 toxic activity, we assayed recombinant APP124–189, which encodes the APP CuBD. Inductively coupled plasma mass spectrometry (ICP-MS) analysis showed that purified APP124–189 had very little metal bound, indicating that it was expressed in the apo form. APP124–189 was subsequently Cu metallated and applied to neurons for 4 d. APP124–189-Cu directly induced elevated LDH release, whereas nonmetallated APP124–189, APP124–189-Zn, APP124–189-Fe, and APP18–146-Cu had no effect on cell survival (*p < 0.0) (Fig. 2A). Titration of APP124–189-Cu revealed that metallated protein concentrations as low as 325 nm induced significant cell toxicity (*p < 0.01) (Fig.2A). APP124–189-Cu could also potentiate neurotoxicity from exogenous LDL (data not shown). We also examined the ability of APP124–189 to induce LPO. Incubation of nonmetallated APP124–189 (0.3 μm) with LDL did not alter LPO levels compared with LDL alone. Interestingly, 3 μm nonmetallated APP124–189 reduced LPO by 21% (Fig. 2B) (**p < 0.05), suggesting that the recombinant apo-protein may be able to chelate residual metals from the LDL and inhibit endogenous LPO. After metallation, significant elevation of LPO was induced by 3 μm APP124–189-Cu (147 ± 4.2% LPO compared with a dialyzed Cu solution; *p < 0.01) (Fig.2B), whereas APP124–189 loaded with Zn or Fe had no effect on LPO (Fig. 2B). In addition, the APP CuBD did not induce LPO after metallation with a large range of metals (data not shown). Cu metallation of the control protein APP18–146, which corresponds to the APP growth factor domain (Rossjohn et al., 1999), also failed to induce LPO because it lacks the CuBD.

A, Cell death induced by Cu-metallated APP124–189. Primary cortical neurons were treated with APP124–189 after metallation with Cu, Fe, or Zn. Cell death was determined with the LDH assay. Nonmetallated APP124–189, APP124–189-Fe, and APP124–189-Zn (1300 nm) had no effect on neuronal cell death. APP124–189-Cu (66–1300 nm) induced a dose-dependent increase in neuronal cell death (*p < 0.01). APP18–146-Cu did not increase neuronal cell death. B, LDL oxidation induced by Cu-metallated human APP124–189. LDL (0.5 mg/ml) was incubated with 0.3 or 3 μm metallated or nonmetallated APP124–189. Nonmetallated APP124–189 (3 μm) induced a significant decrease in LDL oxidation compared with LDL alone (**p < 0.05), whereas 3 μmCu-metallated APP124–189 significantly enhanced LDL oxidation at 3 μm (*p < 0.01). APP124–189 metallated with Fe or Zn did not increase LDL oxidation. APP124–189 pretreated with EDTA induced significantly lower LDL oxidation after Cu metallation when compared with APP124–189-Cu (non-EDTA treated) (***p < 0.05 compared with APP124–189-Cu). Metallation of EDTA-treated APP124–189 with Zn + Cu resulted in LDL oxidation equivalent to APP124–189-Cu (non-EDTA treated). C, LDL oxidation induced by APP124–189 metallated with different concentrations of Cu-Gly. LDL was incubated with APP124–189-Cu after metallation with Cu-Gly at ratios of 0.01:10 (Cu/APP124–189). D, Cu(I) generation from APP124–189-Cu. Nonmetallated and Cu-metallated APP124–189 and APP18–146 (25 μm) were incubated with 250 μm BC, and Cu(I) generation [Cu(I)-BC] was measured by spectrophotometry. The absorbance of BC alone was subtracted from each reading to give Cu(I) levels as absorbance units per milliliter. APP18–146, APP18–146-Cu, and APP124–189 induced negligible levels of Cu(I). APP124–189-Cu induced ∼10-fold higher Cu(I) levels than APP124–189 or APP18–146-Cu (*p < 0.01).
The influence of the Cu concentration used to metallate APP124–189 on subsequent Cu toxicity was examined by pretreating the native protein (50 μm) with increasing amounts of Cu-Gly and measuring LDL oxidation. We observed maximum oxidative activity using 100 μm Cu-Gly (Cu/protein ratio of 2:1), whereas no detectable LDL oxidation was seen at concentrations of Cu-Gly below 25 μm (Cu/protein ratio of 1:2) (Fig. 2C). To confirm that the toxicity of APP124–189-Cu involved generation of Cu(I), we measured protein-associated Cu(I) levels using the BC-Cu(I) detection assay. Measurement of Cu-metallated APP124–189 revealed a 10-fold greater absorbance when compared with nonmetallated APP124–189, whereas both Cu-metallated and nonmetallated APP18–146, which lacks the CuBD, revealed negligible absorbance readings after incubation with BC (*p < 0.01) (Fig.2D). Importantly, these findings establish that the APP CuBD can induce significant neurotoxicity at physiological concentrations of both APP and Cu.
