

Abstract
Insoluble fibrils of amyloid-β peptide (Aβ) are the major component of senile and vascular plaques found in the brains of Alzheimer's disease (AD) patients. Aβ has been implicated in neuronal and vascular degeneration because of its toxicity to neurons and endothelial cells in vitro; some of these cells die with characteristic features of apoptosis. We used primary cultures of murine cerebral endothelial cells (CECs) to explore the mechanisms involved in Aβ−induced cell death. We report here that Aβ25–35, a cytotoxic fragment of Aβ, induced translocation of the apoptosis regulator termed second-mitochondria-derived activator of caspase (Smac) from the intramembranous compartment of the mitochondria to the cytosol 24 hr after exposure. In addition, we demonstrated that X chromosome-linked inhibitor-of-apoptosis protein (XIAP) coimmunoprecipitated with Smac, suggesting that the two proteins bound to one another subsequent to the release of Smac from the mitochondria. Aβ25–35treatment also led to rapid AP-1 activation and subsequent expression of Bim, a member of the BH3-only family of proapoptotic proteins. Bim knockdown using an antisense oligonucleotide strategy suppressed Aβ25–35-induced Smac release and resulted in attenuation of CEC death. Furthermore, AP-1 inhibition, with curcumin or c-fos antisense oligonucleotide, reduced bim expression. These results suggest that Aβ activates an apoptotic cascade involving AP-1 DNA binding, subsequent bim induction, followed by Smac release and binding to XIAP, resulting in CEC death.
- amyloid-β peptide (Aβ)
- Smac
- cerebral endothelial cells
- AP-1
- BH3-only family
- XIAP
- cell death
- Alzheimer's disease
Alzheimer's disease (AD), a common neurodegenerative disease that leads to progressive dementia, results from amyloid-β peptide (Aβ) deposition in senile plaques and the formation of neurofibrillary tangles (Wisniewski and Wegiel, 1995;Yankner, 1996; Selkoe, 1999). Aβ is a 39–43 amino acid peptide fragment derived from the β-amyloid precursor protein, a membrane-bound glycoprotein distributed in many cell types of the nervous system (Glenner et al., 1984; Masters et al., 1985). A large body of literature suggests that Aβ accumulation may be involved in the neuronal degenerative process in AD brains (Yankner et al., 1989;Behl et al., 1994; Yankner, 1996; Selkoe, 1999). Aβ, which also accumulates in cerebrovascular walls, has been shown to induce significant damage to endothelial cells (Thomas et al., 1996) and may contribute to the age-dependent degeneration of cerebral vasculature and the development of cerebral amyloid angiopathy (Perlmutter, 1994;Wisniewski et al., 2000), a major cause of hemorrhagic and ischemic stroke in the elderly with or without AD (Walker, 1997). Although Aβ is toxic to cultured cerebral endothelial cells (CECs) (Price et al., 1997; Preston et al., 1998; Xu et al., 2001a), the underlying molecular mechanism is unclear. Several groups have shown that CECs exposed to Aβ demonstrate features of apoptosis, including DNA condensation and fragmentation, and a requirement for protein synthesis (Blanc et al., 1997; Hase et al., 1997; Suo et al., 1997).
Activation of caspases, a widely recognized feature of apoptosis, occurs either via the death receptor-mediated (tumor necrosis factor–α/Fas ligand mediated) pathway (Ashkenazi and Dixit, 1999;Bratton et al., 2000) or the mitochondrial pathway (Green and Reed, 1998; Bratton et al., 2000). In the mitochondrial pathway and in some cases of the death receptor-mediated pathway, members of the Bcl-2 family of proteins associate with the mitochondria and trigger the release of a number of mitochondrial intermembranous proteins involved in subsequent apoptotic signaling (Gross et al., 1999; Wang, 2001). One subclass of the Bcl-2 family proteins that contains only one BH3 domain (e.g., Bim, Bid, Bik, Bak, Bad) translocates to the mitochondria from other cellular compartments and interacts with other Bcl-2 members to regulate mitochondrial protein release (Kelekar and Thompson, 1998;Huang and Strasser, 2000). These pro-apoptotic “BH3 domain only” proteins appear to be transcriptionally regulated during apoptosis (Dijkers et al., 2000; Whitfield et al., 2001). Included among the proteins released by mitochondria during apoptosis are cytochromec and the novel protein termed second-mitochondria-derived activator of caspase (Smac) (Deveraux and Reed, 1999), which binds to a class of anti-apoptotic proteins known as the inhibitors of apoptosis proteins (IAPs), thereby neutralizing IAP activity to promote caspase activation and apoptotic cell death (Du et al., 2000; Verhagen et al., 2000).
We have demonstrated previously that Aβ induces apoptosis in cultured CECs, resulting from mitochondrial dysfunction and the activation of caspase-8 and –3, and that cell death could be attenuated with broad-spectrum caspase inhibitors (Xu et al., 2001a). In this study, we were interested in understanding regulatory events leading to caspase activation, including regulation of Bim expression, Smac release, and IAP binding in cells exposed to Aβ.
