

Abstract
The cytotoxicity of extracellular amyloid β peptide (Aβ) has been clearly demonstrated in many cell types. In contrast, primary human neurons in culture are resistant to extracellular Aβ-mediated toxicity. Here, we investigate the involvement of p75 neurotrophin receptor (p75NTR) in Aβ-treated human neurons. We find that Aβ1-40 and Aβ1-42, but not the reverse control peptide, Aβ40-1, rapidly increase the levels of p75NTR in a specific and dose-dependent manner. In contrast to observations in cell lines, enhanced expression of p75NTR in human neurons via a herpes simplex virus amplicon vector does not increase the susceptibility of neurons to Aβ. Unexpectedly, inhibition of p75NTR expression with an antisense expression construct or incubation of the cells with an antibody to the extracellular domain of p75NTR sensitizes human neurons to extracellular nonfibrillar or fibrillar Aβ1-42 cytotoxicity. Unlike intracellular Aβ, extracellular Aβ toxicity is independent of p53 and Bax activity. However, Aβ toxicity is inhibited by caspase inhibitors and the glycogen synthase kinase 3β inhibitor lithium. Neuroprotection against Aβ is phosphatidylinositide 3-kinase dependent but Akt independent. These results are consistent with a neuroprotective role for p75NTR against extracellular Aβ toxicity in human neurons.
- Alzheimer
- primary human neurons
- p75 neurotrophin receptor
- amyloid β peptide
- neurotoxicity
- neuroprotection
Introduction
We and others (Mattson et al., 1992; Paradis et al., 1996; Zhang et al., 2002) have consistently observed that primary cultures of human neurons are resistant to extracellular amyloid β peptide (Aβ) cytotoxicity. The lack of extracellular Aβ toxicity in human neurons indicates the absence of a receptor or pathway that mediates Aβ cytotoxicity in other cell types or the presence of mechanisms that promote survival in the presence of Aβ. In the present study, we investigate the relationship between the p75 neurotrophin receptor (p75NTR) and extracellular Aβ toxicity in human neurons.
p75NTR is a member of the tumor necrosis factor (TNF) receptor family that binds neurotrophins nonselectively and mediates both neuronal apoptosis and survival (for review, see Dechant and Barde, 2002). p75NTR promotes neuronal cell death in a ligand-independent or -dependent manner (Rabizadeh et al., 1993; Barrett and Bartlett, 1994; Cassaccia-Bonnefil et al., 1996; Frade et al., 1996; Bredesen and Rabizadeh, 1997; Dechant and Barde, 1997; Friedman, 2000). In a subset of cells, the p75NTR proapoptotic pathway appears to be constitutively active and inhibited by tyrosine kinase A (TrkA) (for review, see Miller and Kaplan, 2001; Dechant and Barde, 2002). Pro-NGF binds with very high affinity to p75NTR and promotes cell death in smooth muscle cells and oligodendrocytes (Lee et al., 2001; Beattie et al., 2002). The proapoptotic role of p75NTR during early development is strongly supported by the presence of excess cholinergic neurons in the basal forebrain of p75NTR-null mice (Naumann et al., 2002). As a neurotrophin-dependent prosurvival protein, p75NTR modulates Trk survival activities (Miller and Kaplan, 2001) or TrkA-independent transduction of the Akt neuronal survival pathway in PC12 cell lines (Roux et al., 2001), ceramide synthesis in neocortical subplate neurons (DeFreitas et al., 2001), T9 glioma, and NIH3T3 cells (Dobrowsky et al., 1994), and nuclear factor κB in Schwann cells (Carter et al., 1996; Gentry et al., 2000). Furthermore, the receptor-interacting protein 2 (RIP2) interacts with p75NTR and modulates p75NTR function in cell death or survival (Khursigara et al., 2001).
