

Abstract
Tumor necrosis factor receptor-I (TNFRI) and TNFRII are two TNFR subtypes in the immune system, but their roles in the brain remain unclear. Here we present a novel interaction between TNFR subtypes and TNF-α in the brain. Our studies on target-depleted TNFR in mice show that TNF-α has little effect on hippocampal neurons in which TNFRI, containing an “intracellular death domain,” is absent (TNFRI −/−), whereas neurons from TNFRII knock-out mice are vulnerable to TNF-α even at low doses. Moreover, little nuclear factor-κB (NF-κB) translocation is induced by TNF-α in neurons of TNFRI −/−, whereas NF-κB subunit p65 is still translocated from the cytoplasm into the nucleus in neurons from wild-type and TNFRII −/− mice. Furthermore, p38 mitogen-activated protein (MAP) kinase activity is upregulated in neurons from both wild-type and TNFRI −/−, but no alteration of p38 MAP kinase was found in neurons from TNFRII. Results from overexpression of TNF receptors further support the above findings. NT2 neuronal-like cells transiently transfected with TNFRI are very sensitive to TNF-α, whereas TNF-α is not toxic and even seems to be trophic to the cells with TNFRII overexpression. Last, our radioligand-binding experiments demonstrate that TNF-α binds TNFRI with high affinity (Kd of 0.6 nm), whereas TNFRII shows lower binding affinity (Kd of 1.14 nm) to TNF-α in NT2 transfected cells. Together, these studies reveal novel neuronal responses of TNF-α in mediating consequences of TNF receptor activation differently. Subsequent neuronal death or survival may ultimately depend on a particular subtype of TNF receptor that is predominately expressed in neurons of the brain during neural development or with neurological diseases.
Cytokines are critical mediators for the initiation of specific inflammatory responses and immune reactions (Baud and Karin, 2001; Baggiolini, 2001; Cavaillon, 2001). Many cytokines have been reported extensively in brains during development or with neurodegenerative disorders (Carr et al., 1997; Collins et al., 2000). Several lines of evidence suggest that tumor necrosis factor-α (TNF-α) and its receptors may contribute significantly to our understanding of neural protection or cell growth and the pathogenesis or therapy of neurodegenerative disorders. First, TNF-α is increased during normal development, as well as in diseased brains, such as Alzheimer's disease, Parkinson's disease, multiple sclerosis, and stroke (Hofman et al., 1989; Fillit et al., 1991; Gary et al., 1998; Barone and Feuerstein, 1999; Kruger et al., 2000; Nagatsu et al., 2000; Lue et al., 2001).
Second, microglial cells and astrocytes are activated in those diseased brains, and TNF-α is secreted from microglia, possibly through TNF receptor signaling (Eikelenboom et al., 1994; D'Souza et al., 1996;Shrikant and Benveniste, 1996; Viviani et al., 1998; Akama and Van Eldik, 2000; Yates et al., 2000; Lue et al., 2001). Third, TNF-α can be trophic or toxic, possibly depending on the types of target cells or receptor subtypes. For example, an excess of TNF-α can kill human cortical neurons and oligodendrocytes (Heller et al., 1992; D'Souza et al., 1995; Rothwell and Luheshi, 1996; Akassoglou et al., 1997;Hisahara et al., 1997; Nawashiro et al., 1997; Shen et al., 1997; Kim et al., 2001), but it is trophic (Cheng et al., 1994; Barger et al., 1995; Liu et al., 1998; Arnett et al., 2001) to rat hippocampal neurons. Fourth, TNF-α does elicit its biological effects through the activation of two distinct receptors, TNFRI, or p55 TNF receptor (p55-TNFR), and TNFRII, or p75 TNF receptor (p75-TNFR) (Tartaglia et al., 1993; Leist et al., 1995; Bruce et al., 1996; Sipe et al., 1996;Shen et al., 1997; Wallach et al., 1997; Akassoglou et al., 1998; Kim et al., 2001). The two receptor subtypes exhibit low amino acid sequence homology (24%) in the extracellular region and <10% homology in the intracellular domain. TNFRI contains an intracellular “death domain” (DD) and contributes to cell death when activated (Tartaglia et al., 1993; Sipe et al., 1996). Conversely, our previous knockdown studies demonstrate that TNFRII plays a trophic or protective role in neuronal survival (Shen et al., 1997). However, the downstream effects of TNF-α on neurons and whether TNF-α-induced neuronal survival and death is mediated through a mechanism that affects receptor-binding abilities to their natural ligand mechanisms remain inconclusive. In the present studies, we hypothesized that TNF-α binds TNF receptor subtypes in neurons with distinct affinities and governs neuron death or survival through different signal transduction pathways. We verified this hypothesis by using gene target deletion or overexpression of each TNF receptor, and we are now able to present evidence for novel TNF-α involvement in “toxic” and “protective” receptor responses for distinct TNFR subtypes in neurons. TNF-α does show higher affinity to TNFRI and lower affinity to TNFRII. We believe that our results may help in understanding the molecular mechanisms of TNF receptors in neurodegeneration and neuroregeneration.