MATERIALS AND METHODS
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO), whereas cell culture supplies were purchased from Invitrogen (Carlsbad, CA) unless specified otherwise.
Mouse CEC primary culture. Mouse CECs were prepared as described previously (Xu et al., 1998). Briefly, fresh mouse brains in ice-cold HBSS with antibiotics were freed of meninges and superficial blood vessels. The gray matter was homogenized and filtered, and the resulting fraction was then digested sequentially with 4 mg/ml collagenase B for 2 hr and 1 mg/ml collagenase/dispase (Roche Molecular Biochemicals, Indianapolis, IN) for 2 hr, followed by centrifugation in a 40% Percoll solution. The second band containing microvessels was collected and washed before plating onto collagen-coated dishes. Mouse CECs migrating from the vessels were pooled to form a proliferating cell culture that was maintained in DMEM supplemented with 10% FBS, 0.5 mg/ml heparin, and 75 μg/ml endothelial cell growth supplements. Mouse CECs of passages 4–15 that were uniformly positive for factor VIII and vimentin (>95% endothelial cell purity) and characteristic bradykinin receptors were grown to 85–95% confluency before use (Xu et al., 1992).
Treatment with Aβ and Bim antisense oligonucleotide. Although Aβ1–40 and Aβ1–42 are the major components of Aβ deposits in the AD brain, in most experimental systems the biological effects of the fragment Aβ25–35 and Aβ1–40 are comparable (Loo et al., 1993; Behl et al., 1994; Xu et al., 2001a,c). We have shown previously that Aβ25–35 has approximately the same potency as Aβ1–40 in inducing cell death in CECs (Xu et al., 2001a) and oligodendrocytes (Xu et al., 2001b). On the basis of these data, only Aβ25–35 was used in this study. CECs were treated with 25 μmAβ25–35 (Sigma, St. Louis, MO) in serum-free growth medium for 1, 2, 4, 8, 24, and 48 hr. In some experiments, mouse CECs were treated with the antisense morpholino oligonucleotide to Bim according to the protocol provided by the manufacturer (Taylor et al., 1996; Summerton et al., 1997; Summerton and Weller, 1997). The oligonucleotides were custom-made by Gene Tools, LLC (Corvallis, OR) with the following sequences: antisense, 5′-TTACATCAGAAGGTTGCTTGGCCAT-3′; and sense, 5′-TACCGGTTCGTTGGAAGACTACATT-3′. Mouse CECs were incubated with a 5 ml solution consisting of 1.4 μm antisense or sense Bim oligonucleotides and 28 μl of 200 μm ethoxylated polyethylenimine for 3 hr and then switched to normal growth medium for at least 24 hr before exposure to Aβ treatment. For c-fos antisense oligonucleotide treatment, separate batches of CECs were pretreated with a 6 ml mixture containing 2 μm c-fos antisense or sense phosphorothioated oligodeoxynucleotide (ODN) and 0.5 μl of 3 mg/ml Superfect (Qiagen, Valencia, CA) for 3 hr before Aβ treatment. The ODNs were custom-made by Invitrogen (Gaithersburg, MD) with the following sequences: sense, 5′-GGTTTGCCCAAACCACGACCATGATG-3′; and antisense, 5′-CATCATGGTCGTGGTTTGGGCAAACC-3′ (Liu et al., 1994; Cui et al., 1999; Xu et al., 2001b).
Subfractionization of cellular proteins from CECs. After treatment, CECs were harvested by centrifugation at 200 ×g for 10 min at 4°C. The cell pellets were washed twice with ice-cold PBS, followed by centrifugation at 200 ×g for 5 min at 4°C, and resuspended with 5 vol of Buffer A [20 mm HEPES, 1.5 mmMgCl2, 10 mm KCl, 1 mm EDTA, 1 mm EGTA, 250 mm sucrose, 0.1 mm PMSF, 1 mm dithiothreitol (DTT), 4 μg/ml pepstatin, 4 μg/ml leupeptin, 5 μg/ml aprotinin, pH 7.9]. After a 10 min incubation on ice, cells were homogenized with a mini-pestle. The lysates were centrifuged at 750 × g for 15 min at 4°C; the supernatant contained mitochondrial and cytosolic proteins, and the pellet contained nuclei. The pellets were resuspended in 45 μl of buffer B (20 mm HEPES, 1.5 mm MgCl2, 20 mm KCl, 0.2 mm EDTA, 0.5 mm DTT, 0.2 mm PMSF, and 1 mg/ml leupeptin and aprotinin, pH 7.9), and 15 μl of buffer C (20 mm HEPES, 1.2 m KCl, 0.2 mm EDTA, 0.5 mm DTT, 0.2 mm PMSF, 1 mg/ml leupeptin and aprotinin, pH 7.9) was then added and mixed. The samples were placed on ice for 30 min and centrifuged at 12,000 × g. Supernatants containing nuclear protein were transferred and stored at −80°C until analysis by SDS-PAGE. The cytosolic/mitochondrial fractions were centrifuged at 10,000 × g for 15 min at 4°C, and the resulting mitochondrial pellets were resuspended in buffer A and frozen in multiple samples at −80°C. The supernatant of the 10,000 ×g spin was further centrifuged at 100,000 ×g for 1 hr at 4°C, and the resulting supernatants (S-100) were removed and stored at −80°C for future analysis. The concentrations of proteins described above were measured by the Lowry method.