Because the ZnBD is contained in APP124–189, the effect of Zn on APP Cu toxicity was measured. We treated 50 μm APP124–189 with EDTA (1 mm) to remove any endogenously bound metals, as confirmed by ICP-MS, followed by dialysis and loading with either Cu or Cu + Zn. The Cu-metallated APP124–189 induced lower Cu-mediated LPO when EDTA-treated protein was used [APP124–189-EDTA-Cu compared with non-EDTA-treated protein loaded with Cu (APP124–189-Cu) (***p < 0.05 compared with APP124–189-Cu)] (Fig.2B). The EDTA-treated APP124–189 loaded with both Zn and Cu (APP124–189-EDTA-Cu/Zn) restored LPO to maximum activity (Fig.2B). These data indicate that Zn is not required to induce LPO by APP124–189-Cu but may have a structural role that can modulate Cu toxicity.
Copper-mediated lipoprotein oxidation and neurotoxicity is induced by APP homologs expressing conserved histidine residues at positions 147, 149, and 151
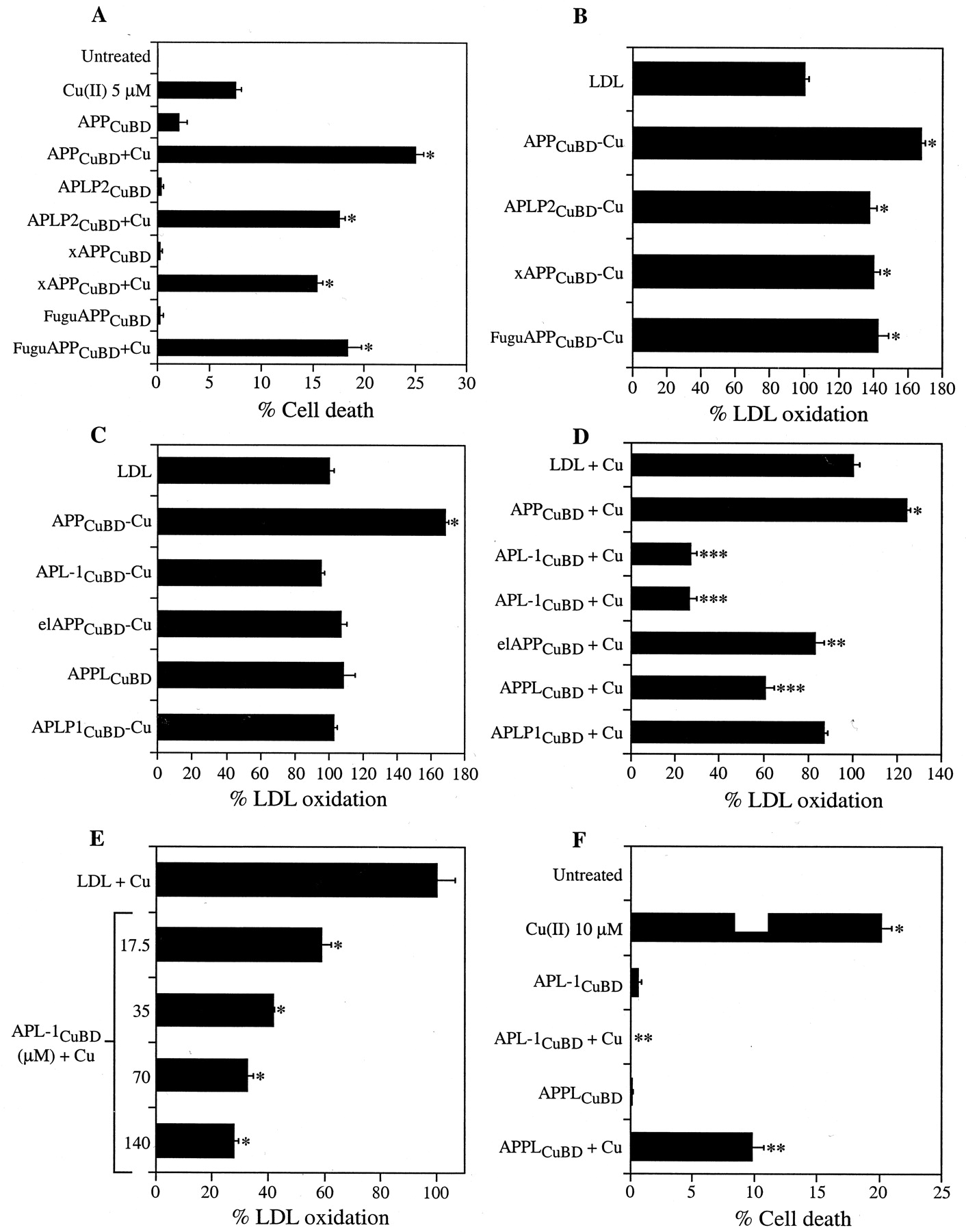
Previous studies have shown that the central histidine region (histidines 147, 149, and 151) of the APP CuBD is important for Cu(II) reduction (Multhaup et al., 1996; Ruiz et al., 1999; White et al., 1999a). To define the role of this region in APP toxicity, we examined two groups of APP homologs (Table1). The first group contained a CuBD sequence from species with a conserved central histidine region but differed in their surrounding residues. Peptides corresponding to residues 135–166 of human APP were synthesized for human APLP2 (APLP2CuBD), Xenopus APP (xAPPCuBD), and F. rubripes APP (FuguAPPCuBD) (Table 1, Group 1). Each homolog significantly potentiated neuronal cell death in cultures exposed to 5 μm Cu(II) (*p < 0.01) (Fig.3A). LPO analysis revealed that Cu-metallated APLP2CuBD, xAPPCuBD, and FuguAPPCuBD all induced a significant increase in LDL oxidation (138 ± 4, 140 ± 4, and 143 ± 6%, respectively) compared with LDL alone (*p < 0.01) (Fig. 3B). These data strongly suggest that the conservation of the central histidine residues in the APP CuBD of diverse species preserves the ability to generate toxic-free radicals and promote neurotoxicity in the presence of Cu.