MATERIALS AND METHODS
Hippocampal neuron cultures from TNFR knock-out mice.Both TNFRI and TNFRII knock-out mice (C57BL/6J background) (Erickson et al., 1994) were purchased from The Jackson Laboratory (Bar Harbor, ME). Hippocampal cultures were prepared as described previously (Kamegai et al., 1990; Malgaroli and Tsien, 1992). Briefly, the CA3–CA1 region of the hippocampus at embryonic day 15 was dissected, and the neurons were recovered by enzymatic digestion with trypsin and mechanical dissociation. Cells were then plated at a density of ∼6 × 105 cells in 3 ml of media per 35 mm dish (Primaria; Falcon, Franklin Lakes, NJ) coated with poly-l-lysine (Sigma, St. Louis, MO). Cultures were maintained at 37°C in a 95% O2–5% CO2 humidified incubator. The culture media was a 1:1 mixture of DMEM and Ham's F12 supplemented with 15 mm hepes, 100 μg/ml human transferrin, 25 μg/ml bovine insulin, 20 nm progestenne, 20 nmhydrocortisine-21-phosphate, 10 mm l-carnitine, 30 nm 3,3′, 5-triiodo-l-thyronine, 7 ng/ml α-tocopherol, 7 ng/ml retinol acetate, 1 μm thioctic acid, and 100 μm putrescine. No serum was used in the dissociation or the cultures. Neurons were used for experiments 5 d after plating.
Molecular cloning and plasmid construction. We isolated poly(A)+ RNA from human brain tissue and synthesized cDNA using reverse-transcriptase methodology. PCR was used to amplify the TNFRI or TNFRII coding region. The forward oligonucleotide primers for TNFRI and TNFRII are adapted with anXbaI restriction site on the 5′ end, and the reverse primers are adapted with an XhoI restriction site on the 3′ end. For transient transfection, the cDNA was inserted into a pcDNA3.1 expression vector (Invitrogen, San Diego, CA) that directs expression from both the cytomegalovirus (CMV) promoter and SV40 ori. These constructs were transfected into human NT2 neurotypic cells following a modified calcium phosphate precipitation procedure (Stratagene, La Jolla, CA).
Reverse transcription-PCR for TNFRI and TNFRII mRNA detection. The PCR technique was used to amplify cDNA derived from RNA extracted from nontransfected and transfected NT2 cells with TNFRI or TNFRII. The PCR forward oligonucleotide primer for TNFRI was 5′-TCG ATT TGC TGT ACC AAG TG-3′, and the backward oligonucleotide primer for TNFRI was 5′-GAA AAT GAC CAG GGG CAA CAG-3′. The PCR forward oligonucleotide primer for TNFRII was 5′-GGT CAC GCA ACC TGT CTT-3′, and the backward oligonucleotide primer for TNFRII was 5′-GGC TTC ATC CCA GCA TCA-3′. After an initial denaturation step at 94°C for 5 min, the cycle was initiated, which consisted of denaturing for 1 min at 94°C, annealing for 2 min at 53°C, and extending for 2 min at 72°C. The cycle was repeated 35 times.
Nuclear and cytoplasmic isolation. To prepare nuclear extracts, 4 × 106 or 107 treated cells were trypsinized and incubated for 20 min in a hypoosmotic buffer (10 mm HEPES, pH 7.8, 10 mmKCl, 2 mm MgCl2, 0.1 mm EDTA, 10 mg/ml aprotinin, 0.5 mg/ml leupeptin, 3 mg/ml PMSF, and 3 mm DTT) and 25 ml of 10% NP-40. The nuclei were pelleted by centrifugation for 5 min in a microcentrifuge. The supernatants containing the cytoplasmic proteins were removed and stored at −70°C. The pelleted nuclei were resuspended in a high-salt buffer (50 mm HEPES, pH 7.4, 50 mm KCl, 300 mmNaCl, 0. 1 mm EDTA, 10% v/v glycerol, 3 mm DTT, and 3 mm PMSF) to solubilize DNA binding proteins. The resuspended nuclei were gently shaken for 30 min at 4°C. The extracts were spun in a microcentrifuge for 10 min, and the clear supernatant, containing nuclear protein, was aliquoted for either determination of the protein concentration or stored at −70°C for assays.