Western blot analysis. Samples (20–40 μg of protein) were electrophoresed onto a 10–15% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked in TBST buffer containing 20 mm Tris-HCl, 5% nonfat milk, 150 mm NaCl, and 0.05% Tween 20, pH 7.5, for 1 hr at room temperature. Thereafter the blot was incubated with primary rabbit anti-Bim antibody (1:1000; Calbiochem, La Jolla, CA), rabbit anti-XIAP antibody (1:500; BD Biosciences, San Diego, CA), goat anti-Smac antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), or mouse anti-actin antiserum (1:500; Santa Cruz Biotechnology), respectively, for 1–2 hr at room temperature. The membrane was washed with TBST three times at 10 min intervals, incubated with the second antibody (1:5000; anti-rabbit, anti-mouse, or anti-goat IgG conjugated with alkaline phosphatase; Promega, Madison, WI) at room temperature for 1 hr, and then washed three times each at 10 min intervals with TBST and two times each for 10 min with TBS (TBST without Tween 20). The color reaction was developed by the Blot AP System according to the technical manual provided by Promega.
Immunofluorescence staining. Mouse CECs grown on coverslips were fixed with 4% paraformaldehyde for 30 min and washed three times with 0.1 m PBS, pH 7.4. The cells were then incubated with a primary goat anti-Smac antibody (1:50; Santa Cruz Biotechnology) overnight at 4°C. On the following day, the cells were incubated with fluorescein-conjugated anti-goat IgG (1:100; Vector Labs, Burlingame, CA) for 1 hr. CECs were counterstained with 1 μg/ml propidium iodide (Molecular Probes, Eugene, OR) to visualize nuclear morphology. Slides were washed, wet mounted, and examined with an Olympus fluorescence microscope.
Coimmunoprecipitation. A total of 2 × 107 CECs were pelleted and then lysed in lysis buffer (1% Triton X-100, 150 mm NaCl, 10 mm Tris, pH 7.4, 1 mm EDTA, 1 mm EGTA, pH 8.0, 0.2 mmsodium orthovanadate, 0.5% NP-40, 0.2 mm PMSF, 4 μg/ml pepstatin, 4 μg/ml leupeptin, 5 μg/ml aprotinin) on ice for 30 min. Cellular debris was removed by centrifugation at 16,000 × g for 20 min, and the supernatant was incubated with goat anti-Smac antibody (1:200; Santa Cruz Biotechnology) at a concentration of 2 μg/ml at 4°C for 3 hr. Protein G Sepharose 4 Fast Flow (50% beads in lysis buffer; Amersham Biosciences) was added to the antigen–antibody mixture and incubated with gentle agitation for another 1–2 hr. The immunoprecipitate was washed five times with 500 μl of lysis buffer at 4°C, resuspended in SDS loading buffer, separated on an SDS-polyacrylamide gel, transferred to PVDF membrane, and further analyzed by Western blot using rabbit anti-XIAP antibody (1:500; BD Biosciences) as described previously. Affinity-purified normal goat IgG was used as a negative control for immunoprecipitation.
Electrophoretic mobility shift assay. Electrophoretic mobility shift assay (EMSA) to assess AP-1 binding activity has been described in detail elsewhere (An et al., 1993; Xu et al., 2001b). The following AP-1 consensus oligonucleotide was used: 5′-CGCTTGATGAGTCAGCCGGAA-3′ (Promega), end-labeled with γ-32P-ATP by T4 polynucleotide kinase. The binding reaction was performed in a total volume of 20 μl containing binding buffer (10 mm Tris-HCl, 20 mm NaCl, 1 mm DTT, 1 mm EDTA, 5% glycerol, pH 7.6), 0.0175 pmol of labeled probe (>10,000 cpm), 10–20 μg of nuclear protein, and 1 μg of poly(dI-dC). After incubation for 20 min at room temperature, the mixture was subjected to electrophoresis on a nondenaturing 6% polyacrylamide gel at 180 V for 2 hr under low ionic strength conditions. The gel was dried and subjected to autoradiography. For supershift assays, samples were incubated with anti-c-Jun or anti-c-Fos antibody (1:4; Santa Cruz Biotechnology) for 1 hr before addition of γ-32P-ATP AP-1 oligonucleotide probe. The specificity of AP-1 DNA binding activity was also demonstrated by the complete inhibition of AP-1 binding in the presence of a 100-fold molar excess of cold AP-1 oligonucleotide (data not shown).
