

Abstract
We hypothesized that cation-dependent Cl- transport protein Na-K-Cl cotransporter isoform 1 (NKCC1) plays a role in the disruption of ion homeostasis in cerebral ischemia. In the current study, a role for NKCC1 in neuronal death was elucidated in neurotoxicity induced by glutamate and oxygen and glucose deprivation (OGD). Incubation of cortical neurons cultured for 14–15 d in vitro (DIV) with 100 μm glutamate for 24 hr resulted in 50% cell death. Three hours of OGD followed by 21 hr of reoxygenation led to 70% cell death. Inhibition of NMDA receptors with dizocilpine hydrogen maleate (1 μm) prevented both OGD- and glutamate-mediated cell death. Moreover, blocking of NKCC1 activity with bumetanide (5–10 μm) abolished glutamate- or OGD-induced neurotoxicity. Bumetanide was ineffective if added after 10–120 min of glutamate incubation or 3–6 hr of OGD treatment. Accumulation of intracellular Na+ and 36Cl content after NMDA receptor activation was inhibited by bumetanide. Blockage of NKCC1 significantly attenuated cell swelling after OGD or NMDA receptor activation. This neuroprotection was age dependent. Inhibition of NKCC1 did not protect DIV 7–8 neurons against OGD-mediated cell death. In contrast, cell death in DIV 7–8 neurons was prevented by the protein-synthesis inhibitor, cycloheximide. Taken together, the results suggest that NKCC1 activity is involved in the acute excitotoxicity as a result of excessive Na+ and Cl- entry and disruption of ion homeostasis.
Introduction
Cl- movement has been shown to be a central component of the acute excitotoxic response in neurons (Rothman, 1985; Olney et al., 1986; Nicklas et al., 1987). The acute excitotoxicity is thought to be mediated by excessive depolarization of the postsynaptic membrane. This results in an osmotic imbalance when countered by an influx of Cl-, Na+, and water, leading to eventual lysis. Removal or reduction of Cl- from extracellular medium during excitatory amino acid exposure completely eliminates the acute excitotoxic response in hippocampal (Rothman, 1985) and retinal (Olney et al., 1986; Nicklas et al., 1987) neurons. A significant increase in intracellular Cl- concentration ([Cl-]i) is observed in hippocampal neurons during OGD (Inglefield and Schwartz-Bloom, 1998). Although the mechanisms underlying the Cl--dependent excitotoxicity are not yet understood, blockage of Cl- entry through the Cl-/HCO3- exchanger or GABA receptor effectively protects cells against the acute excitotoxicity (Zeevalk et al., 1989; Chen et al., 1998; Inglefield and Schwartz-Bloom, 1998).
We hypothesized that Na-K-Cl cotransporter isoform 1 (NKCC1) may also contribute to the Cl- movement during excitotoxicity. The electroneutral NKCC1 transports Na+, K+, and Cl- into cells under physiological conditions (Russell, 2000). NKCC1 is characteristically inhibited by the sulfamoylbenzoic acid “loop” diuretics, such as bumetanide and furosemide (Russell, 2000). NKCC1 is important in the maintenance of intracellular Cl- in neurons and contributes to GABA-mediated depolarization in immature neurons (Misgeld et al., 1986; Sun and Murali, 1998; Sung et al., 2000; Alvarez-Leefmans, 2001). Thus, NKCC1 may affect neuronal excitability through regulation of [Cl-]i (Sung et al., 2000; Jang et al., 2001). In addition, activation of the ionotropic glutamate NMDA receptor, the AMPA receptor, and the metabotropic glutamate receptor (group I) significantly stimulates NKCC1 activity in cortical neurons (Schomberg et al., 2001).
Our recent study reveals an upregulation of NKCC1 protein expression in the cortex at 24 hr reperfusion after focal ischemia (Yan et al., 2001). Pharmacological inhibition of NKCC1 with bumetanide results in a significant reduction of infarction volume (Yan et al., 2001). In addition, water content increase is 70% less in the bumetanide-treated brains (Yan et al., 2001). These results strongly suggest that NKCC1 is important in ischemic neuronal damage. However, the cellular mechanisms by which NKCC1 contributes to ischemic cell death have not been defined. In this study, we report that inhibition of NKCC1 activity abolished glutamate-mediated neurotoxicity and significantly attenuated OGD-induced neuronal death. Thus, NKCC1 may be involved in ischemic cell death through an association with excitotoxicity.
A preliminary report was presented at the Ninth International Symposium on Pharmacology of Cerebral Ischemia (Beck et al., 2002).
Materials and Methods
Cortical neuron and astrocyte cultures. Primary rat neurons were cultured using techniques established in our laboratory as described previously (Schomberg et al., 2001). Embryonic day (E) 17 pregnant rats were anesthetized with halothane and killed. Fetuses were removed and rinsed in cold HBSS. The cortices were removed, minced, and rinsed in HBSS. The tissues were treated with 0.2 mg/ml trypsin at 37°C for 25 min. The cells were centrifuged briefly, and the pellet was washed and triturated thoroughly in HBSS. Cell suspension was filtered through a 70 μm cell strainer. The cell suspension was diluted in Eagle's minimal essential media (EMEM) containing 100 U/ml penicillin and 100 μg/ml streptomycin, 10 mm glutamine, 10% fetal bovine serum, and 10% horse serum. Live cells (6.25 × 10 5) were added to each well of a poly-D-lysine-coated 24-well plate and incubated at 37°C in an incubator (model 3110, Thermo Forma, Marietta, OH) with 5% CO2 and atmospheric air. After 96 hr in culture, cultures were refed with EMEM containing 4 μm cytosine arabinofuranoside (Ara-c) for 72 hr. Ara-c is a mitotic inhibitor that prevents growth of glial cells. After the Ara-c treatment, cultures were refed with EMEM containing both 10% fetal bovine and 10% horse sera.