Electrophoretic mobility shift assay. Electrophoretic mobility shift assay (EMSA) was performed using a double-stranded 15 base pair oligonucleotide (5′-CTAGGGGGACTTTCC-3′) containing the nuclear factor-κB (NF-κB) consensus sequence, radiolabeled with [32P]ATP. For the binding reaction, the nuclear protein extract (10 μg) and 0.25 ng (25,000 cpm) of labeled oligonucleotide was incubated in a total volume of 30 μl in binding buffer (10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 50 mm KCl, 1 mm EDTA, 5 mm DTT, 5% glycerol, and 2 μg of poly dI-dC) for 15 min at 4°C. DNA protein complexes were separated from unbound probe on native 4.5% polyacrylamide gels in 250 mm Tris borate EDTA buffer at 200–250 V for 2–3 hr. The resultant gel was vacuum dried and exposed to Kodak film (Eastman Kodak, Rochester, NY) for 8–15 hr at −70°C. The visual inspection of the free probe band at the bottom of the gel confirms that equivalent amounts of radiolabeled probe was used for each sample. The amount of DNA–protein complex present is analyzed using densitometry or a PhosphorImager. Cold competition is performed using 100-fold excess unlabeled NF-κβ consensus 5′-GGGGACTTTCCC-3′ or mutant NF-κβ binding site 5′-GGCGACTTTCCC-3′ for identification of specific bands.
Lactate dehydrogenase release and statistical analysis. For quantitative assessment of neuronal cell damage, lactate dehydrogenase (LDH) release from degenerating neurons was measured using a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI). Percentage of LDH release was calculated as the ratio of LDH contained in the supernatant relative to total LDH contained in both the supernatant and cell lysate. Data were analyzed by ANOVA with Student's paired t test.
Reagents. Antibodies for TNF receptors were purchased from R & D Systems (Minneapolis, MN), and the antibody for NF-κB p65 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Western blot analysis. The samples of neurons and cells were lysed in buffer containing 10 mm Tris-HCl, pH 7.4, 25 mm NaCl, 50 mmEDTA, 1 mm EGTA plus 0.5% Triton X-100, 10% SDS, and a protease inhibitor cocktail [1 mmphenylmethylsulfonyl fluoride, 1 μg/ml pepstatin A, 5 μg/ml leupeptin, and 2 μg/ml aprotinin (Boehringer Mannheim, Indianapolis, IN)]. For each sample, 10 μg of protein was separated on a 15% SDS tricine gel and transferred to a polyvinylidene difluoride (PVDF) membrane electrophoretically for 2 hr with 0.1% Tween 20 in TBS. NF-κB p65 or p38 kinase was detected with antibodies against NF-kB p65 or p38 kinase from Santa Cruz Biotechnology at a 1:1000 dilution for 16 hr at 4°C, followed by incubation with an HRP-conjugated secondary antibody and processed using ECL detection (Amersham Biosciences, Arlington Heights, IL). β-Actin and lamin were examined as housekeeping proteins in the cytoplasm and nucleus, respectively.
Transfection. Plasmid DNA used for transfections was purified using Qiagen (Hilden, Germany) Maxiprep kits. Exogenous DNA transfection followed the procedures of LiopofectAMINE (Invitrogen). For transient transfection, the TNFRI or TNFRII cDNA was inserted into a pcDNA3.1 expression vector that directs its expression from both CMV and SV40 promoters. The vector alone was used as a control. These constructs were transfected into NT2 cells. For stable transfection, the cDNA was subcloned into the pcDNA3.1 vector and transfected into NT2 cells using LipofectAMINE PLUS (Invitrogen). Forty-eight hours later, the cells were seeded into 96-well plates at 103 cells per well, selected with G418 (600 μg/ml; Invitrogen), and maintained with 350 μg/ml G418. The selected cells were screened by Western dot blotting to confirm TNFRI or TNFRII expression.
TNF receptor binding in whole cells. TNFRI or TNFRII stable transfected NT2 cells (1 × 106 per assay) were incubated with increasing concentrations of125I-TNF-α alone or with 100-fold excess of unlabeled TNF-α for 2 hr at 4°C.125I-TNF-α receptor complexes were precipitated at 4°C by the addition of 500 μl of 25% polyethylene glycol and 500 μl of 0.1% rabbit gamma globulin. Cells were incubated with increasing concentrations of125I-labeled TNF-α alone or with 200-fold excess of unlabeled TNF-α for 2 hr at 4°C and then washed three times. Specific binding (the difference between binding of125I-TNF-α in the absence or presence of cold TNF-α) was usually >80% of the total125I-TNF-α binding activity. After centrifugation, 3 ml of ice-cold buffer was added to the tubes, followed by filtration through a Brandell cell harvester, using GF/C glass fiber filters soaked previously in 0.05% polyethylenimine. Radioactivity on the filters was determined in a liquid scintillation counter with a counting efficiency of ∼40% (Wallac Oy, Turku, Finland). The data preprocessing was performed using the Prism program, and the actual nonlinear curve-fitting was performed by LIGANDS software.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling and DNA fragmentation. Neurons were grown on poly-l-lysine-coated chamber slides. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was used to visualize neurons with fragmented DNA. Neurons were harvested 16 hr after TNF-α treatment, fixed in 4% paraformaldehyde for 20 min, washed in three changes of PBS, and then incubated for 1 hr at 37°C with 75 μl of a cocktail (Boehringer Mannheim) consisting of 0.5 μl of terminal transferase, 0.95 μl of biotin-dUTP, 6.0 μl of CoCl2, 15.0 μl of terminal deoxynucleotidyl transferase buffer, and 52.55 μl of distilled water. The reaction was stopped by incubation in 4× SSC buffer, followed by three washes in PBS. Neurons were then labeled with a streptavidin Cy2 secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 45 min at room temperature and counterstained with Hoechst 33258 (0.5 μg/ml) for 5 min. The fraction of TUNEL-positive cells as a percentage of total cell number was determined. To analyze DNA integrity, DNA was extracted from neurons with or without TNF-α treatment from TNFRI −/− or TNFRII −/− mice, and its optical density was measured at 260 nm and separated on a 1% agarose gel to confirm DNA loading after staining the gel with ethidium bromide. Equal amounts of DNA were separated by conventional agarose gel electrophoresis and Southern blotted, and apoptotic DNA ladders were visualized by hybridization with digoxigenin-labeled total mouse genomic DNA probe (Boehringer Mannheim).