Assessment of mouse CEC death. The extent of CEC death was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) and LDH assays as described previously (Xu et al., 1998,2000).
Statistical analysis. Quantitative data are expressed as mean ± SD based on at least three separate experiments of triplicate samples. Difference among groups was statistically analyzed by one-way ANOVA followed by Bonferroni's post hoc test. Comparison between two experimental groups was based on a two-tailedt test. A p value <0.05 was considered significant.
RESULTS
Smac release from mitochondria and binding to XIAP in CECs
Smac release from mitochondria appears to be a general feature of apoptosis involving the mitochondrial pathway of cell death (Du et al., 2000; Adrain et al., 2001; Carson et al., 2002; Deng et al., 2002;Madesh et al., 2002; Verhagen and Vaux, 2002). We have shown previously that Aβ25–35 is toxic to CECs, inducing several events in the apoptotic cascade, including cytochromec release from mitochondria, and subsequent caspase activation (Xu et al., 2001a). To explore whether Smac redistribution from mitochondria to the cytosol in CECs occurs during Aβ25–35-induced apoptosis, we used Western blotting to analyze the content of Smac in different cellular fractions. As shown in Figure1A, Smac protein levels increased in the cytosol, at a time when the protein decreased in the mitochondrial fraction (24–48 hr after Aβ25–35 treatment); the nuclear fraction remained devoid of Smac during this period of time, suggesting that Smac translocated from mitochondria to cytosol. The cellular redistribution of Smac was confirmed by immunofluorescent staining with anti-Smac antibody, which showed that the normal punctate mitochondrial pattern was changed to a more diffuse cytosolic pattern after Aβ25–35 treatment (Fig. 1B). An immunoprecipitation study with anti-Smac antibody revealed that released Smac bound primarily to XIAP, a member of the IAP family of apoptosis regulators, in the cytosol (Fig. 1C).

Aβ25–35-induced mitochondrial Smac release into the cytosol and Smac binding to XIAP. A, Western blot analysis shows the time course of Smac translocation from the mitochondrial (Mt.) to the cytosolic (Cyt.) fraction after Aβ25–35 exposure for 24 hr. Note the absence of Smac in the nuclear fraction (Nuc.) at all times. Actin immunoblotting serves as a control. B, Immunofluorescent staining with anti-Smac antibody confirms the translocation of Smac from mitochondria (punctate staining) to cytosol (diffuse staining) after Aβ25–35exposure for 0 or 24 hr. C, Smac binding to XIAP is demonstrated by coimmunoprecipitation (IP) with anti-Smac but not control (Ctl; normal goat IgG) antibody. Representative data from three separate experiments with similar results are shown.
Regulation of Smac release
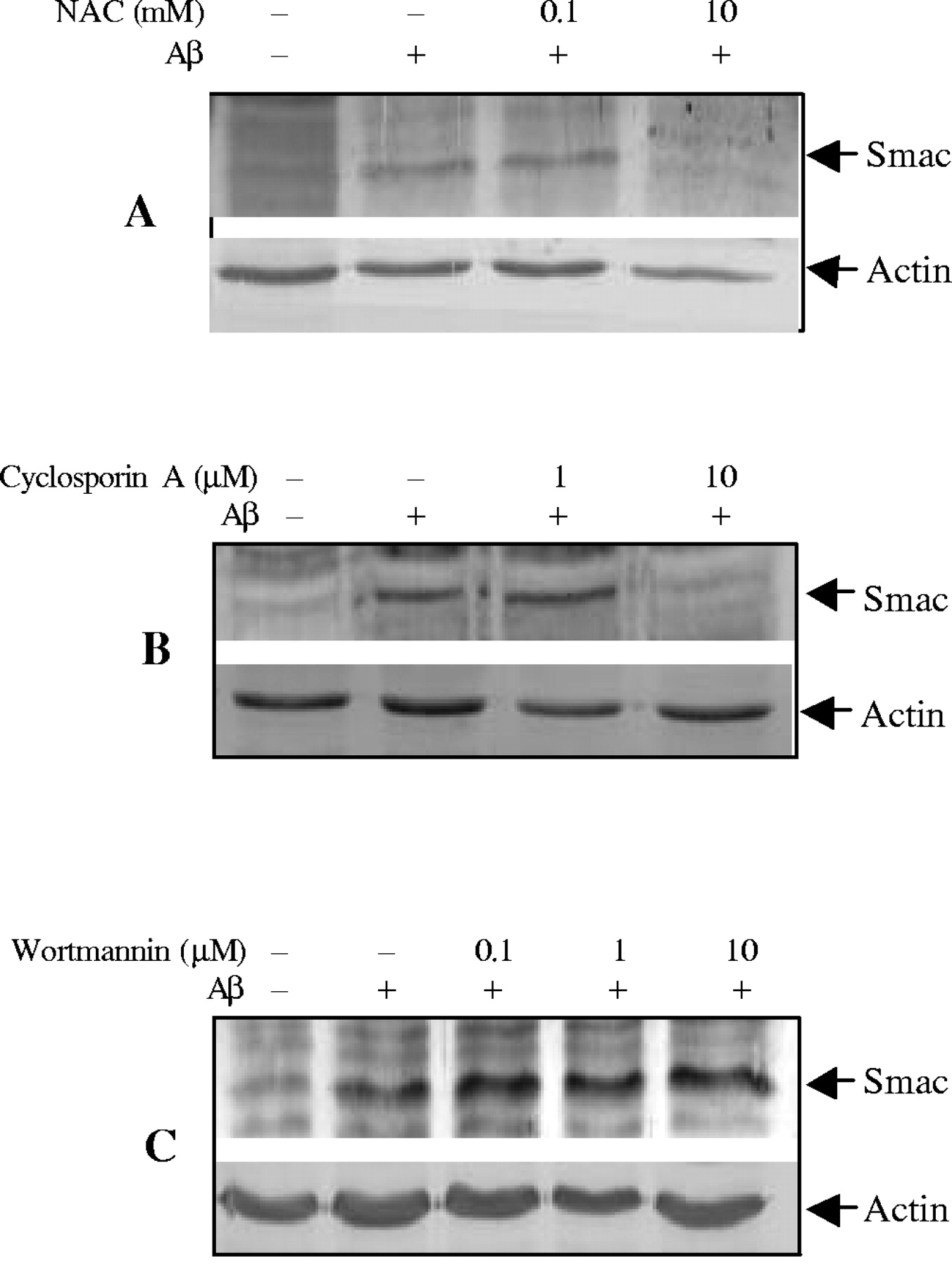