Astrocytes were prepared from the same cortical cell suspension as described above with the following changes. The cell suspension was diluted in EMEM containing 100 U/ml penicillin and 100 μg/ml streptomycin, 10 mm glutamine, and 10% fetal bovine serum; 1.2 × 10 4 live cells per well were seeded in collagen-coated 24-well plates.
OGD treatment. DIV 7–8 and DIV 14–15 neuronal cultures were grown in 24-well plates. The cells were rinsed twice with an isotonic OGD solution, pH 7.4, containing (in mm): 0 glucose, 20 NaHCO3, 120 NaCl, 5.36 KCl, 0.33 Na2HPO4, 0.44 KH2PO4, 1.27 CaCl2, 0.81 MgSO4. Cells were incubated in 0.5 ml of the OGD solution in a hypoxic incubator (model 3130, Thermo Forma) containing 94% N2, 1%O2, and 5% CO2. The oxygen level in the medium of cultured cells in 24-well plates was monitored with an oxygen probe (model M1-730, Microelectrodes, Bedford, NH) and decreased to ∼2–3% after 60 min in the hypoxic incubator. The OGD incubation was 3 hr for DIV 14–15 neurons or 8 hr for DIV 7–8 neurons. For reoxygenation, the cells were incubated for 24 hr in 0.5 ml of EMEM containing 5.5 mm glucose at 37°C in the incubator with 5% CO2 and atmospheric air. Normoxic control cells were incubated in 5% CO2 and atmospheric air in a buffer identical to the OGD solution except for the addition of 5.5 mm glucose. In the drug treatment studies, cells were pretreated with 10 μm bumetanide or 1 μm dizocilpine hydrogen maleate (MK801) for 30 min before the OGD treatment. Bumetanide or MK801 was present in all subsequent washes and incubations. For post-ischemic treatment, bumetanide (10 μm) was added 3, 4, and 6 hr after induction of OGD treatment.
Glutamate treatment. DIV 14–15 neuron cultures were incubated in EMEM containing 100 μm glutamate at 5% CO2 and atmospheric air for 24 hr at 37°C. For the drug pretreatment study, 0.5, 1.0, 5.0, or 10 μm bumetanide or 1 μm MK801 was added in cultures 30 min before the addition of glutamate, and cells were subsequently incubated for 24 hr at 37°C. For the time course studies, bumetanide was added either concurrently with glutamate or 10–120 min after the glutamate treatment, as indicated.
Measurement of cell death. Cell viability was assessed by propidium iodide (PI) uptake and retention of calcein using a Nikon TE 300 inverted epifluorescence microscope. Cultured neurons were rinsed with the isotonic control buffer and incubated with 1 μg/ml calcein-AM and 10 μg/ml PI in the same buffer at 37°C for 35 min. For cell counting, cells were rinsed with buffer and visualized using a Nikon 20× objective lens. Calcein fluorescence was visualized using FITC filters [excitation (Ex) 488 nm; emission (Em) 515 nm], and PI fluorescence was visualized using Texas Red filters (Ex 536 nm; Em 645 nm). Images were collected using a Princeton Instruments MicroMax CCD camera. In a blind manner, a total of 1000 cells per condition were counted using MetaMorph image-processing software (Universal Imaging, Downingtown, PA). Cell mortality was expressed as the ratio of PI-positive cells to the sum of calcein-positive and PI-positive cells.
Intracellular Cl-content measurement. Cells on 24-well plates were preincubated at 37°C for 30 min in HEPES–MEM 36Cl (0.4 μCi/ml), as described previously (Schomberg et al., 2003). In our previous study, steady-state intracellular 36Cl was reached by 10 min incubation and maintained through 60 min (Schomberg et al., 2003). Thus, a 30 min preincubation with 36Cl was used in the current study. The cells were then incubated in HEPES–MEM (Schomberg et al., 2001) containing 36Cl (0.4 μCi/ml) in either the presence or absence of 10 μm bumetanide, 1.0 μm MK801, and 100 μm NMDA for 15 min. Thus, the specific activity of 36Cl in HEPES–MEM was constant under all conditions. Intracellular 36Cl content measurement was terminated by three washes with 1 ml ice-cold washing buffer containing (in mm): 118 NaCl, 26 NaHCO3, 1.8 CaCl2, pH 7.40. Radioactivity of the cellular extract in 1% SDS was analyzed by liquid scintillation counting (Packard 1900CA, Downers Grove, IL). In each experiment, specific activities (counts per micromole per minute) of 36Cl were determined for each assay condition and used to calculate intracellular Cl - content (micromole per milligram per protein). Protein content was measured in each sample using the bicinchoninic acid method (Schomberg et al., 2003).