RESULTS
Neuronal viability in knock-outs of TNF receptor subtype TNFRI or TNFRII mice
To verify whether TNFR subtypes contribute to neurodegeneration or neuroregeneration and to identify specific TNFR subtypes linked to neuron death or survival, we analyzed cytotoxic effects in cultures of primary hippocampal neurons from mice with the absence of either TNFRI (TNFRI −/−) or TNFRII (TNFRII −/−). Studies from our Southern blot and Western blot hybridization confirmed no expression of TNFRI or TNFRII in either TNFRI −/− or TNFRII −/− mice, respectively (data not shown). Morphologically, TNFRI −/− neurons grow rather healthily, with multiple and long processing neurites even with TNF-α treatment (Fig. 1A). To further confirm our morphological observation, we used LDH as a cell death marker to quantitatively measure changes of LDH release from neurons in each condition. We found that treatment with TNF-α at various doses in TNFRI −/− neurons produced no changes of LDH release compared with neurons from wild-type or TNFRII −/− mice, even at high doses of TNF-α (100–1000 nm) (Fig.1B). These data suggest that TNFRI gene deletion might shut down a cellular death-related signal pathway, and neurons containing no intracellular death domain may become “insensitive” to insults. Likewise, neurons with TNFRII −/− alone seem to grow fine, suggesting that TNFRII may not be required for normal neuronal survival. However, our results further demonstrate that knock-out of TNFRII increased neuronal vulnerability toward injury induced by TNF-α in terms of neuronal degeneration or neurite loss, even at the low dose of TNF-α (100 pm) (Fig.1A). This result was further supported by quantitative studies of LDH release (Fig. 1B). We found that LDH release was significantly increased after the TNFRII −/− hippocampal neurons were treated with TNF-α for 48 hr, even at 100 pm TNF-α (Fig. 1B). Furthermore, we found that, by using techniques of TUNEL and DNA fragmentation, most of these neurons with TNFRII −/− were apoptotic cells after TNF-α treatment (Fig.2A,B, respectively).

Hippocampal neurons from knock-out TNFRI or TNFRII exhibit neuronal death or survival after exposures of TNF-α.A, Morphological changes of hippocampal neurons from TNFRI knock-out (TNFRI −/−) or TNFRII knock-out (TNFRII −/−) mice after exposure to TNF-α for 48 hr. Nikon (Tokyo, Japan) microscope at 200×. Scale bar, 100 μm. TNFRI −/− neurons show resistance to TNF-α insults, but TNFRII −/− demonstrated degeneration after TNF-α treatment. B, The primary cultured neurons from four mice from each type of TNFR knock-out mice; TNFRI −/−, TNFRII −/−, and wild type were incubated for 48 hr with TNF-α. The dead or detached damaged neurons were removed with the supernatant. The remaining neurons were lysed. LDH levels were measured in both supernatant and cell lysates. The data in B are from three independent experiments and represent mean ± SE, which are the percentage of supernatant LDH value relative to total LDH contained in both the supernatant and cell lysates, as described in Materials and Methods.

Hippocampal neuronal apoptosis induced by TNF-α treatment is TNFR dependent. TNF-α-induced apoptosis in hippocampal neurons in the absence of TNFRII is demonstrated in TUNEL (A) and DNA fragmentation (B). The hippocampal neurons from TNFRI −/− or TNFRII −/− mice were incubated in TNF-α at 100 pg/ml for 18 hr. The cells were fixed and analyzed for TUNEL reactivity, and neurons were analyzed for apoptosis using TUNEL for fragmented nuclear DNA (A). These images are typical of apoptotic single neurons cultured with TNF-α at 100 pm. For DNA fragmentation, Southern analysis of DNA isolated from neurons from TNFRI −/− or TNFRII −/− mice. Neurons from TNFRI −/−, TNFRII −/−, and wild type were treated with TNF-α (100 pm) for 48 hr, and cychlohexmide was used as positive control (B). The results from TUNEL and Hoechst staining and DNA fragmentation indicate that neurons with TNFRI deletion were resistant to TNF-α-induced apoptosis, whereas neurons with TNFRII deletion were more vulnerable.