Oxidative stress plays a critical role in mediating Aβ−induced apoptotic cell death in multiple cell types (Harris et al., 1995a,b;Davis, 1996; Thomas et al., 1996; Misonou et al., 2000; Xu et al., 2000), and anti-oxidants can protect cells from Aβ toxicity (Xu et al., 2001a). To determine whether inhibition of oxidative stress contributes to improvement of cell survival by inhibiting Smac release, we examined the effects of the antioxidant N-acetyl-cysteine (NAC) on Smac release induced by Aβ25–35. As illustrated in Figure2A, NAC effectively inhibited Smac release from mitochondria.

Regulation of Aβ25–35-induced Smac release from mitochondria in CECs. CECs were treated with 25 μm Aβ25–35 in the presence of NAC (A), cyclosporin A (B), or wortmannin (C) at indicated concentrations, and cytosolic fractions were analyzed by immunoblotting with anti-Smac antibody. NAC and cyclosporin A inhibited, whereas wortmannin increased, Smac release. Representative data from three separate experiments with similar results are shown.
It has been proposed that the formation of the mitochondrial permeability transition pore (PTP) may play a role in the release of mitochondrial caspase activators during apoptosis (Pritchard et al., 2000; Buckman and Reynolds, 2001). Cyclosporin A (CsA), which inhibits PTP formation, was used to examine its effect on Aβ25–35-induced Smac release. CsA significantly prevented Smac release from mitochondria to cytosol (Fig.2B).
Multiple signal pathways may modulate mitochondrial intermembrane protein release (Imaizumi et al., 1999; Kaltschmidt et al., 1999;Bozyczko-Coyne et al., 2001; Martin et al., 2001; Xu et al., 2001b). To determine whether the phosphotidylinositol 3-kinase (PI3K)/Akt pathway regulates Smac release in CECs, we treated CECs with a PI3K/Akt inhibitor, wortmannin. As shown in Figure 2C, wortmannin increased Smac release.
AP-1 binding activity
Proto-oncogenes belonging to the c-fos and c-jun families are immediate-early genes that are rapidly and transiently induced in response to various stimuli (Kruijer et al., 1984; Kornhauser et al., 1992). These gene products can interact via a leucine zipper to form heterodimers that act as a transcription factor by binding specifically to the AP-1 binding sites in the promotor region of selected genes (Curran and Franza, 1988; Beato, 1991), transactivating downstream genes that constitute the delayed genetic response. In this study, AP-1 binding activity was determined by EMSA. As illustrated in Figure 3A, treatment with Aβ25–35 resulted in a substantial increase in AP-1 binding activity in a time-dependent manner, starting as early as 1 hr and persisting up to 24 hr after Aβ exposure; low basal levels of AP-1 binding activity were detected in control CECs. Addition of excess of unlabeled AP-1 oligonucleotides abolished the observed DNA–protein complex, demonstrating the specificity of AP-1 DNA binding activity (data not shown). Preincubation of the nuclear extract with anti-c-Jun antibody or anti-c-Fos antibody also reduced AP-1 DNA binding activity (Fig.3B).