Intracellular Na+measurement. Intracellular Na+ concentration ([Na+]i) was measured with the fluorescent dye sodium binding benzofuran isophthalate (SBFI-AM) as described previously (Su et al., 2002a). Cultured neurons grown on coverslips were loaded with 2.5 μm SBFI-AM at room temperature in HEPES–MEM containing 0.05% pluronic acid for 45 min. The coverslips were placed in an open-bath imaging chamber (Model RC24, Warner Instruments, Hamden, CT) containing HEPES–MEM at ambient temperature. To reduce phototoxicity, the HEPES–MEM in these experiments was supplemented with (in mm): 1.0 pyruvate, 0.4 ascorbic acid, 0.01 troloxC, and 0.1 butylated hydroxyanisole (Dent et al., 1999). Using a Nikon TE 300 inverted epifluorescence microscope and a 40× Super Fluor oil immersion objective lens, neurons were excited every 30 sec at 345 and 385 nm, and the emission fluorescence at 510 nm was recorded. Images were collected and analyzed with the MetaFluor image-processing software (Universal Imaging Corporation) as described previously (Su et al., 2002a). The ratios of 340/380 were recorded for 10 min in HEPES–MEM, HEPES–MEM plus 1 μm MK801, or HEPES–MEM plus 5 μm bumetanide to establish baselines, and then the bath chamber buffer was changed to HEPES–MEM with 60 μm NMDA, 60 μm NMDA plus 1 MK801, or 60 μm NMDA plus 5 μm bumetanide for 20 min. In the bumetanide experiments, cells were preincubated in 5 μm bumetanide for 10 min to ensure completed inhibition of the NKCC1. At the end of each experiment, absolute [Na+]i was determined for each cell by calibrating the SBFI fluorescence ratio with solutions containing 0, 10, 20, 40, or 80 mm [Na+]o plus monensin (10 μm) and gramicidin (3 μm) to equilibrate extracellular and intracellular [Na+], as described previously (Su et al., 2002a).
In our preliminary study, we observed severe phototoxicity and excitotoxicity in neurons under 37°C conditions. Cells failed to exhibit concentration-dependent changes in the SBFI fluorescence ratios during calibration after NMDA treatment at 37°C. Thus, in the current study, intracellular Na+ measurements were performed at room temperature.
Measurement of cell swelling. Concurrent with the [Na+]i measurement after NMDA exposure, relative cell volume changes were monitored using video-enhanced differential interference contrast (DIC) microscopy, as described in our previous study (Su et al., 2002a). Cells were visualized every 1.5 min using a Nikon 40× Plan Fluor DIC objective lens, and the images were recorded. The mean cross-sectional area (CSA) was calculated after tracing the perimeter of the cell body with Meta-Morph image-processing software.
For cell-swelling measurement after OGD, a region of neurons on a plate (DIV 14–15) was marked with a marking pen, and images were recorded as described above. This culture then underwent 3 hr of OGD treatment. After 3 hr of the incubation, neurons in the same marked regions were visualized, and images of the same group of cells were recorded. The control CSA values were obtained with a 20× plan objective before the 3 hr incubation treatment. Sister cultures were treated with the isotonic normoxic buffer in 5% CO2 and atmospheric air for 3 hr. In the bumetanide studies, bumetanide (10 μm) was added to cultures immediately before the OGD treatment, and cultures were treated with OGD for 3 hr. Relative changes of mean CSA (CSAr) values were obtained in cells at the end of 3 hr exposure to normoxic, OGD, or OGD plus bumetanide. CSAr values were calculated as experimental values/control values.
Materials. Bumetanide, glutamic acid, NMDA, NMDA receptor antagonist MK801, cytosine-1-β-D arabinofuranoside, pyruvate, l-ascorbic acid, troloxC, butylated hydroxyanisole, propidium iodide, and cycloheximide were purchased from Sigma (St. Louis, MO). Calcein-AM was from Dojindo Molecular Technologies (Gaithersburg, MD). SBFI was from Molecular Probes (Eugene, OR). EMEM and HBSS were from Mediatech Cellgro (Herndon, VA). Fetal bovine and horse sera were obtained from Hyclone Laboratories (Logan, UT). Chloride-36 was purchased from Amersham Biosciences (Piscataway, NJ).
Results
Pharmacological inhibition of NKCC1 reduces neuronal death after glutamate and OGD treatment
We hypothesized that stimulation of NKCC1 activity may lead to disruption of ion homeostasis and contribute to neuronal death. In this study, we first examined whether inhibition of NKCC1 is neuroprotective in excitotoxicity. A low level of cell death occurred in control conditions (Fig. 1A,B). Glutamate treatment (100 μm) for 24 hr led to ∼50% cell death in rat cortical neurons (DIV 14–15), as shown in Figure 1, C and D. In the presence of glutamate (100 μm) and bumetanide (10 μm), the glutamate-mediated cell damage was abolished (Fig. 1E,F). NMDA receptor antagonist MK801 (1 μm) blocked the glutamate-mediated cell death (Fig. 1G,H). Figure 2A illustrates the quantitative analysis of this study. Glutamate-mediated cell death was abolished by either bumetanide or MK801. NMDA (100 μm) led to 60% cell death that was inhibited by bumetanide. Blocking of both NKCC1 and NMDA receptors did not further improve cell survival. Neither bumetanide nor MK801 affected the basal levels of cell death (Fig. 2A).

Inhibition of NKCC1 reduces cell mortality after glutamate-mediated neurotoxicity. A—H, Cell mortality was assessed in DIV 14–15 neurons by PI and calcein-AM staining after 24 hr of glutamate (100 μm) treatment. Neurons were preincubated in EMEM in the presence or absence of 10 μm bumetanide or 1 μm MK801 for 30 min at 37°C. Cells were then incubated with 100 μm glutamate, glutamate plus bumetanide, glutamate plus MK801, or glutamate plus MK801 and bumetanide for 24 hr. Cells were stained with PI and calcein-AM for 30 min at 37°C. Positively stained cells were visualized, and cell images were acquired. A, C, E, G, Calcein-AM. B, D, F, H, PI. A, B, Control. C, D, Glutamate. E, F, Glutamate plus bumetanide. G, H, Glutamate plus MK801. Scale bar, 256 μm.