Neuronal survival and death in neurons with overexpression of TNF receptor subtypes
Likewise, we assume that neurons overexpressing TNFRI have high levels of TNFRI containing the death domain and might be more vulnerable to insults. In contrast, neurons overexpressing TNFRII might be more resistant to insults. To support this hypothesis and further prove the above findings, we overexpressed each TNFRI and TNFRII subtype in human NT2 neurotypic cells (Fig.3A) and used LDH release to evaluate cytotoxicity in each TNF receptor cDNA-transfected human NT2 neurotypic cells. We found that LDH release from neurons overexpressing TNFRI is significantly increased in a dose-dependent manner, even at the very low doses of TNF-α (10–100 pm) concentrations, which are generally not toxic to neurons with no transfection (Fig. 3B). However, neurons overexpressing the TNFRII receptor have much less cytotoxicity, even at higher doses of TNF-α (Fig. 3B). Interestingly, the cells cotransfected with both TNFRI and TNFRII were more vulnerable to TNF-α compared with TNFRI-transfected cells alone but have less toxicity than that of TNFRII alone (Fig. 3B), suggesting that TNFRI functional expression may override TNFRII functions even if they have similar protein levels. This is consistent with results from TNFR double knock-out mice (Bruce et al., 1996). Moreover, we also found that cells with TNFRI overexpression (cell death domain) exhibit DNA fragmentation, whereas TNFRII-overexpressing cells or TNFRI- and TNFRII-coexpressing cells show no or little DNA fragmentation (Fig.3C).

Overexpressing either TNFRI or TNFRII reveals different vulnerability to TNF-α. Human neurotypic NT2 cells were transiently transfected with either TNFRI or TNFRII cDNA plasmid.A, The protein expression at each TNF receptor subtype after transfection was confirmed by Western blot. B, Overexpression of TNFRI resulted in neuronal vulnerability to TNF-α death even at a low dose (10–100 pm), whereas cells overexpressing TNFRII survive in the presence of TNF-α even at high doses (100–1000 nm). All results were repeated four times from independent experiments. *p < 0.05; **p < 0.01. C, DNA fragmentation was observed in NT2 cells with TNFRI overexpression but not found in the cells with TNFRII overexpression or no transfection.
Distinct signal transductions induced by TNF-α are TNF receptor subtype dependent
To further study the possible mechanisms by which TNF-α promotes or prevents neuronal death, we further examined TNF receptor-related signal transduction pathways. It is well established that TNF-α induces the activity of NF-κB DNA binding, which contributes to cell death and is TNFRI dependent in the peripheral immune system (Tartaglia et al., 1993; Sipe et al., 1996; Yu et al., 1999). To determine whether NF-κB was involved in TNF-α-induced TNF receptor expression in neurons, an experiment to examine expression of p65, a subunit of NF-κB, by using Western blot was performed. Nuclear or cytoplasmic extracts were prepared from hippocampal neurons of wild-type, TNFRI −/−, or TNFRII −/− mice after a 30 min incubation with TNF-α at 0, 1, 10, 100, and 1000 pm. We found that TNF-α can induce an increase in nuclear NF-κB p65 in a dose-dependent manner, whereas decreased cytoplasmic NF-κB p65 was observed in hippocampal neurons of wild-type mice (Fig.4A), suggesting that, in general, TNF-α-induced NF-κB in the cytoplasm is translocated into the nucleus. However, no NF-κB p65 alteration or translocation was observed in either the cytoplasm or nucleus in hippocampal neurons of TNFRI −/−, whereas the translocation of NF-κB was still observed in hippocampal neurons of TNFRII −/− (Fig. 4B). This result indicates that translocation of NF-κB p65 in neurons induced by TNF-α is TNFRI dependent. To further support this finding, we conducted NF-κB binding studies using EMSA and found low NF-κB binding activity in TNFRI −/− neurons, whereas in neurons from TNFRII −/− mice, there still remain high levels of NF-κB binding activity (Fig. 4C). Moreover, although studies in signal transduction for TNFRII remain inconclusive, p38 mitogen-activated protein (MAP) kinase plays a role in neurodifferentiation and neuronal survival. Thus, we studied effects of TNF-α on p38 MAP kinase expression in hippocampal neurons of TNFR −/− mice. As illustrated in Figure5A, we found that TNF-α treatment resulted in an increase of the p38 MAP kinase expression in a dose-dependent manner in hippocampal neurons of either wild-type or TNFRI −/− mice. However, no changes of p38 MAP kinase were observed in hippocampal neurons of TNFRII −/− mice (Fig. 5A). Furthermore, to confirm these results, we transfected the TNFRI- or TNFRII-encoding gene to NT2 cells. We found that no significant changes of p38 MAP kinase expression were observed in neurons with treatment of TNF-α in TNFRI-transfected NT2 cells (Fig. 5B). However, after TNFRII was transfected, TNF-α treatment increased the p38 MAP kinase expression in the cells (Fig. 5B). Because p38 MAP kinase has been reported to be essential in neuronal survival and differentiation, our data are consistent with studies that show that TNF-α might be trophic to certain types of cells or receptor dependent (Cheng et al., 1994). The result supports our hypothesis that p38 MAP kinase expression is related to neuronal survival in a TNFRII-dependent manner. We believe that different types of neurons have different expression ratios of the two TNF receptors. Together, these data suggest that TNFRII may play a supportive role in the ability of neurons to respond to insults, whereas TNFRI may contribute neuronal death.