Aβ25–35 activation of AP-1.A, EMSA study demonstrates a time-dependent elevation of AP-1 binding activity after Aβ25−35 treatment in CECs. Note the low level of AP-1 binding in cultures unexposed to Aβ25–35 (0h). B, The specificity of AP-1 binding is confirmed by the effects of anti-c-Jun or anti-c-Fos antibodies in reducing AP-1 binding activity. Representative data from three separate experiments with similar results are shown. h, Hour.
Bim expression is required for Smac release
Bim belongs to the BH3-only subclass of the Bcl-2 family of apoptosis regulators. These proteins contain only one of the Bcl-2 homology regions (BH3) and are essential initiators of apoptotic cell death (Kelekar and Thompson, 1998; Huang and Strasser, 2000). To determine the possible role of Bim in Aβ−induced cell death, Bim expression was examined in CECs. Western blot analysis showed that Bim expression was increased after Aβ exposure, persisting for up to 24 hr after exposure (Fig.4A,B).
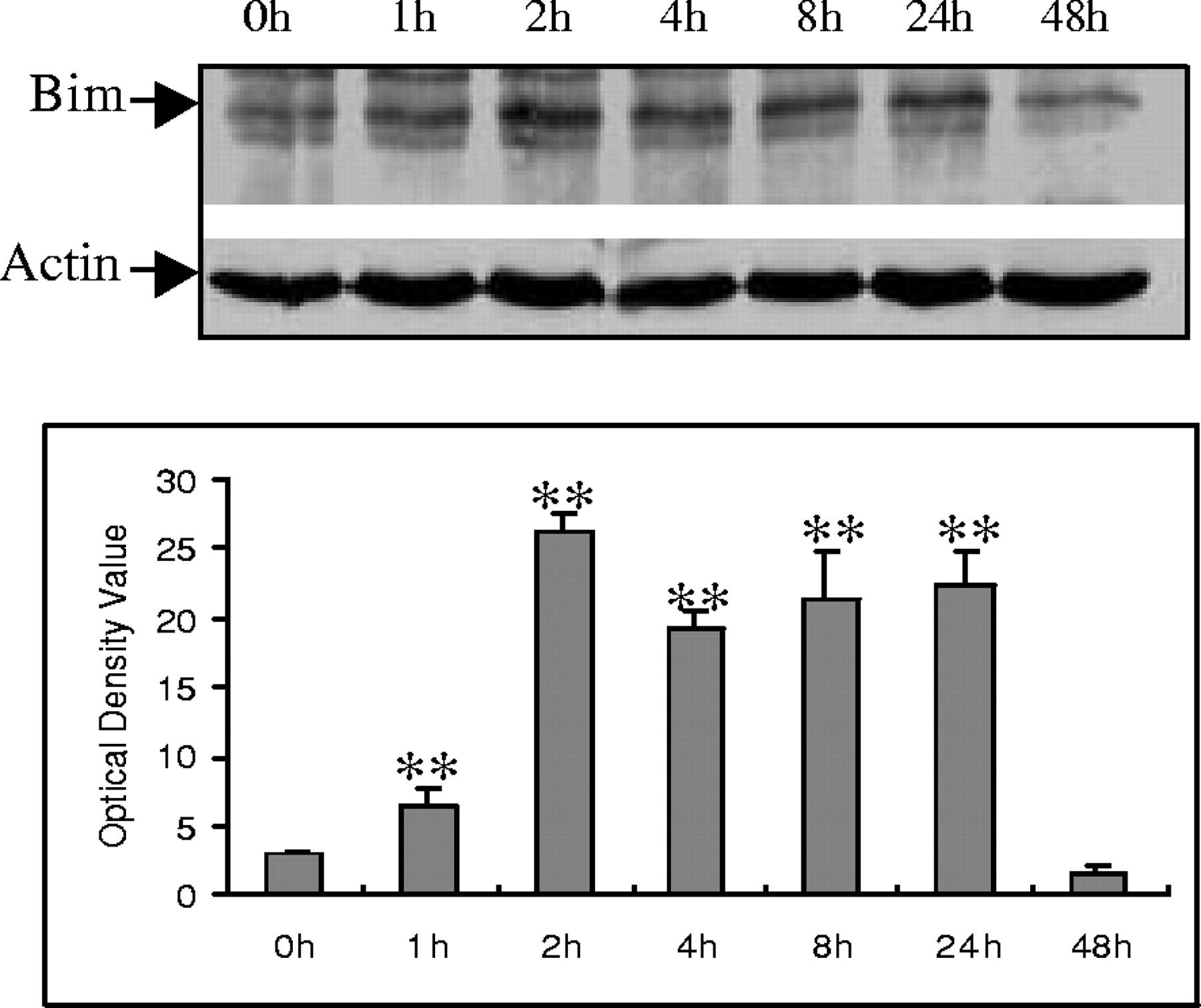
Aβ25–35 induction of Bim protein expression in CECs. A, A representative Western blot demonstrates that Aβ25–35 induced Bim protein expression at the indicated time points. B, Quantitative analysis of three Western blots (normalized to actin expression) using the NIH image system graphically illustrates Bim protein induction by Aβ25–35. Data in B are expressed as mean ± SD. **p < 0.05 versus 0 hr (0h) exposure.