Dose-dependent inhibition of NKCC1 decreases glutamate-mediated neurotoxicity. A, Summary data of Figure 1. In the case of NMDA, 100 μm NMDA was used. Data are means ± SEM; n = 5–8. *p < 0.05 versus control; **p < 0.05 versus NMDA; #p < 0.05 versus glutamate by ANOVA (Bonferroni/Dunn). B, Neurons were preincubated in EMEM in the presence or absence of 0.5–10 μm bumetanide for 30 min at 37°C. Cells were then incubated with 100 μm glutamate or glutamate plus bumetanide for 24 hr. Cells were stained with PI and calcein-AM for 30 min at 37°C. Data are means ± SEM; n = 3–8. *p < 0.05 versus control; #p < 0.05 versus glutamate by ANOVA (Bonferroni/Dunn). Glu, Glutamate; Bum, bumetanide; Con, control.
Bumetanide is a potent inhibitor for NKCC1 and has an IC50 of 0.2 μm (O'Grady et al., 1987; Russell, 2000); however, bumetanide at high concentrations (2 mm) has been reported to inhibit NMDA receptor activity (Lerma and Martin del Rio, 1992). To further establish that the bumetanide-mediated protection observed above is via inhibition of NKCC1 activity, we conducted a dose-dependent study of bumetanide at 0.5, 1.0, 5.0, and 10 μm. As shown in Figure 2B, 0.5 and 1.0 μm bumetanide significantly reduced glutamate-mediated cell death. A nearly complete protective effect of bumetanide was found at 5.0 and 10 μm. Either 5 or 10 μm bumetanide was used in the rest of the study.
Activity of NKCC1 is associated with the initial cell death events of glutamate-mediated neurotoxicity
We hypothesized that activation of NKCC1 contributes to glutamate-mediated neurotoxicity by intracellular Na+ and Cl- overload during the initial stage of excitotoxicity. To test this possibility, we examined whether inhibition of NKCC1 is neuroprotective after glutamate receptors have been activated. As shown in Figure 3, blocking of NKCC1 either before glutamate treatment or concurrently with the addition of glutamate significantly reduced glutamate-mediated cell death (p < 0.05). In contrast, inhibition of NKCC1 at 10, 30, 60, or 120 min of glutamate treatment had no effect on neuronal death (Fig. 3) (p > 0.05). These results suggest that NKCC1 plays an important role during the initial stage of the glutamate-mediated neurotoxicity.

Time-dependent effect of bumetanide on glutamate-mediated neurotoxicity. Cell mortality was assessed in DIV 14–15 neurons by PI and calcein-AM staining after 24 hr of glutamate treatment. Neurons were incubated in EMEM in the presence of 100 μm glutamate for 24 hr at 37°C; 10 μm bumetanide was added 30 min before, concurrently, or 10, 30, 60, or 120 min after the glutamate treatment. Data are means ± SEM; n = 3–5. *p < 0.05 versus control; #p < 0.05 versus glutamate by ANOVA (Bonferroni/Dunn). Con, Control; Glu, glutamate; Bum, bumetanide.
NKCC1 is involved in OGD-mediated cell death
Previous findings suggest that OGD-mediated cell death is essentially a result of NMDA ionotropic receptor-triggered excitotoxicity (Gwag et al., 1997). Rat cortical neuron cultures (DIV 14–15) exhibited 70% cell death after 3 hr of OGD and 21 hr of reoxygenation compared with the cells in control conditions (Fig. 4A). However, OGD-mediated cell death was significantly attenuated by 10 μm bumetanide (Fig. 4A)(p < 0.05), consistent with the results of the glutamate study described above. Inhibition of the NMDA receptor with the antagonist MK801 (1 μm) abolished OGD-induced cell death (p < 0.05). Moreover, no additional neuroprotection was found when both NMDA receptors and NKCC1 were blocked (Fig. 4A) (p > 0.05). Furthermore, inhibition of protein synthesis by cycloheximide had no effects on OGD-induced cell death (p > 0.05), suggesting that apoptosis is not a prominent mechanism in this model. As shown in Figure 4B, inhibition of NKCC1 before or concurrently with the induction of OGD significantly reduced OGD-mediated neuronal death. However, neuroprotection was absent if bumetanide was added 3, 4, or 6 hr after OGD treatment. This finding is consistent with the glutamate-mediated toxicity as described above.

Inhibition of NKCC1 reduces cell mortality after OGD. A, Cell mortality was assessed in DIV 14–15 neurons by PI and calcein-AM staining after 3 hr OGD and 21 hr reoxygenation. Neurons were preincubated in EMEM containing 10 μm bumetanide, 1 μm MK-801, bumetanide plus MK801, or 1 μm cycloheximide (CHX) for 30 min. The drugs were present during the rest of the incubation period. Data are means ± SEM; n = 3–5. *p < 0.05 versus control; #p < 0.05 versus OGD by ANOVA (Bonferroni/Dunn). B, Bumetanide (10 μm) was added 30 min before, concurrently, or 3, 4, or 6 hr after the OGD treatment. Data are means ± SEM; n = 5–8. *p < 0.05 versus control; #p < 0.05 versus OGD by ANOVA (Bonferroni/Dunn). C, Bicuculline (10 μm) was added 30 min before the OGD treatment. Data are means ± SEM; n = 3. *p < 0.05 versus control; #p < 0.05 versus OGD by ANOVA (Bonferroni/Dunn). Bum, Bumetanide; Bic, bicuculline.