p75NTR binds Aβ and mediates Aβ toxicity in PC12, SK-NMC, NIH 3T3, and SK-N-BE cell lines, suggesting that p75NTR acts as a death receptor for Aβ toxicity (Rabizadeh et al., 1994; Yaar et al., 1997, 2002; Kuner et al., 1998; Perini et al., 2002). In support of this hypothesis, p75NTR immunoreactivity increases in cholinergic hippocampal projection neurites (Mufson and Kordower, 1992). However, p75NTR decreases in the cell bodies and neurites of the nucleus basalis of Meynert (NBM) of Alzheimer disease (AD) brains (Salehi et al., 2000), and lower levels of p75NTR in cholinergic basal forebrain neurons correlate with greater cognitive impairment in mild cognitively impaired and early AD individuals, thus supporting the opposite hypothesis that p75NTR may be protective in human adult brains (Mufson et al., 2002). This hypothesis is further corroborated by observations that amyloid precursor protein (APP) Swedish and presenilin I M146L transgenic AD models exhibit increased levels of p75NTR in the presence of extracellular Aβ deposits, but fail to show neuronal loss (Jaffar et al., 2001).
In the present study, we observe that low levels of extracellular Aβ increase the levels of p75NTR in primary cultures of human neurons. Unexpectedly, we find that p75NTR protects primary human neurons against Aβ-induced toxicity. The neuroprotective ability of p75NTR against Aβ may open a new avenue for therapeutic intervention against Aβ toxicity in AD patients.
Materials and Methods
Antibodies. The antibodies used were rabbit polyclonal antisera to the cytoplasmic domain of human p75 NTR (Promega, Madison, WI), mouse monoclonal anti-primate NGF receptor of 75 kDa (ATCC #HB8737; American Type Culture Collection, Manassas, VA) (Levi et al., 1994), rabbit anti-human soluble TNF receptor I (sTNF-RI) [TNF-BPI from R & D Systems (Minneapolis, MN)], monoclonal anti-β-actin AC-15 (Sigma, St. Louis, MO), and NGF H-20 (Santa Cruz Biotechnology, Santa Cruz, CA; sc-548), rabbit polyclonal to phospho-Akt (Ser 473), phospho-Akt (Thr 308), and total Akt (all from the phospho-Akt pathway sampler kit; Cell Signaling Technology, Beverly, MA), polyclonal to total and phospho-phosphatidylinositol 3-kinase (PI3K) (Santa Cruz Biotechnology), rabbit polyclonal N20 anti-Bax (Santa Cruz Biotechnology), and monoclonal YTH-2D2 (Trevigen, Gaithersburg, MD) and 6A7 (Phar Mingen, San Diego, CA) anti-Bax. Secondary HRP-conjugated antibodies were either donkey anti-rabbit HRP or goat anti-mouse HRP (Amersham Biosciences, Dorval, Quebec, Canada).
DNA constructs. For p75 sense (p75S) expression, the pMVE1-containing human p75 NTR cDNA sequence was obtained from Dr. M. Chao (New York University School of Medicine) (Johnson et al., 1986), a 3.8 kb insert removed with EcoR1 and cloned in the EcoR1 site of pHSVPrPUC vector (Geller et al., 1990) under the control of the herpes simplex virus (HSV) immediate-early 4/5 promoter. For the p75 antisense (p75AS) expression construct, the 3.8 kb antisense-inserted construct in pHSVPrPUC was cleaved with HindIII to remove a 2.7 kb fragment and religated. The p53 wild-type (WT) and p53 dominant-negative (DN) (R273H) cDNAs were cloned into pCMV-NEO vector by Dr. A. Levine (Rockefeller University, New York, NY) (Hinds et al., 1990). The Akt constructs in pcDNA3 under the cytomegalovirus promoter were obtained from Dr. J. Woodgett (Ontario Cancer Institute, Toronto, Ontario, Canada). The wild-type and triple-mutant, dominant-negative (K179A, T308A, S473A) Akt constructs were made from bovine PKBα (protein kinase Bα)-Akt 1 and tagged at the N terminus with a hemagglutinin epitope (Coffer and Woodgett, 1991). Constitutively active Akt construct was cloned from human Akt1 and has an Src myristoylation sequence to confer constitutive activation [J. Jin from Dr. J. Woodgett's laboratory (University of Toronto, Toronto, Ontario, Canada)].