Peptides corresponding to the copper binding ectodomain (CuBD) of human APP and APP orthologs and paralogs

A, Cell death induced by APP CuBD homologs with a highly conserved central histidine region. Primary cortical neurons were incubated with subtoxic Cu(II) (5 μm) and 70 μm nonmetallated APPCuBD, APLP2CuBD, xAPPCuBD, or FuguAPPCuBD (2 exposures of each over 4 d). Cell death was determined with the LDH assay. All peptides induced significantly elevated neuronal cell death compared with Cu or peptides alone (*p < 0.01).B, LDL oxidation induced by APP CuBD homologs with a highly conserved central histidine region. LDL (0.5 mg/ml) was incubated with APPCuBD or with the APP homologs APLP2CuBD, xAPPCuBD, or FuguAPPCuBD (50 μm) (Cu metallated). All peptides induced significant increases in LDL oxidation compared with LDL alone (*p < 0.01). C, LDL oxidation induced by APP CuBD homologs with a nonconserved central histidine region. LDL (0.5 mg/ml) was incubated with APPCuBD or with APL-1CuBD, elAPPCuBD, APPLCuBD, or APLP1CuBD (50 μm) (Cu metallated). Only APPCuBD Cu induced significantly elevated LDL oxidation (*p < 0.01). D, Effect of APP CuBD homologs on LDL oxidation induced by Cu-Gly. LDL (0.5 mg/ml) was incubated with 20 μm Cu-Gly with or without nonmetallated APP CuBD peptides (140 μm). APPCuBD induced a significant elevation of LDL oxidation compared with LDL + Cu-Gly (*p < 0.01). APL-1CuBD, elAPPCuBD, and APPLCuBD significantly decreased LDL oxidation induced by Cu-Gly (**p < 0.05, ***p < 0.01), whereas APLP1CuBDhad no significant effect on LDL oxidation. E, Effect of APL-1CuBD on LDL oxidation induced by Cu-Gly. LDL was incubated with 20 μm Cu-Gly with or without APL-1CuBD (17.5–140 μm). APL-1CuBD induced a dose-dependent decrease in Cu-Gly-mediated LDL oxidation (*p < 0.01).F, Effect of APL-1CuBD and APPLCuBD on Cu-induced neuronal cell death. Primary cortical neurons were incubated with a toxic concentration of Cu-Gly (10 μm) (*p < 0.01) with or without APL-1CuBD or APPLCuBD (70 μm) (2 treatments of each over 4 d). Cell death was determined with the LDH assay. APL-1CuBD and APPLCuBD significantly inhibited Cu-induced cell death (**p < 0.01 compared with Cu).