Because 10 μm bumetanide inhibits GABAA receptors (Sung et al., 2000), we investigated whether bumetanide-mediated protection is via inhibition of GABAA receptors, instead of NKCC1. We compared the effects of the GABAA receptor antagonist bicuculline and bumetanide on OGD-mediated cell death. As shown in Figure 4C, although bicuculline attenuated OGD-mediated cell death, protection was significantly lower than the one mediated by bumetanide. No additive effect was observed in the presence of both bicuculline and bumetanide. This implies that bumetanide mediates its effect via blocking of NKCC1.
Neither a short treatment of OGD nor glutamate induces cell death in astrocytes
The cortical neuron culture preparation in the current study contained 20–30% astrocytes. To rule out a possible error in determination of cell mortality of neurons by counting contaminating astrocytes, we investigated whether astrocytes die after OGD or glutamate treatment. Compared with a control group of cultured astrocytes (Fig. 5A,B), treatment of cells (DIV 14–15) with either glutamate (100 μm) for 24 hr (Fig. 5C,D) or OGD for 6 hr (Fig. 5E,F) failed to induce cell death. In addition, neither 8 hr OGD in DIV 7–8 astrocytes nor 3 hr OGD in DIV 14–15 astrocytes caused significant cell death (2.1 ± 1.1 and 1.3 ± 0.4%, respectively). Therefore, the cells observed in the cultures throughout the study that were positively stained with PI were neurons.

Neither OGD nor glutamate causes cell death in astrocytes. Cell mortality was assessed in DIV 14–15 astrocytes by PI and calcein-AM staining after 24 hr of glutamate (100 μm) treatment or 6 hr of OGD and 18 hr of reoxygenation. A, C, E, Calcein-AM. B, D, F, PI. A, B, Control. C, D, Glutamate. E, F, OGD. Scale bar, 256 μm.
NKCC1 is not involved in OGD or glutamate-mediated cell death in immature neurons
Immature neurons are less susceptible to glutamate-mediated neurotoxicity because of a low level of glutamate receptors (McDonald et al., 1997). However, functional NKCC1 protein is expressed in DIV 7–8 neurons (Sun and Murali, 1998). Thus, if the role of NKCC1 in cell death is associated with NMDA glutamate receptor-mediated excitotoxicity, inhibition of NKCC1 would not provide significant neuroprotection in immature neurons. As shown in Figure 6, 8 hr of OGD and 16 hr of reoxygenation led to 60% cell death in DIV 7–8 neurons (p < 0.05). Inhibition of NKCC1 with bumetanide (10 μm) had no significantly protective effects on cell death in DIV 7–8 neurons. In contrast, cycloheximide (1 μm) significantly attenuated OGD-induced cell death in DIV 7–8 neurons (Fig. 6). Moreover, glutamate did not induce cell death in DIV 7–8 neurons (Fig. 6, inset). There were no significant neuroprotective effects of either MK801 (1 μm) or bumetanide (10 μm) on cells at DIV 7–8 (Fig. 6, inset). These data further suggest that NKCC1 is specifically involved in glutamate-mediated excitotoxicity.

Inhibition of NKCC1 did not protect DIV 7–8 neurons from cell death after OGD or glutamate exposure. Cell mortality was assessed in DIV 7–8 neurons by PI and calcein-AM staining after 8 hr OGD and 16 hr reoxygenation or 24 hr of glutamate treatment (100 μm) (inset). For drug-treated groups, neurons were preincubated in EMEM containing bumetanide (10 μm), MK801(1 μm), or cycloheximide (1 μm) for 30 min and remained during the rest of the incubation period. Data are means ± SEM; n = 3–5. *p < 0.05 versus control; #p < 0.05 versus OGD by ANOVA (Bonferroni/Dunn). Con, Control; Bum, bumetanide; CHX, cycloheximide; Glu, glutamate.
NKCC1 contributes to intracellular Na+ and Cl- accumulations and cell swelling during excitotoxicity