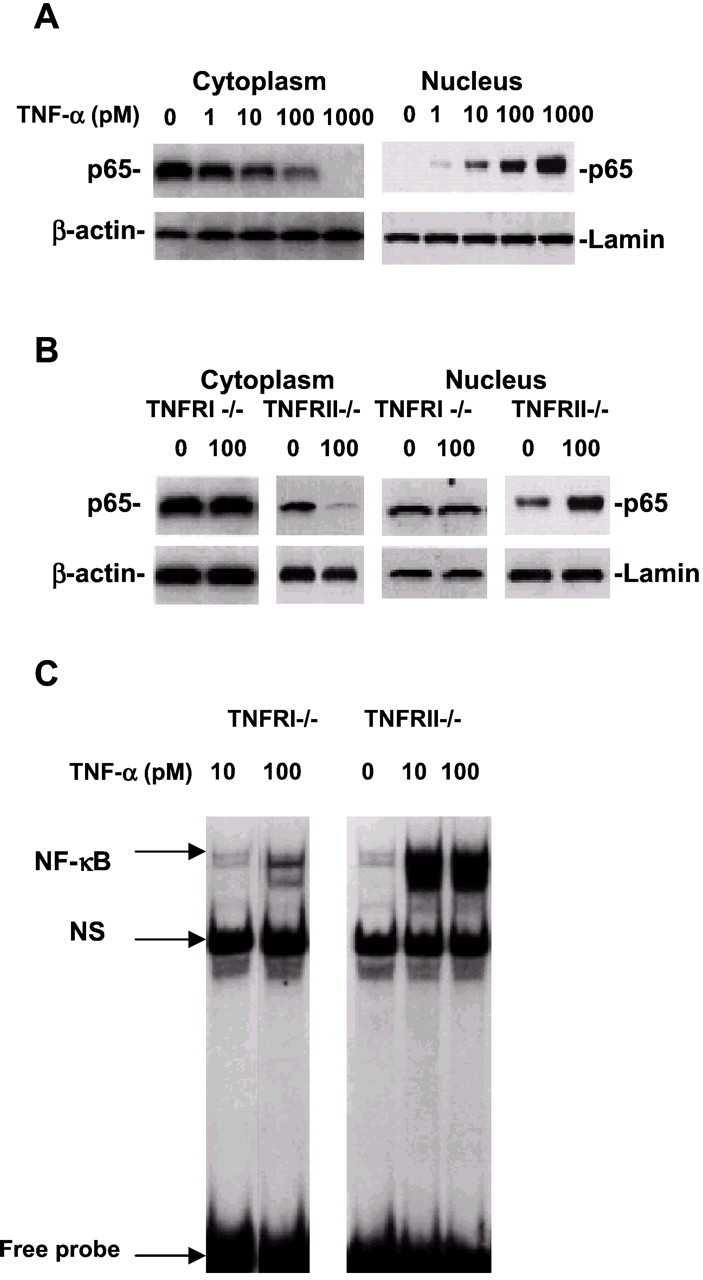
NF-κB p65 translocation and activity in hippocampal neurons are TNFRI dependent. A, Western blot of cytoplasmic or nuclear NF-κB p65 in hippocampal neurons of wild-type mice. Neurons were treated with soluble TNF-α at 0, 1, 10, 100, and 1000 pm for 30 min. At the indicated various doses of TNF-α treatment, cell cytoplasmic or nuclear samples were subjected to SDS-PAGE, and the blots were probed with the antibody to NF-κB p65 or β-actin and lamin as the cytoplasmic and nucleic housekeeping proteins, respectively. Visualization of the proteins was performed with ECL. The result demonstrates that cytoplasmic NF-κB p65 was at high levels and nuclear NF-κB p65 was low without TNF-α treatment. However, TNF-α treatment resulted in low levels of cytoplasmic NF-κB p65 and high levels in the nucleus, suggesting that NF-κB p65 was translocated from the cytoplasm to the nucleus in cortical neurons after TNF-α treatment. B, Western blot of nuclear NF-κB p65 in hippocampal neurons from TNFRI knock-out mice (TNFRI −/−). The neurons from TNFRI knock-out brains were treated with TNF-α at 100 pm for 30 min. The cytoplasmic and nuclear samples were subjected to SDS-PAGE, and the blots were probed with the antibody to NF-κB p65 or β-actin and lamin. We found that little NF-κB p65 was induced by TNF-α at 100 pm compared with that in wild-type or TNFRII −/− hippocampal neurons, whereas NF-κB p65 induced by TNF-α was still translocated from cytoplasm to nucleus in neurons from TNFRII −/− mice. C, EMSA analysis of NF-κB binding activity in hippocampal neurons from TNFRI −/− and TNFRII −/− mice. EMSA assay with nuclear extracts prepared from hippocampal neurons of TNFRI −/− and TNFRII −/− with TNF-α treatment (10 and 100 pm) for 30 min.32P-labeled oligonucleotide probe used contained the NF-κB p65 subunit and 50-fold molar excess of unlabeled AP2 (nonspecific competitor; NS) probe. The NF-kB activity was abolished in TNFRI-deleted neurons but not in neurons with the TNFRII deletion.