The C. elegans APL-1 CuBD strongly protects against Cu-induced lipoprotein oxidation and neurotoxicity
The second group of APP homologs examined (Table 1, Group 2) revealed single or multiple amino acid variations within the central histidine binding site but maintained the adjacent cysteines. Peptides corresponding to the human APP residues 135–166 were synthesized for APLP1 (APLP1CuBD), Drosophila APPL (APPL CuBD), electric ray APP (elAPPCuBD), and C. elegans APL-1 (APL-1 CuBD) (Table 1). The peptides were metallated with Cu(II) and tested for LPO activity. There was no change in LPO compared with LDL alone with any of the metallated peptides, whereas parallel treatment with human APPCuBD Cu elevated LPO levels (Fig.3C). Interestingly, the exposure of LDL to 20 μm Cu-Gly with 140 μmnonmetallated elAPPCuBD, APPLCuBD, or APL-1CuBDpeptide actually inhibited LDL oxidation by ∼20, 40, and 80%, respectively (**p < 0.05, ***p < 0.01) (Fig. 3D). In contrast, 140 μmhuman APPCuBD or other Group 1 peptides increased LPO by a further 25% or more (*p < 0.01) (Fig. 3D). Titration of APL-1CuBD against 20 μm Cu(II) revealed a clear dose-responsive protection by APL-1CuBD (Fig.3E).
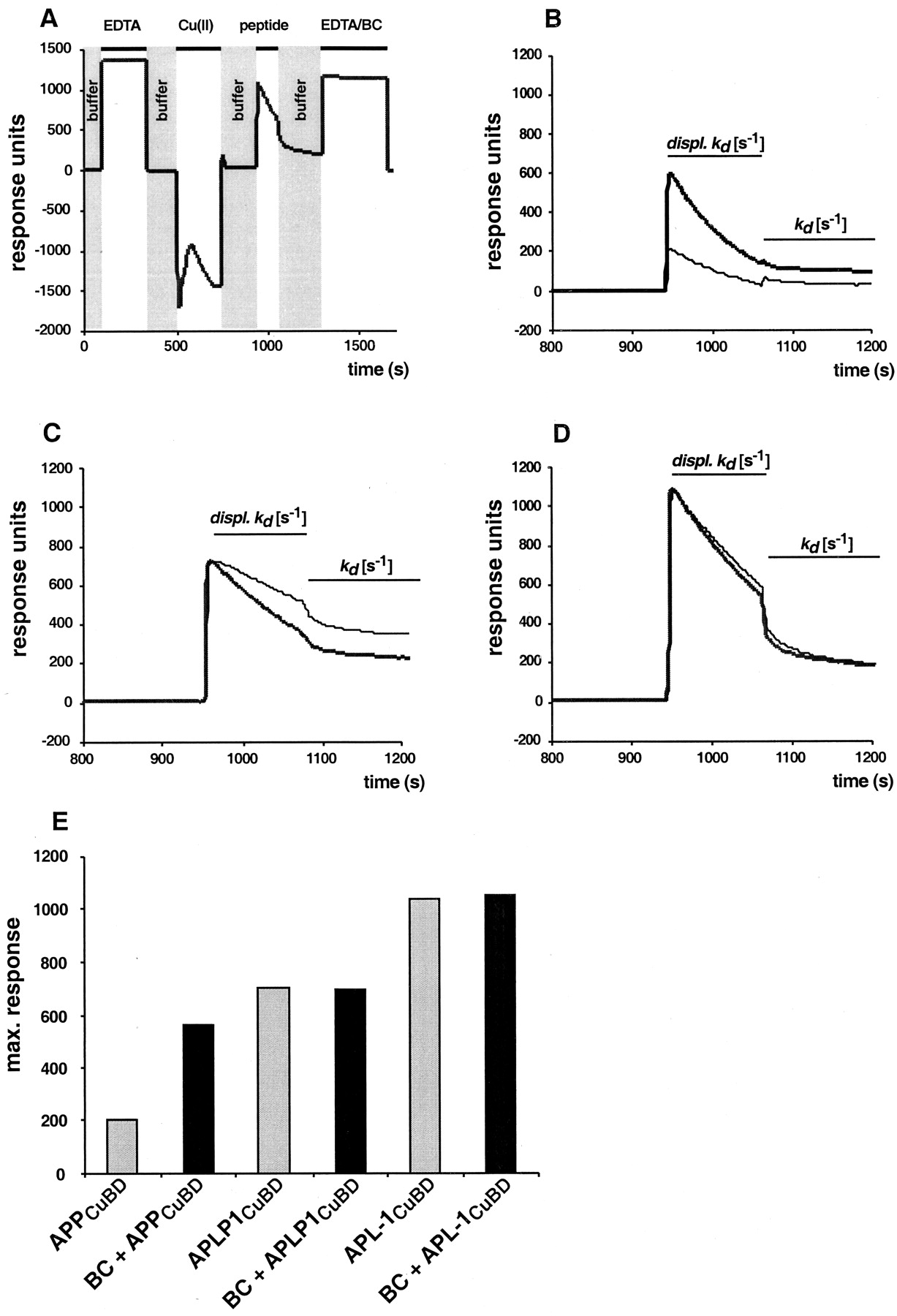
A, Sensorgram showing the profile for the sensor surface treatment for the cycle of APPCuBD peptide (and of its derivatives) binding to an NTA sensor chip and its regeneration with EDTA/BC. The profile shows that an intermediate ternary complex of NTA Cu(II) peptide is formed (peptide). B,C, D, The dissociation kinetics for APPCuBD (B), APLP1CuBD(C), and APLP1CuBD is interpreted as the displacement of peptide Cu(I) complexes from the sensor surface (displ. Kd ) and peptide elution from the Cu(II) NTA surface (Kd ) (D). Thebold dark line represents the peptides being injected in the presence of 10 μm of the Cu(I)-specific chelator BC. The thin line represents the peptides being injected in the presence of running buffer. E, Maximum response units were reached at 980 sec and taken from the curvesin B, C, and D obtained in the presence of running buffer (gray bars) or BC (black bars).