Chemicals, peptides, and recombinant proteins. Staurosporine (Sigma) was dissolved at 1 mm in DMSO and diluted in culture medium for treatments. Propidium iodide (Sigma) was dissolved in distilled (d)H2O at 1 mg/ml and used at 0.1 μg/ml in PBS. Hoechst dye (Intergen, Purchase, NY) was dissolved in dH2O at 200 μg/ml and diluted at 0.4 μg/ml in PBS. Wortmannin (Sigma) was dissolved in 100% DMSO at 1 mm and diluted to 200 nm and 10 μm in culture medium. 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) (Calbiochem, La Jolla, CA) was dissolved at 10 mm in dH2O. Lithium chloride (Sigma) was dissolved in dH2Oat2 m and diluted in culture medium at 20 mm. Boc-Asp-fluoromethylketone (Boc-D-fmk) (Bio-Rad, Mississauga, Ontario, Canada) was dissolved in 100% DMSO at 20 mm and diluted to 5 μm in the culture medium. Aged Aβ peptides [Aβ1-40, Aβ1-42, and Aβ42-1 from Bachem (Torrence, CA) and Aβ40-1 from Sigma] were solubilized at 25 μm in dH2O,aged 5 d at 37°C, and diluted at 100 nm in culture medium. Unless indicated otherwise, the aged peptides were used and represent the fibrillar form of the peptides. To compare the fibrillized and nonfibrillized peptides, these were prepared as described previously (Zhang et al., 2002). Disaggregated peptides were prepared by dissolving peptides (American Peptide Company, Santa Clara, CA) at 2 mg/ml in 1,1,1,3,3,3-hexafluoro-2-propanol. The solution was degassed in nitrogen, sealed, sonicated for 30 min, filtered in an Anotop 25 Plus 20 nm filter, and aliquots were stored in polypropylene tubes at -80°C until use. The concentration of peptide was measured by Bradford assay (Bio-Rad). The nonfibrillar Aβ peptides were made by dissolving disaggregated peptides at 25 μm in 5 mm Tris buffer, pH 7.4, diluting the peptide to 0.25 μm, and freezing immediately at -20°C in aliquots of 50 μl. For fibrillized peptides, 25 μm peptide solution was incubated at 37°C for 1 hr under continuous mixing in Eppendorf tubes. The samples were vortexed and sonicated twice for 1 min in a bath-type sonicator before freezing at -20°C in 50 μl aliquots. Each aliquot was used once to avoid changes during freeze-thaw cycles. Aβ treatments were performed as described previously (Paradis et al., 1996; Zhang et al., 2002). Neurons were incubated with 100 nm Aβ unless indicated otherwise. Recombinant p75 NTR (R-p75 NTR) (Sigma) was resuspended at 1 μg/ml (11 μm) in PBS containing 0.1% BSA. Recombinant NGF 2.5S was from Boehringer Mannheim (Indianapolis, IN). Recombinant active caspase-6 (PharMingen) was prepared in caspase-6 active buffer containing 20 mm PIPES, 100 mm NaCl, 10 mm DTT, 1 mm EDTA, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid, and 10% sucrose, pH 7.2 (Stennicke and Salvesen, 1997).
Cell cultures. Primary neurons were obtained from fetal brains under ethical approval from McGill Institutional Review Board and cultured as described previously (LeBlanc, 1995; Paradis et al., 1996; LeBlanc et al., 1997). Briefly, fetal brains are dissociated in trypsin (Invitrogen, San Diego, CA) and deoxyribonuclease I (Boehringer Mannheim), filtered through nylon mesh, and plated on poly-lysine-coated tissue culture dishes at a density of 3 × 10 6 cells/ml. Neurons attach within 12-24 hr and rapidly elaborate an intricate neuritic network within 3 d of plating. Neurons are treated with 100 mm deoxyfluorouridine at days 4 and 6 of plating to prevent proliferation of contaminating dividing cell types (<10%). These neurons are used at day 11 of plating.
Immunostaining of cell cultures. Neurons were fixed in 4% paraformaldehyde-4% sucrose for 20 min, permeabilized in 0.25% Triton X-100, blocked with 10% goat serum in 0.1% saponin, and incubated with anti-choline acetyltransferase (ChAT) monoclonal antibody (Chemicon, Temecula, CA) or Promega anti-p75 NTR in goat serum. The primary antibody was revealed with anti-mouse or anti-rabbit conjugated to FITC, and cells were counterstained with propidium iodide or Hoechst to stain the nuclei. Control staining without primary antibody produced no detectable signal.