p38 MAP kinase expression in hippocampal neurons is TNFRII dependent. Hippocampal neurons from TNFRI −/−or TNFRII −/− mouse brains were treated with either glutamate (0, 10, or 100 μm) or TNF-α (0, 10, or 100 pm) for 24 hr. Extracts were fractionated by SDS-PAGE on 12% Tris-glycine gels and transferred to PVDF membranes; 10 μg of cell lysates were loaded per lane. The p38 MAP kinase was detected with affinity-purified antibody using a horseradish peroxidase-conjugated secondary antibody and processed using ECL detection. We found that glutamate decreased p38 MAP kinase expression, whereas TNF-α increased p38 MAP kinase activity in hippocampal neurons from wild-type or TNFRI −/− mice. These effects are dose dependent. However, no alteration of p38 MAP kinase expression was observed in hippocampal neurons of TNFRII −/− by either glutamate or TNF-α, suggesting that p38 MAP kinase expression induced by either TNF-α or glutamate is TNFRII dependent.
Distinct TNF-α radioligand binding affinities to TNF receptor subtypes in neurons
Last, to study how TNF-α interacts with its two different receptor subtypes and whether TNF-α binds the two receptors differentially, we subcloned cDNA for each TNF receptor into the expression vector pcDNA3.1 containing the human CMV promoter and SV40 ori. These constructs were stably transfected into human NT2 neurotypic cells (Pleasure et al., 1992), which have few endogenous TNF receptors (Fig. 6A), using a modified calcium phosphate transfection technique. The expression in transfected cells was measured by125I-specific binding. We then characterized specific 125I-TNF-α binding to the surface of the TNFRI or TNFRII cDNA-transfected NT2 cells in the absence and presence of a 200-fold excess of cold TNF-α. As shown in Figure 6, the Kd averages, 0.6 nm for TNFRI (B) and 1.14 nm for TNFRII (C), as determined with the NT2 cell transfectants, are comparable with theKd of the native TNFRI or TNFRII expressed constitutively on CHO cells and endogenous TNF receptor in HeLa cells (Loetscher et al., 1990; Schall et al., 1990). The endogenous TNF binding sites of NT2 cells have affinities comparable with those of the human TNFRI and TNFRII subtypes (data not shown).

Detection of TNFRI and TNFRII in NT2 cells and saturation analysis of specific 125I-TNF-α binding to transfected human NT2 neurotypic cells at various TNF-α concentrations. A, Detection of mRNA and protein expression of both TNFRI and TNFRII by using reverse transcription (RT)-PCR and Western blot techniques. For RT-PCR, the negative control reaction in the absence of RT with RNA template yielded no detectable product, whereas the positive control reaction in the TNFRI or TNFRII transfection with RNA template yielded a highly abundant expected band. For Western blot analysis, the same TNFRI- and TNFRII-transfected NT2 cells were used for confirmation of these two-receptor expression. Both results demonstrate little expression of both endogenous TNFRI and TNFRII in NT2 cells (A). Scatchard analyses of the 125I-TNFα binding in TNFRI- and TNFRII-transfected NT2 cells are illustrated in Band C, respectively. The assays with transfected and control cells contained 1 × 106 cells per assay. Five independent binding assays were performed, and theKd averages for TNFRI and TNFRII are 0.6 and 1.14 nm, respectively.
DISCUSSION
The results presented in this paper suggest a novel and specific interaction between TNF-α and the extracellular domains of TNF receptors in neurons, indicating novel TNF-α involvement in TNFRI and TNFRII. It is very helpful to explain discrepancy effects of TNF-α (trophic and toxic) on neurons. That is, survival or death roles that TNF-α plays depend on which TNF receptor subtype is activated.