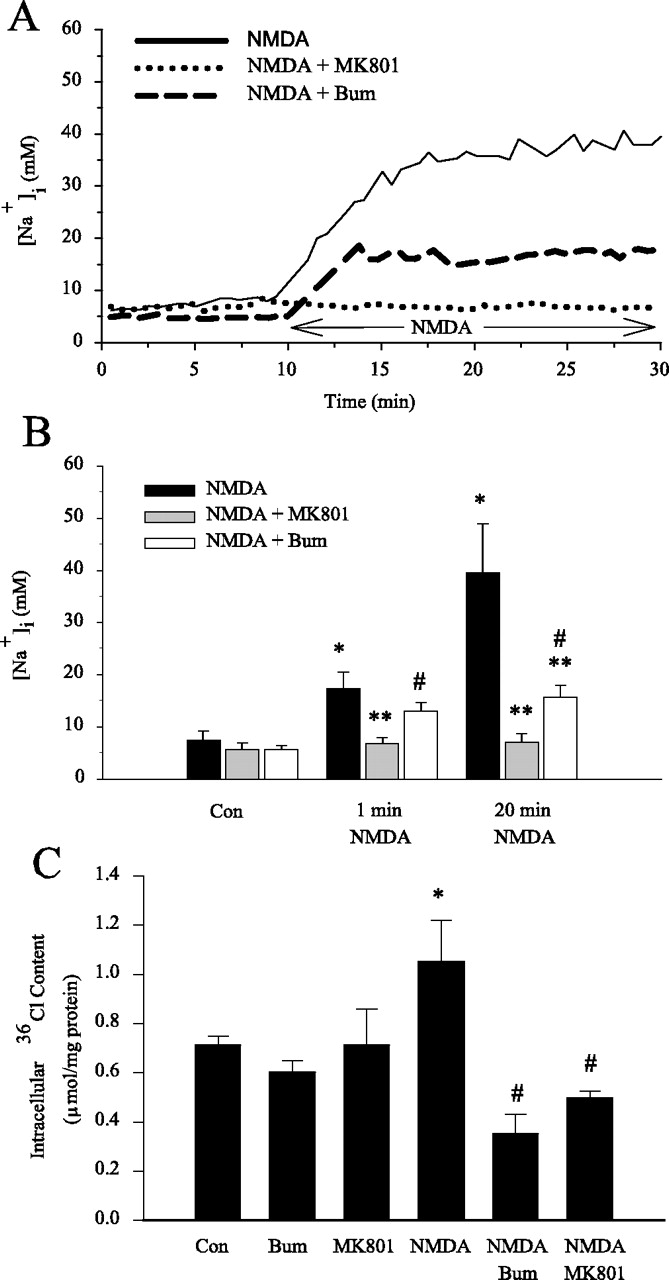
Changes of [Na+]i and intracellular Cl- content were monitored during activation of NMDA receptors. The mean resting [Na+]i level was 7.4 ± 1.8 mm in cortical neurons (Fig. 7A,B). On exposure to 60 μm NMDA and activation of NMDA receptors, [Na+]i increased abruptly by nearly 10 mm at the first minute ([Na+]i of 17.3 ± 3.7 mm). [Na+]i increased further and reached 39.5 ± 9.5 mm after 20 min exposure to NMDA. These increases in [Na+]i can be blocked by MK801. In the presence of 1 μm MK801, 60 μm NMDA did not trigger a significant rise of [Na+]i (6.8 ± 1.1 mm at 1 min and 7.1 ± 1.6 mm at 20 min). Interestingly, inhibition of NKCC1 activity with bumetanide had no significant effect on the initial rise of intracellular Na+ accumulation (13.0 ± 1.7 mm at 1 min); however, bumetanide significantly attenuated the rise of [Na+]i by 52% after 20 min of exposure of NMDA ([Na+]i of 15.6 ± 2.3 mm). This study further suggests that activation of NKCC1 activity contributes to NMDA-mediated rise in [Na+]i.

Inhibition of NKCC1 activity by bumetanide reduces intracellular Na+ and 36Cl accumulations. A, Representative trace of [Na+]i in a single neuron during perfusion of HEPES—MEM (10 min) followed by 60 μm NMDA (20 min), 1 μm MK801 (10 min) followed by 60 μm NMDA + 1 μm MK801 (20 min); or 5 μm bumetanide (10 min) followed by 60 μm NMDA + 5 μm bumetanide (20 min). B, Summary data. Data are means ± SEM; n = 4–5 from three different cultures. *p < 0.05 versus control; **p < 0.05 versus 60 μm NMDA; #p < 0.05 versus 60 μm NMDA + 1 μm MK801 (Mann—Whitney rank sum test). C, DIV 14–15 neurons were preincubated in HEPES—MEM for 10 min at 37°C. Cells were then equilibrated in HEPES—MEM with 36Cl (0.4 μCi/ml) for 30 min. After a 30 min equilibration with 36Cl, neurons were treated with HEPES—MEM containing 36Cl (0.4 μCi/ml) in either the presence or absence of 10 μm bumetanide, 1 μm MK801, 100 μm NMDA, NMDA plus bumetanide, NMDA plus MK801 for 15 min. Data are means ± SEM; n = 6–7. *p < 0.05 versus control; #p < 0.05 versus NMDA-treated by ANOVA (Bonferroni/Dunn). Bum, Bumetanide; Con, control.
Furthermore, activation of NMDA receptors by 100 μm NMDA for 15 min induced a significant increase in intracellular 36Cl content (Fig. 7C). This NMDA-mediated rise in intracellular 36Cl content was abolished by NMDA receptor antagonist MK801 (1 μm). Inhibition of NKCC1 with 10 μm bumetanide also blocked the intracellular 36Cl accumulation. Neither blocking of NKCC1 activity nor NMDA receptors affected the basal levels of intracellular 36Cl content (Fig. 7C). Taken together, the results suggest that NKCC1 contributes to Na+ and Cl- influx during the NMDA-mediated excitotoxicity.
Activation of NKCC1 activity secondary to activation of NMDA receptors will inevitably lead to cell swelling. We examined whether inhibition of NKCC1 activity would attenuate cell swelling as well as ionic influx. As shown in Figure 8A, concurrent measurement of cell volume with [Na+]i measurements in the same neurons (at room temperature) revealed that 60 μm NMDA induced significant swelling that progressed with time. After 20 min of NMDA exposure, cells had swelled by ∼27% (CSAr = 1.27 ± 0.05) (Fig. 8A,B). This NMDA-mediated swelling was sensitive to MK801. In the presence of both NMDA and MK801, swelling was abolished, and the average CSAr values were 1.00 ± 0.03 after 20 min of NMDA exposure (Fig. 8A,B). Moreover, the NMDA-induced swelling was significantly reduced when NKCC1 was inhibited. Average CSAr values were 1.07 ± 0.05 in the presence of both NMDA and 5 μm bumetanide (Fig. 8A,B).