Viral production and transduction of human primary neuronal cultures. HSV amplicon vectors were packaged and purified as described previously (Bowers et al., 2001). Viral pellets were resuspended in 100 μl of PBS and stored at -80°C until use. Vectors were titered as described previously (Bowers et al., 2000). Transductions were performed at a multiplicity of infection of 0.5.
Microinjections. Neurons were injected as described previously (Zhang et al., 2000; Zhang and LeBlanc, 2002). The injected volume was 25 pl. The cDNAs were purified by UltraClean 15 DNA Purification kit (MoBio Laboratories, Solana Beach, CA) and injected into neurons at 30 ng/μl with 100 μg/ml dextran Texas Red (DTR) (Molecular Probes, Eugene, OR). Two hundred injections (90% survival rate) were performed per experimental condition in each of a minimum of three independent neuron cultures.
HB8737 anti-p75NTR (antibody directed against the extracellular segment of p75NTR) treatments. HB8737 cell (American Type Culture Collection)-conditioned culture media was equilibrated with 1/10 vol of 1 m Tris, pH 8.0, and proteins were precipitated with a saturated solution of ammonium sulfate at 4°C (Sigma). The precipitate was centrifuged at 10,000 rpm on a Sorvall RC-5B centrifuge and resuspended in a total volume of 2.5 ml of 20 mm sodium phosphate, pH 7.0. The solution was desalted through a PD10 column (Amersham Biosciences). Finally, the antibody was purified using a Hi-Trap protein A affinity column (Amersham Biosciences) according to the instructions of the manufacturer. Antibody purity was verified by SDS-PAGE followed by Coomassie staining. Cells were treated for 24 hr on coverslips in MEM without serum in the presence or absence of 0.1 μm Aβ1-42 and 1 μg/ml (14 nm) antibody directed against the extracellular segment of p75 NTR (antip75ec). An antibody directed against the intracellular segment of p75 NTR (anti-p75ic) (Promega) was used at 1 μg/ml as a control. Anti-p75ec was competed with 14 or 28 nm R-p75 NTR. The potential of anti-p75ec to induce cell death in cells microinjected with p75S or p75AS was done by treating the microinjected neurons with 1 μg/ml anti-p75ec for 24 hr.
Cell death assays. Neurons plated on aclar coverslips were treated as described above or as noted in the figure legend, fixed in 4% paraformaldehyde-4% sucrose for 20 min, permeabilized with 0.1% Triton X-100 in 0.75 mm sodium citrate, and stained for apoptotic neurons by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) using the Cell Death kit I as described by the manufacturer (Boehringer Mannheim). The percentage of cell death was calculated as the number of TUNEL-positive cells over the total number of DTR-positive microinjected neurons. For antibody-treated neurons, the number of TUNEL-positive neurons was counted over ∼100 cells in five areas of each coverslip (∼500 cells/sample). The percentage of apoptotic neurons was determined by calculating the number of TUNEL-positive neurons over the total number of neurons visualized by propidium iodide or Hoechst counterstaining. Counts for both assays were done on blinded coverslips.
Treatment of neurons with Aβs and kinase inhibitors. Aβ treatments were performed as described previously (Paradis et al., 1996; Zhang et al., 2002). Neurons were incubated for the indicated time with 100 nm or indicated concentration of Aβs. Treatments with kinase inhibitors were performed with a 1-6 hr preincubation followed by the addition of 100 nm Aβ peptides to freshly diluted inhibitor. Neurons were incubated for 24 hr before performing the cell death assay.