To determine whether the ability to inhibit LPO by these APP homologs was reflected in increased neuronal survival against Cu toxicity, we treated cortical cultures with a toxic concentration of Cu(II) (10 μm) together with either APPLCuBDor APL-1CuBD (two exposures at 70 μm). The APL-1CuBD peptide completely abrogated Cu toxicity to background levels (0%) (**p < 0.01) (Fig. 3F), whereas APPLCuBD lowered Cu-induced cell death from 20.1 ± 0.9 to 9.8 ± 1.0%. These findings demonstrate that C. elegans APL-1, and to a lesser extent the APPL CuBDs, has a potent inhibitory effect on Cu toxicity in vitro.
Inhibition of Cu-mediated lipid peroxidation by the APL-1 CuBD is mediated by tyrosine 147 and lysine 151
Because the central histidines at positions 147, 149, and 151 are important for APPCuBD CuBD activity, we proposed that the APL-1 protective phenotype would be caused by the sequence differences at the corresponding residues in APL-1CuBD, which are tyrosine at 147 and lysine at 151. To test this, mutagenesis studies were performed on the human APPCuBD and C. elegansAPL-1CuBD. Human and C. eleganspeptides were synthesized containing the amino acids of the opposing peptide at positions 147 and 151 (APPCuBDY147.K151 and APL-1CuBDH147.H151) (Table2). The numbering of the mutations in the APL-1CuBD mutant peptides is based on the human APP sequence to simplify the presentation of the data (Table 2). Nonmetallated APPCuBDY147.K151 was added to LDL + 20 μm Cu-Gly and inhibited Cu-induced oxidation to a similar level as wild-type APL-1CuBD (*p < 0.01) (Table 2). Conversely, mutation of the Y147 and K151 residues in APL-1 to histidines, (APL-1CuBDH147.H151) converted it into a toxic peptide with an LPO activity similar to wild-type human APPCuBD (*p < 0.01) (Table 2). Single amino acid substitutions of either histidine 147 with tyrosine or histidine 151 with lysine (APPCuBDY147 or APPCuBDK151) (Table 2) produced inactive peptides that induced only background levels of LPO (*p < 0.01) (Table 2). Similarly, the single mutated APL-1 peptides APL-1CuBDH147 and APL-1CuBDH151 revealed significantly reduced protective effects against Cu in nonmetallated peptide assays (*p < 0.01) (Table 2).