Inhibition of NKCC1 activity by bumetanide reduces cell swelling. A, At room temperature, mean CSAr value in a single neuron was determined during 10 min of normal HEPES—MEM perfusion, followed by 20 min of 60 μm NMDA perfusion, or 10 min of normal HEPES—MEM + 5 μm bumetanide and 20 min of 60 μm NMDA + 5 μm bumetanide perfusion, or 10 min HEPES—MEM + 1 μm MK801 perfusion, and 20 min of 60 μm NMDA + 1 μm MK801 perfusion. B, The average CSAr value in single neurons after 20 min of exposure to HEPES—MEM, 60 μm NMDA, 60 μm NMDA + 1 μm MK801, or 60 μm NMDA + 5 μm bumetanide. Data are means ± SEM; n = 4–5 from three different cultures. *p < 0.05 versus control; #p < 0.05 versus 60 μm NMDA (Mann—Whitney rank sum test). C, Relative change in CSAr value of single neurons from its initial values were determined after 3 hr of incubation in normoxic control, OGD, or OGD plus bumetanide (10 μm) conditions. The bars represent neurons with -10, 10, 10–30, or > 30% of control CSAr value. Data are means ± SEM; n = 3–4. *p < 0.05 versus OGD by ANOVA (Bonferroni/Dunn). Bum, Bumetanide.
As shown in Figure 8B, activation of NMDA receptors at 37°C led to an increase in CSAr values in a similar manner as observed under room temperature conditions. In the presence of NMDA and bumetanide, the rise in CSAr values was significantly attenuated. At 20 min of NMDA incubation at 37°C, CSAr values increased by 25%, but only an 8% increase in CSAr values occurred when NKCC1 was blocked by bumetanide.
We then investigated whether NKCC1 contributed to cell swelling in OGD conditions. After 3 hr of incubation under normoxic control conditions, ∼70% of neurons were swollen by 0–10% (Fig. 8C). A small population of control cells increased in cell volume by 10–30%. These changes are likely a result of a temporary change of environmental conditions during imaging (difference in temperature and atmosphere between the incubator and microscope stage, or the absence of serum and growth factors, etc.). No cells increased in volume by 30% or more under these control conditions. In contrast, 3 hr of OGD led to significant cell swelling. Approximately 80% of neurons exhibited an increase of CSAr values by >30%; however, inhibition of NKCC1 activity reduced the number of swollen cells (>30%) to 43% (p < 0.05). The results imply that blocking of NKCC1 activity prevents neuronal swelling during OGD and thereby may reduce subsequent cell death. No significant effect of bumetanide on CSAr values was observed under control conditions (data not shown).
Discussion
Specificity of the pharmacological inhibitor bumetanide against NKCC1
A role for Cl- ion transporters in neuroexcitotoxicity has been examined previously. Zeevalk et al. (1989) reported that excitotoxicity is attenuated by the Cl- channel blocker disodium 4,4′-diisothiocyanatostilbene-2,2′-disulfonate (DIDS) (600 μm), and cellular edema was reduced by the ion transporter antagonist furosemide (1 mm). These pharmacological approaches are problematic because of the lack of inhibitor specificity at high concentrations. Lerma and Martin del Rio (1992) reported that NMDA-induced currents were reduced by furosemide with an IC50 of 1.23 ± 0.06 mm, whereas 2 mm bumetanide inhibited 54% of the current. DIDS has also been observed to block GABA release (Zeevalk et al., 1989) and kainate-induced currents (Chen et al., 1998). In the present study, we performed a dose-dependent experiment with 0.5–10 μm bumetanide. We believe that bumetanide at 0.5–10 μm exerts its action solely on NKCC1. This view is based on the following data. First, we conducted dose–responses with bumetanide on NKCC1 activity inhibition in neurons and found an IC50 of ∼0.2 μm bumetanide for NKCC1 activity inhibition (data not shown). This value is remarkably similar to IC50 values in other cell types in which bumetanide has proved to be 10–100 times more potent for inhibition of NKCC1 than furosemide in almost all cases (O'Grady et al., 1987; Russell, 2000). Bumetanide at 10 μm has no discernible effects on other membrane transport processes such as Na+/H+ or Cl-/HCO3- exchangers (O'Grady et al., 1987). On the other hand, GABAA receptors in dorsal root ganglion neurons were inhibited by 10 μm bumetanide (Sung et al., 2000). Our results, however, demonstrate that 10 μm bumetanide abolished the OGD-mediated cell death, whereas the GABAA receptor antagonist bicuculline only attenuated the cell death by ∼27%. This further implies that bumetanide acts primarily on NKCC1.
Second, in our recent studies, we have taken both pharmacological inhibition and genetic ablation approaches to block the NKCC1 activity in astrocytes. We reported that blocking of NKCC1 activity in astrocytes with 10 μm bumetanide abolishes high-potassium-mediated glial swelling and significantly decreases the release of excitatory amino acids from astrocytes (Su et al., 2002a,b). Genetic ablation of NKCC1 also blocks high-potassium astrocyte swelling (Su et al., 2002b). On the basis of the consistent agreement between the pharmacological inhibition and genetic knockout data of the previous studies, as well as a low concentration of bumetanide used in this study, we believe that the bumetanide-mediated effects in the present study represent NKCC1 activity. It is important to confirm these findings using a genetic knock-out model in a future study.
Inhibition of NKCC1 activity in neurons is neuroprotective
NKCC1 is important in cell volume regulation and maintenance of intracellular Na+, K+, and Cl- ion homeostasis under physiological conditions. Overstimulation of NKCC1 may lead to an imbalance of ion homeostasis and contribute to neuronal damage during ischemia (Staub et al., 1994). In neurons with injured axons, Cl- accumulation is attributable to influx of Cl- mediated by NKCC1 and a decrease in Cl- extrusion by reduced expression of K-Cl contransporter (Nabekura et al., 2002). This leads to a switch of GABA action potential from inhibitory to excitatory and may contribute to excitotoxicity (Nabekura et al., 2002). Recent work from this laboratory (Yan et al., 2001) has shown an upregulation of expression of NKCC1 in brain after focal cerebral ischemia. Pharmacological inhibition of NKCC1 significantly decreases brain edema and infarct volume in focal cerebral ischemic model (Yan et al., 2001).