Previous studies have reported that the translocation of mitochondrial intermembranous proteins may be under the control of some BH3-only family members (Gross et al., 1999; Li et al., 2001; Wang, 2001; Madesh et al., 2002). To explore the possibility that mitochondrial Smac release was regulated by Bim activation in Aβ−induced cell death, we treated CECs with bim antisense oligonucleotides. Treatment with antisense oligonucleotide diminished the Aβ25–35-induced increase in bimexpression (Fig. 5A). Furthermore, Bim knockdown reduced Smac release from mitochondria (Fig.5B) and decreased cell death 24 hr after Aβ25–35 exposure (Fig.5C,D). To confirm the specificity ofbim antisense oligonucleotide, the bim sense oligonucleotide was also tested and had no effect on Bim activation, Smac release, or subsequent cell death (Fig.5A–D).

Effects of Bim suppression on Smac release and CEC death. CECs were cultured in the presence or absence of bimsense or antisense oligonucleotide with or without Aβ25−35 treatment for 24 hr.A, A representative immunoblot demonstrates thatbim antisense oligonucleotide reduced bimexpression in CECs after Aβ25−35exposure. Sense oligonucleotide treatment had no effect.B, bim antisense oligonucleotide reduced mitochondrial Smac release in CECs after Aβ25–35treatment, as demonstrated in this representative Western blot, and increased CEC survival, assessed by MTT assay (C) and LDH release (D). Data are expressed as mean ± SD from three separate experiments in quadruplicate.Control, Control without oligonucleotide treatment;AS, antisense oligonucleotide; S, sense oligonucleotide. **p < 0.05 versus control group.
AP-1 inhibition decreases Bim expression
To determine whether AP-1 activity directly regulatesbim expression, curcumin, an AP-1 inhibitor, was applied to CECs. As illustrated in Figure 6, treatment with curcumin effectively inhibited AP-1 DNA binding activity (Fig. 6A), resulting in decreased Bim protein levels after 4 hr of exposure to Aβ25–35 (Fig.6B). No effects of curcumin on AP-1 activation andbim induction were observed at 2 mmcurcumin (Fig. 6A,B). To confirm the effect of AP-1 DNA binding inhibition on bim expression, CECs were treated with c-fos antisense oligonucleotides. This strategy inhibited AP-1 DNA binding (Fig. 6C) by disrupting c-Fos–c-Jun dimerization (Liu et al., 1994; Cui et al., 1999; Xu et al., 2001b) and resulted in decreased Bim expression (Fig.6D).

The effect of AP-1 inhibition on Aβ25–35-induced Bim expression. Curcumin (A) or c-fos antisense (AS) oligonucleotide (C) reduced AP-1 binding activity as shown by EMSA. Curcumin (B) or c-fos antisense oligonucleotide (D) also reduced Bim expression as shown by immunoblotting. C-fos sense (S) oligonucleotide had no effect. Representative data from three separate experiments with similar results are shown.
DISCUSSION
The results demonstrate that CECs undergo apoptosis when exposed to Aβ25–35. This event is accompanied by the translocation of Smac from the mitochondrial intermembranous compartment to the cytosol, where it appears to bind to XIAP. Furthermore, we have provided evidence that Smac release is regulated by expression of the BH3-only protein, Bim. Suppression of Bim expression using a Bim antisense oligonucleotide effectively inhibited Aβ-induced mitochondrial Smac release and reduced subsequent cell death. Bim expression, in turn, is regulated by AP-1 binding activity, because inhibition of AP-1 with curcumin or c-fos antisense oligonucleotide reduced Bim expression. On the basis of these results, we propose that Aβ25–35 activates AP-1, which transactivates Bim expression. Bim expression then leads to Smac release from mitochondria and subsequent binding to anti-apoptotic XIAP, resulting in CEC apoptosis. This proposed sequence of events is consistent with previous work which demonstrated that Aβ induced CECs to die with features of apoptosis, including DNA condensation and fragmentation (Xu et al., 2001a), and involved mitochondrial dysfunction, mitochondrial DNA damage, and activation of caspase-8 and –3 (Xu et al., 2001a).