It has been recognized and established that TNF-α binds to responsive cells with both high and low affinities, suggesting two distinct classes of TNF receptors. The gene for the high-affinity receptor was shown to be TNFRI, a 55 kDa membrane-spanning cell-surface protein with a 150 residue intracellular domain (Tartaglia et al., 1993). No enzymatic activity has been attributed to this receptor, which, with a cytoplasmic domain containing four cysteine repeats, shares sequence homology with the p75 NGF receptor gene (p75NGFR) (Chao, 1994). Our unpublished data from neurodegenerative diseased brains demonstrate that different types of neurons may have different expression ratios of the two TNF receptors. For example, in neurodegenerative disorders, including Alzheimer's and Parkinson's diseases, we hypothesize that the two receptor ratios could be changed such that more membrane-bound TNFRI is decreased as a result of increased neuron death. Alternatively, the expression level of TNFRII could be affected–downregulated such that there is less TNFRII expression, leading to decreased neuron survival (Dowling et al., 1996). Additional studies are aimed at determining whether more rigorous techniques will reveal single neuron expression by using double-labeling in situ hybridization, laser capture, and human primary neuron culture techniques.
Importantly, we noted that results from transfected cell lines and spinal cord in TNFRI and TNFRII (Liu et al., 1996; Kim et al., 2001) are different from our current findings in the hippocampus from these animals. We believe that this discrepancy may show that there is more than just the “death domain” contributing to apoptosis and that other downstream factors, such as caspase or MAP kinase, in these different types of neurons are involved. The TNF receptor superfamily contains several members with homologous cytoplasmic domains known as death domains. The intracellular DD is critical in initiating apoptosis and other signaling pathways after ligand binding by receptors (Tartaglia et al., 1993). In the absence of a ligand, DD-containing receptors are maintained in an inactive state. However, under pathological conditions in which glial cells are activated in the brain and TNF-α is secreted, TNFRI is then activated. TNFRI contains a cytoplasmic DD required for signaling pathways, which is responsible for NF-κB activation and apoptosis (Hsu et al., 1995). Our results from knock-out studies support this notion and are consistent with and expand on these findings. Morphologically, neurons of TNFRII −/− are much more vulnerable to TNF-α than that of TNFRI −/− and wild type (Fig. 1A). This observation is supported by the results from LDH release (Fig. 1B). However, there is no significant difference of LDH release from hippocampal neurons between TNFRI −/− and wild-type mice at lower doses of TNF-α. This suggests that neurodegeneration or neuronal death induced by TNF-α from TNFRI −/− mice are partially involved an apoptotic process, and LDH release might not be able to clearly differentiate necrotic and apoptotic processes. When these molecules activate TNFRII, we found that p38 MAP kinase expression is increased, which enhances neuronal survival as a compensatory effect (Fig. 5). However, TNF-α also has protective effects, because when gene targeting the TNF-α gene in mice, they develop a more severe disease (Liu et al., 1998). Indeed, TNF also protects neurons from glutamate injury (Bruce et al., 1996) and mice suffering from other autoimmune diseases, such as systemic lupus erythematosus (Jacob et al., 1990). Thus, we hypothesize that the biological rationale for having two TNF receptors is that TNFRI may provide a molecular mechanism for ensuring rapid and active apoptosis when a neuron is injured or sick and unsuccessful in rescuing its life. During the developmental period, when a neuron encounters an appropriate target and sequesters TNF-α, TNFRII is significantly activated, and occasional TNFRI activation is insufficient to override this survive signal. Conversely, if a neuron is sick and reaches an inappropriate target, TNFRI is robustly activated by TNF-α secreted by surrounding glial cells and promotes apoptosis.
Together, these results are the first to demonstrate that deletion of different TNF receptor subtypes signals neuron death and survival. Second, functions of these two receptor subtypes might then be involved in different signal transduction pathways. Third, the binding affinity of TNF-α to TNFRI is higher than the binding affinity to TNFRII. The translocation and binding activity of NF-κB are partially TNFRI activation dependent, whereas p38 MAP kinase activation contributes to TNFRII activation (Guo et al., 2001). Additional studies on structural features specific to the TNF receptor subtypes by site-directed mutagenesis and their differential expression in brains with neurodegenerative disorders such as Alzheimer's disease and multiple sclerosis using immunohistochemistry need to be conducted. The results from these studies may lead to alternative therapeutic targets of TNF receptors to those diseases, providing the basis for developing agonist and antagonist systems for TNF receptor subtypes and also encouraging better strategies for treatments of brain disorders related to neuroinflammation.
Footnotes
This work was supported by grants from the Alzheimer's Disease Association, Arizona Disease Control Commission, and the Edward Johnson Foundation. We thank Dr. David V. Goeddel from Tularik Inc. for his constructive and helpful discussion and encouragement.
Correspondence should be addressed to Yong Shen, Haldeman Laboratory of Molecular and Cellular Neurobiology, Sun Health Research Institute, 10515 W. Santa Fe Drive, Sun City, AZ 85351. E-mail:yong.shen{at}sunhealth.org.
L. Yang's present address: Earle Chiles Institute of Molecular Biology, Oregon Health Sciences University, Portland, OR 97213.
Y. Konishi's present address: Department of Physiology, Ehime University School of Medicine, Ehime, Japan 791-0295.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}