PI3K activity assays. Neurons were collected in lysis buffer (1% Nonidet P-40, 10% glycerol, 137 mm NaCl, 20 mm Tris-HCl, pH 7.4, 1 mm phenylmethylsulfonyl fluoride, 20 mm sodium fluoride, 1 mm sodium pyrophosphate, 1 mm sodium vanadate, and 2 μg/ml aprotinin and leupeptin), centrifuged to remove detergent-insoluble products, immunoprecipitated with anti-total PI3K antisera (Santa Cruz Biotechnology) overnight at 4°C, and 30 μl of the immunoprecipitated Sepharose beads were incubated with 45 μl of kinase buffer (10 mm MgCl2, 50 mm Tris-HCl, pH 7.4), 5 μl of lipid substrate [1 mg/ml phosphatidylinositol/phosphatidyl serine (1:1)], 2 μlof[32P]ATP (10 μCi/μl), and 0.075 μlof cold 100 mm ATP for 20 min at room temperature. The reaction was stopped with 100 μl of 1N HCl. Lipids were extracted with a 1:1 mixture of ChCl3/MeOH, and unreacted ATP was extracted with MeOH-100 mm HCl in 2 mm EDTA. The lipid was separated on a Whatman (Maidstone, UK) 250 μm silica flexible gel thin layer chromatography plate (pre-coated with 5% potassium oxalate in 1.2 mm EDTA and 40% MeOH) with a basic solvent made of CHCl3/MeOH/H2O/NH4OH at a ratio of 45:35:8.5:1.5. The plate was dried and exposed to autoradiography.
Western blot analyses. Proteins were extracted in NP-40 lysis buffer as described previously (LeBlanc, 1995). For analysis of phosphorylated kinases, proteins were extracted with 2× sample buffer made of 0.81 mm Tris, pH 6.8, 16.25% glycerol, 2.0% SDS, quantitated for protein, and then adjusted to 5% mercaptoethanol and 0.25% bromophenol blue before PAGE. Proteins from 6 ml of conditioned media (24 hr serum) were immunoprecipitated with 10% trichloroacetic acid and loaded. Proteins were transferred to Immobilon-P polyvinylidene difluoride membranes. After a 1 hr blocking period in Blotto A (5% low-fat milk in Tris-buffered saline with Tween 20), antibodies were added in Blotto A at the concentration recommended by the manufacturer and incubated for 1 hr at room temperature or 4°C overnight. Development of immunoreactivity was performed with anti-rabbit or anti-mouse HRP-conjugated secondary antibodies and ECL (Amersham Biosciences). The level of immunostaining was measured by densitometric analysis (Molecular Dynamics, Sunnyvale, CA).
Statistical evaluations. One-way or two-way ANOVAs with post hoc tests (Statview 5.01) determined the statistical significance of the difference between treatments. Dunnett's test was used when comparing several groups with the control group (e.g., comparison between different treatment groups vs untreated group). Scheffé's test was applied when comparing between every other group (e.g., comparison between each treatment group). A value of p < 0.05 was taken as the criterion for statistical significance.
Results
Aβ upregulates p75NTR levels in primary cultures of human neurons
Approximately 50% of the human neurons in primary cultures immunostain positively for ChAT (54.2 ± 3%). Anti-p75NTR immunostained 90-95% of neurons in the cultures (data not shown). Western blot analysis for p75NTR reveals a unique band at ∼75 kDa in human neurons and fetal brain, but not in the erythroleukemia cell line K562 protein extracts used as a negative control (Fig. 1A). Aged Aβ1-40 and Aβ1-42, but not Aβ40-1, peptides increase p75NTR levels twofold to threefold in serum (p < 0.05). To eliminate possible effects of serum proteins that could interact with Aβ or possible effects of serum on cellular response, Aβ treatments were also performed in the absence of serum. In serum-free conditions, Aβ1-40 and Aβ1-42 increase the levels of p75NTR by sixfold to eightfold compared with serum-treated neurons (p < 0.05) or threefold to fourfold compared with serum-deprived neurons (p < 0.04) (Fig. 1C). The increase in p75NTR is dose dependent (Fig. 1B), occurs within 3-6 hr, and peaks at 24 hr of treatment (Fig. 1D). In contrast, treatment with Aβ40-1 does not alter the levels of p75NTR in either serum-treated or serum-deprived neurons (Fig. 1C)(p = 0.11). Aβ1-42 did not increase the levels of another member of the tumor necrosis receptor family, TNF-R1, indicating a specific effect on p75NTR (Fig. 1B). Serum deprivation appears to reduce TNF-R1 levels, but the effect is independent of Aβ1-42 treatments. Furthermore, apoptosis-inducer and protein kinase-inhibitor staurosporine decreases rather than increases p75NTR levels (Fig. 1E). These results show that p75NTR levels specifically increase in neurons exposed to extracellular Aβ.