Caspase activity has been shown recently to be under the influence of the IAP class of proteins, consisting of NAIP, cIAP-1, cIAP-2, XIAP, and Survivin, which directly bind to and inactivate caspases (Deveraux et al., 1997, 1998; Roy et al., 1997; Shin et al., 2001; Verhagen et al., 2001). The IAPs are regulated by Smac. Several studies have demonstrated that Smac is released from the mitochondria by apoptotic stimuli and then binds to and inactivates the IAPs, disinhibiting caspase activation (Du et al., 2000; Verhagen et al., 2000). In this study we have shown that Smac, in fact, does bind to XIAP. It is likely that Smac binding of XIAP promotes the activation of downstream effector caspases, as observed in our earlier work (Xu et al., 2001a). Because XIAP exists exclusively in the cytosol (Du et al., 2000;Verhagen et al., 2000), interaction with Smac is likely to occur only after its release from the mitochondria.
There is growing evidence that a critical checkpoint in the regulation of apoptosis occurs at the level of the mitochondria and that mitochondrial disruption may lead to the stimulation of apoptosis (Green and Reed, 1998). We examined several interventions that altered mitochondrial susceptibility to damage to determine their effect on Smac release. In general, those interventions that have demonstrated cytoprotective properties reduced Smac release in CECs. For example, the antioxidant NAC, which was reported previously to protect CECs from Aβ25−35-induced CEC death (Xu et al., 2001a), blocked Smac release in the present study. Likewise, the permeability transition pore inhibitor cyclosporin A reduced Smac release from CECs treated with Aβ25−35.
Smac release after apoptotic stimuli is controlled by the interaction of Bcl-2 family members on the mitochondrial membrane. For example, several groups have shown that Bcl-2 overexpression inhibited Smac release and attenuated death in cells stimulated by death ligands or via the mitochondrial pathway (Adrain et al., 2001; Fulda et al., 2002;Sun et al., 2002). The involvement of Bax has also been implicated, at least in receptor-mediated apoptosis, because Bax knock-out prevented Smac release and subsequent cell death in TRAIL-induced human colon cancer cells (Deng et al., 2002). In addition, there is growing evidence that receptor-mediated Smac release may require the activation of caspase-8, which cleaves the cytosolic BH3-only protein, Bid, into a truncated form (tBid); tBid then translocates to the mitochondria where it triggers the release of Smac and other apoptotic signals (Li et al., 1998; Luo et al., 1998; Madesh et al., 2002). Another BH3-only protein, Bim, normally associates with cellular microtubule complexes but translocates to the mitochondria shortly after apoptotic stimuli (Putcha et al., 2001). Li et al. (2001) has reported that recombinant Bim alone is as efficient as tBid in releasing cytochrome cand endonuclease G when incubated with mitochondria in vitro. Mice lacking Bim show defects in apoptotic response in their immune system (Bouillet et al., 1999). Because the requirement for mitochondrial processing to activate Smac may be analogous to the requirement for mitochondrial processing of cytochrome c, we explored the relationship between Bim expression and Smac release in Aβ-treated CECs. The results shown here demonstrate that Bim knockdown using an antisense strategy effectively inhibited the release of Smac from mitochondria and reduced CEC cell death, implying that Bim expression regulates Smac release and subsequent cascade activation in Aβ25–35-induced apoptosis.
Recent reports suggest that the expression of BH3-only proteins such as Bim may primarily involve transcriptional regulation. For example, the forkhead transcription factor induced Bim expression in T lymphocytes and is thought to be suppressed by survival-promoting cytokines via the PI3K/Akt anti-apoptotic pathway (Dijkers et al., 2000). Our finding that the PI3K/Akt inhibitor, wortmannin, increased Smac release in Aβ25−35-treated CECs raises the possibility that this anti-apoptotic pathway may regulate Bim expression, which in turn influences Smac release. Dominant-negative constructs of c-jun preventedbim upregulation and inhibited mitochondrial cytochromec release in cultured neurons after NGF withdrawal, suggesting that activation of the JNK/c-Jun pathway may also participate in the transcriptional regulation of bim(Whitfield et al., 2001). In this study, we demonstrated that Aβ25–35 exposure caused a significant elevation of AP-1 binding activity. AP-1, composed of a heterodimer of c-Fos and c-Jun, is likely to be involved as shown by EMSA. Aβ25−35activation of AP-1 is likely to lead to the transactivation ofbim. This contention is supported by the finding that inhibition of AP-1 by curcumin or a c-fos antisense oligonucleotide resulted in bim downregulation. In conclusion, the present study suggests a central role for Bim-mediated mitochondrial Smac release in Aβ25–35-induced CEC apoptosis. This cascade is regulated by upstream c-Jun–c-Fos/AP-1 activity. The elucidation of mechanisms involved in Aβ-induced CEC death may be important for understanding the pathogenesis of cerebral amyloid angiopathy and cell death in Alzheimer's disease.
Footnotes
This work was supported by National Institutes of Health Grants NS37230, NS40162, and NS40525 (C.Y.H.), American Heart Association (AHA) Grant 0050597N (J.X.), and AHA Postdoctoral Fellowship 0120652Z (K.J.Y.).
Correspondence should be addressed to Dr. Chung Y. Hsu, Department of Neurology, Box 8111, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail:hsuc{at}neuro.wustl.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}