Modulation of Cu-induced LDL oxidation by CuBD peptides with amino acid substitutions within the central histidine region
To examine whether the histidine–tyrosine and histidine–lysine substitutions could alter other APP homologs, we measured the LPO activity of APLP2CuBDY147.K151 and xAPPCuBDY147.K151. Consistent with the human APPCuBDY147.K151 results, these peptides also induced significantly protective effects similar to wild-type APL-1CuBD in nonmetallated peptide assays (*p < 0.01 compared with wild-type APLP2CuBD and xAPPCuBDpeptides) (Table 2). Because the APLP1CuBDpeptide is inactive, we also examined the effect of substituting the central histidines of human APPCuBD with the S147 and R149 present in APLP1CuBD. The APPCuBDS147.R149.H151 peptide was inactive in the nonmetallated peptide assays (*p < 0.01 compared with wild-type APPCuBD) (Table 2). These findings demonstrated that the toxic and protective activities of the CuBD is dependent on the amino acids present in positions 147 and 151 in human APP or their equivalent position in APP orthologs and paralogs.
The phenotype of the human APP and C. elegans APL-1 CuBDs correlates with Cu(II) binding and reduction
To understand the mechanism underlying the contrasting activities of the human APPCuBD and APL-1CuBD peptides, we used SPR to analyze specific binding to Cu(II) and determined the dissociation kinetics of the peptides (Table 3). Immobilized NTA on sensor chips in conjunction with SPR was used to assess directly the affinity of APP CuBD peptides for metal-ion binding. Our studies revealed that an intermediate ternary complex of NTA Cu(II) peptide is formed on the chip surface. The decreasing response signal (Fig.4B, Displ. kd ), just before the real dissociation phase when the chip is washed with buffer, represents the dissociation of the complex when the peptide binding capacity of Cu(II) NTA is exceeded. The peptides exhibited displacement activities by competing with NTA for Cu(II) binding (A. Simons, T. Ruppert, C. Schmidt, A. Schlicksupp, R. Pipkorn, J. Reed, A. R. White, T. A. Bayer, C. L. Masters, R. Cappai, G. Multhaup, unpublished observations). The dissociation phases of three representative peptides are shown in Figure 4. Three peptides were examined: the APPCuBD peptide (toxic), APLP1CuBD (inert), and APL-1CuBD (protective). The six sensorgrams were evaluated for mean dissociation rate constants. There was a clear difference between APPCuBD and APL-1CuBD, with the latter showing higher maximum response units attributable to a higher affinity for the Cu(II) NTA chip surface (Table 3, Fig. 4B–E). These data indicate that Cu(II) binding and reduction are affected by the amino acid side chains at positions 147 and 151, correlating with the toxic or protective phenotypes.
Kinetic parameters of the wild-type APP CuBD peptides measured by surface plasma resonance
When the peptides were injected in the presence of 10 μmCu(I)-specific chelator BC (Fig. 4B–D,bold curves), the maximum response increased threefold for APPCuBD but remained unchanged for the inert and protective peptides (Fig. 4E). The displacement of the ternary complex was unaffected for APPCuBD(Fig. 4B) and APL-1CuBD (Fig.4D) but increased twofold for APLP1CuBD (Fig. 4C). The dissociation from NTA Cu(II) was specifically decreased for APPCuBD (Fig. 4B) in the presence of BC when compared with the other peptides (Fig.4C,D) that do not show significant alterations in their dissociation constants. These data clearly show that BC increases the Cu(II) binding capacity of APPCuBD but not of APLP1CuBD or APL-1CuBD(Fig. 4E). APL-1CuBD was the most effective in Cu(II) binding, and its maximal binding remained unaffected by BC.
These kinetic results suggest that APPCuBDreduces Cu(II) NTA as fast as it binds to Cu(II) NTA. After reduction, Cu(I) is immediately released from the NTA (Simons, Ruppert, Schmidt, Schlicksupp, Pipkorn, Reed, White, Masters, Cappai, Multhaup, unpublished observations), most likely as an APPCuBD Cu(I) complex. This is supported by our earlier study analyzing Cu binding of APPCuBD by liquid chromatography electrospray ionization mass spectrometry (Multhaup et al., 1996). We could identify APP Cu complexes without being able to differentiate between Cu(II) and Cu(I) binding. The rate of Cu(II) to Cu(I) reduction by APLP1CuBD seems to be much slower, because maximum binding was reached first before Cu(II) was reduced and peptide Cu(I) complexes were displaced from the NTA surface. This is in agreement with previous results showing that the APLP1 peptide has significantly less ability to produce Cu(I) as measured by the BC assay (Multhaup et al., 1996). There was no significant difference in the displacement of the ternary complex including APL-1CuBD in the presence or absence of BC (Fig. 4D). The slight difference between both constants (Table 3) derived from Figure 4Dmight be attributable to the Cu-reducing activity of APL-1CuBD being lower than for APLP1CuBD or being totally absent. These data indicate that the toxic phenotype of the CuBD peptides examined here correlates with Cu(II) binding and reduction kinetics. The toxic activity of human APPCuBD may reflect lower Cu(II) binding and high Cu(II) reduction, whereas protection by APL-1CuBD is mediated through high Cu(II) binding and limited Cu(II) reduction.