To elucidate the cellular mechanisms underlying the role of NKCC1 in ischemic damage, we examined the effect of inhibition of NKCC1 activity on excitotoxicity mediated by glutamate and OGD. As illustrated in the current study, blockage of NKCC1 activity by bumetanide abolished glutamate-mediated excitotoxicity and significantly decreased OGD-induced neurotoxicity in DIV 14–15 neurons. In contrast, no neuroprotection was observed in DIV 7–8 neurons, which abundantly express NKCC1 (Sun and Murali, 1998) but lack high levels of glutamate receptor (McDonald et al., 1997). Inhibition of protein synthesis by cycloheximide significantly reduced cell death in DIV 7–8 neurons but was ineffective in DIV 14–15 neurons. This is consistent with the early findings that OGD-induced cell death is mediated primarily by apoptosis in immature neurons and is sensitive to cycloheximide (McDonald et al., 1997). This study provides a first line of evidence that NKCC1 is involved in neuronal death in an in vitro ischemic model. Taken together, the findings from both in vitro and in vivo studies strongly suggest that NKCC1 plays an important role in ischemic necrotic cell damage.
NKCC1 plays a role in acute excitotoxicity
Numerous reports indicate that acute excitotoxic neurodegeneration after glutamate receptor activation is dependent on Na+ and Cl- entry (Olney, 1971; Rothman, 1985; Choi et al., 1987). Excitotoxic dendritic injury is characterized by the formation of focal swellings along the length of the dendritic arbor (Hasbani et al., 1998). Blockage of Na+ and Cl- entry by removal of extracellular Na+ and Cl- abolishes excitotoxic dendritic injury, but removal of extracellular Ca2+ has no effect (Hasbani et al., 1998). In rat hippocampal slices, Na+ entry blockade by lidocaine and the Na+ channel inhibitor tetrodotoxin (TTX) attenuates hypoxia-induced loss of intracellular ATP, inhibition of protein synthesis, and damage of CA1 pyramidal neurons (Raley-Susman et al., 2001). Removal of extracellular Na+ or Cl- abolishes NMDA-mediated neurodegeneration in retinal ganglion cells (Chen et al., 1998).
The cellular pathways for the Na+ or Cl- entry described above, however, are not fully understood. One mechanism for the excessive Cl- entry is by GABA and glycine receptors. An activation of a Cl- conductance, which is sensitive to GABA and glycine receptor antagonists, is found in retinal ganglion cells after kainate treatment (Chen et al., 1998). Blockage of GABA and glycine receptors with the antagonists (bicuculline, strychnine) only decreases the NMDA-mediated lactate dehydrogenase release from retina by 50% (Chen et al., 1998). On the other hand, inhibition of Na+ channels by lidocaine and TTX attenuates the hypoxia-mediated Na+ accumulation in hippocampal slices by 28 and 20%, respectively (Raley-Susman et al., 2001). These findings suggest that multiple entry pathways for Na+ and Cl- govern the influx of Na+ or Cl- during excitotoxicity, in addition to Na+ and Cl- channels.
Our study suggests that NKCC1 may act as another mechanism for an overload of intracellular Na+ and Cl- during excitotoxicity. This hypothesis is based on the following findings. First, NKCC1 transports Na+, Cl-, and K+ into cells driven by the chemical gradients of three ions. It is well established that the rapidly triggered excitotoxic cell death depends on both Na+ and Cl- entry. Thus, NKCC1 is a likely candidate to transport an influx of Na+ and Cl- concurrently. Second, NKCC1 has been demonstrated to play an important role in regulation of intracellular Na+ and Cl- concentrations and cell volume in many types of cells (Russell, 2000; Su et al., 2002a,b). Activation of NMDA receptors stimulates NKCC1 activity in neurons (Schomberg et al., 2001). In the present study, a rapid increase in neuronal volume occurred after OGD or NMDA treatment. Neuronal swelling was significantly attenuated when NKCC1 was blocked. Activation of NMDA receptors triggered a significant increase in [Na+]i and intracellular 36Cl content accumulation. Blocking of NKCC1 activity abolished the Cl- accumulation and attenuated the Na+ rise by 52%.
It is generally believed that acute excitotoxic cell damage after glutamate receptor activation is dependent on Na+ and Cl- entry, whereas mechanisms leading to delayed cell death are primarily dependent on Ca2+ (Olney, 1971; Rothman, 1985; Choi et al., 1987). We found that blocking of NKCC1 is neuroprotective only when bumetanide was added concurrently with or 30 min before the glutamate or OGD treatment. The protective effect of bumetanide disappeared if added 10–120 min after the glutamate treatment or 3–6 hr OGD. These findings further suggest that NKCC1 is involved in the initial stage of cell damage during excitotoxicity that depends on extracellular Na+ and Cl-.
In summary, pharmacological inhibition of NKCC1 protected neurons from OGD- and glutamate-mediated toxicity. The NKCC1 inhibitor bumetanide reduced cell swelling during excitotoxicity. No protective effect of bumetanide was observed in immature neurons after OGD and glutamate treatment. The results of the study imply that NKCC1 may contribute to ischemic neuronal damage by facilitating excessive Na+ and Cl- entry.
Footnotes
This work was supported in part by National Institutes of Health Grant RO1NS38118 and National Science Foundation Career Award IBN9981826 to D.S.
Correspondence should be addressed to Dr. Dandan Sun, Department of Neurological Surgery, Medical School, University of Wisconsin, H4/332, Clinical Sciences Center, 600 Highland Avenue, Madison, WI 53792. E-mail: sun{at}neurosurg.wisc.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235061-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}