DISCUSSION
A growing body of data supports a significant role for redox active metals, such as Cu and Fe, as key modulators of the pathogenic pathways that underlie neurodegenerative disorders (for review, seeWaggoner et al., 1999; Bush, 2000; Sayre et al., 2000). A delineation of the interactions between these metals and their molecular partners is needed to understand their role in the disease process. In relation to AD, the interaction among Cu, APP, and Aβ can result in ROI generation and subsequent oxidative stress. We have shown that APP-deficient neurons have increased resistance to Cu toxicity and that a CuBD peptide can induce toxicity from Cu added to culture medium (White et al., 1999a). In vivo studies have revealed increased Cu levels in brain and liver of APP−/− mice, and we have observed alterations to Cu metabolism in a transgenic mouse model of AD that expresses high levels of human APP (our unpublished observations). These data demonstrate an important role for APP in Cu homeostasis. The present findings, however, provide unequivocal evidence that the full-length APP molecule can induce neurotoxicity at physiologically relevant concentrations of APP and Cu. Both membrane-associated and soluble APP purified from human brain as well as recombinant APP ectodomain induced Cu(I)-mediated LPO in vitro. These findings contrast with previous studies demonstrating neuroprotective and neuritogenic activity for soluble APP (Milward et al., 1992; Mattson et al., 1993; Small et al., 1994; Cappai et al., 1999). However, the difference can be explained by the need to metallate Cu to APP to mediate neurotoxic effects because nonmetallated did not induce LPO or neuronal cell death in this study.
In vivo, cell-associated APP Cu could induce neurotoxicity through its prolonged exposure on the surface of neurites (Storey et al., 1999) and its juxtaposition to membrane lipids, whereas soluble APP directly binds to neuronal membrane receptors and to fibrillar Aβ on the cell surface (Melchor and Van Nostrand, 2000). The interaction of APP Cu with lipoproteins provides an additional mechanism for APP-mediated neurotoxicity. Whether APP Cu is able to induce direct or oxidized lipoprotein-mediated neuronal damage in vivo would depend on the availability of both Cu and lipoproteins. A potential role for oxidized lipoproteins in mediating neurodegeneration is supported by LDL being present in the brain as a result of cholesterol metabolism (Keller et al., 1999, 2000), whereas high-density lipoprotein (HDL) is synthesized by glial cells and associated with amyloid plaques in AD (Harr et al., 1996; Markesbery, 1997; Yamada et al., 1997). In addition, the expression of the apolipoprotein E4, a risk factor for AD, can promote APP secretion (Howland et al., 1998). Interestingly, lipoproteins derived from AD CSF fluid reveal a higher level of oxidation than control lipoproteins (Bassett et al., 1999, 2000; Schippling et al., 2000) and Cu-oxidized LDL and HDL induce neuronal cell death in vitro (Dubbing 1963; Kabara 1973;Pitas et al., 1987; Keller et al., 1999, 2000). Significantly, cerebral cortex and hippocampus from a transgenic mouse model of AD amyloidosis reveal increased LPO compared with wild-type mice well before the appearance of Aβ plaques (Praticò et al., 2001). Our finding that APP Cu can induce LDL oxidation and subsequent neuronal deathin vitro suggests that similar mechanisms could mediate oxidative damage in AD.
A key finding from this study came from the analysis of the CuBD from different species and APP family members. We showed that the amino acids at positions 147 and 151 of the central histidine region can dramatically influence the activity of the CuBD. This is supported byRuiz et al. (1999) who showed that Cu reduction was diminished in mutant APP147–151 peptides with histidine–alanine substitutions. APP homologs with conserved histidine residues at positions 147 and 151 all induced significant neurotoxicity. However, the APLP1, APL-1, APPL, and elAPP genes all contained different residues within the histidine region and consequently revealed nontoxic or protective activities in the presence of Cu. Particularly striking was the high level of protection against Cu toxicity afforded by APL-1CuBD. The substitution of the APL-1 Y147 and K151 for histidines, as found in all toxic APP homologs, completely reversed the APL-1 phenotype from protective to toxic. Conversely, substitution of H147 and H151 for tyrosine and lysine in human APP, APLP2, or xAPP resulted in a protective phenotype, whereas human APPCuBD was converted into an inert phenotype by substituting histidine 147 for serine and histidine 149 for arginine as present in APLP1. Biophysical analysis suggested that the mechanism responsible for the human and C. elegansphenotypes could be correlated to the Cu binding and reduction activity of the peptides. There was a clear distinction in these activities between the human and C. elegans sequences and the inert APLP1 peptide having an intermediate value. This demonstrates that residues 147 and 151 can markedly influence APP Cu chemistry, although these studies will need to be extended to all APP homologs to confirm the findings. Significantly, the CuBD of human APP is able to modulate both Cu neurotoxicity, as shown here, and Aβ production (Borchardt et al., 2000). Our studies identify histidines 147 and 151 as key targets for therapeutic modulation of these toxic processes.
The species differences may reflect significant evolutionary changes in APP Cu chemistry. Our data support a general evolutionary trend toward a decreased need for Cu protection in higher order species. Alternatively, there is a gain in activity toward promoting Cu(II) reduction. APP (including APLP2) from humans, Xenopus, and pufferfish induces significant Cu toxicity, whereas APP from lower order species (Drosophila, electric ray, and C. elegans) either has little effect on Cu toxicity or is protective. Although APLP1 has no net effect on Cu toxicity and is expressed in humans, this molecule is considered to be the ancestral form of APP (Coulson et al., 2000). The general increase in APP Cu toxicity of higher order species may reflect a decrease in environmental Cu levels or adaptation of additional mechanisms for detoxifying Cu, such as ceruloplasmin and transcuprein. The APP CuBD may have a function in sensing and responding to environmental (extracellular) Cu, a hypothesis supported by the fact that cells transfected with human APP cDNA can respond to increased Cu levels by enhancing APP secretion (Borchardt et al., 1999), and APP−/− mice have increased Cu levels in brain and liver (White et al., 1999b). This is further supported by neurons from C. elegans being involved in sensing and avoiding Cu (Sambongi et al., 1999). Whether APL-1 is involved in this process is unknown. APP could also act as a cell membrane Cu(II) reductase similar to Fre1 in yeast (Hassett and Kosman, 1995;Georgatsou et al., 1997) providing Cu(I) for subsequent uptake by Cu transport proteins or delivery to a recipient cuproprotein. This in turn could shift APP processing toward secretion to enhance Cu removal.
Regardless of the primary function of APP Cu reduction, it is clear that perturbations to Cu and/or APP metabolism can potentially result in Cu(I)-mediated neurotoxicity from cell-associated or soluble APP (Multhaup et al., 1998; White et al., 1999a). Amyloidogenic Aβ can mediate APP and APLP2 accumulation (Cribbs et al., 1995; Schmitt et al., 1997; White et al., 1998) and could result in Cu-mediated toxicity, particularly if associated with additional changes to Cu or lipoprotein metabolism (Keller et al., 1999, 2000). Our findings may also have implications for other neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS). Motor neuron loss, in some familial cases of ALS, involves point mutations that promote the interaction of aggregated superoxide dismutase-bound Cu with exogenous molecules, resulting in free radical toxicity (Yim et al., 1996; Waggoner et al., 1999; Azzouz et al., 2000). Our work supports this hypothesis by showing that altering the Cu chemistry of a redox active protein can markedly increase its oxidative potential. Further studies are needed to determine the role of APP Cu interactions in vivo and whether aberrant generation of Cu(I) by APP contributes to the neurodegenerative process in AD.
Footnotes
This work was supported in part by grants from the National Health and Medical Research Council of Australia to C.L.M. and R.C. G.M. and K.B. were supported by the Deutsche Forschungsgemeinschaft and the Bundesministerium für Forschung und Technologie. We thank Dr. Robert Cherny and Irene Volitakis for assistance with ICP-MS analyses.
Correspondence should be addressed to Dr. Roberto Cappai, Department of Pathology, The University of Melbourne, Victoria 3010, Australia. E-mail: r.cappai{at}unimelb.edu.au.





{kind=link}
{kind=link}
{kind=link}
{kind=link}