Aβ upregulates p75NTR in primary cultures of human neurons. A, Western blot analysis of p75NTR or β-actin in proteins from untreated neurons (Control) or neurons treated with 100 nm Aβ1-40, Aβ1-42, or reverse peptide control Aβ40-1 for 48 hr and in fetal brain or K562 protein extracts (10 μg/lane). B, Western blot analysis of p75NTR and TNF-R1 with 0.1, 1.0, or 10 μM Aβ1-42 treatments of 48 hr. The point zero represents untreated neurons cultured in serum before the addition of peptide. C, Quantitative analysis of p75NTR levels after 48 hr of treatment measured by densitometric analysis of ECL-Western blots. Data represent the means and SEMs of experiments in three independent neuron cultures. D, Quantitative analysis of p75NTR levels with time of 100 nm Aβ treatments in four independent experiments. For Aβ1-40 orAβ1-42 treatments of ≥3 hr, p < 0.01 compared with untreated control or Aβ40-1. E, Western blot analysis of p75NTR in neurons treated with 0.1 or 10 μm staurosporine in serum-containing conditions.
p75NTR protects human neurons against Aβ
To address the role of p75NTR levels in survival and cell death of neurons exposed to Aβ peptides, we constructed HSV amplicon vectors expressing the human p75 cDNA (p75S for sense) or an 800 bp antisense p75 cDNA (p75AS). Transduction of human primary neurons with the p75S construct shows high expression of p75NTR in neurons 48 hr postinfection (Fig. 2A). p75AS reduces the level of p75NTR slightly (Fig. 2B). Also, treatment with Aβ1-42 at 100 nm concentration did not increase the levels of p75NTR in p75AS-transduced neurons, indicating that the p75AS was functional. To confirm the antisense function of p75AS, cultures were cotransduced with p75S and p75AS constructs. The results show that p75AS effectively inhibits expression from the p75S construct (Fig. 2C). p75AS had no effect on the expression of eGFP (enhanced green fluorescent protein) from another pHSV construct (results not shown). Therefore, these results indicate that newly synthesized p75NTR expression is inhibited by p75AS in the presence of p75S transduction or Aβ.

p75NTR protects human neurons against extracellular Aβ1-42 toxicity. Western blot analysis of p75NTR in two independent experiments of pHSVPrPUCp75 sense (p75S)-infected primary neurons (A) or pHSVPrPUCp75 antisense (p75AS)-infected primary neurons in the presence and absence of 100 nm Aβ1-42 (B). BE, Proteins from a brain extract. Ctl, Control. C, Interference of p75S-mediated expression of p75NTR by cotransduction of neuron cultures with p75AS shown by Western blotting of the transduced cultures with anti-p75NTR and β-actin. D, TUNEL-positive cell death in neurons microinjected with DTR, pHSVPrPUC empty construct (vector), p75S, p75AS, or PrPAS, and either untreated (Ctl) or treated with 100 nm Aβ1-42 or Aβ42-1 in the presence of serum. Data represent the mean and SEM of three independent experiments. The difference between microinjection of p75S, p75AS, and PrPAS versus vector microinjections was assessed by one-way ANOVA followed by Scheffé's test; *p < 0.0001. There was no other statistically significant difference between the different groups.
Unfortunately, we are unable to assess cell death with high levels of confidence in these transduced cells because of variability in transduction rates (our unpublished results) and the formation of a nonspecific deposit on neurons (likely derived from the amplicon-packaging cell line debris) that may affect the ability of the TUNEL reagents to reach the nuclei. To specifically address the role of p75NTR in cell death, we microinjected the neurons with amplicon vectors as described previously (Zhang et al., 2000, 2002; Bounhar et al., 2001), and assessed the effect of p75NTR on the resistance of neurons to extracellular Aβ1-42 toxicity. Because the increase in p75NTR was higher with Aβ1-42, all of the additional treatments were done with 100 nm Aβ1-42. In the presence or absence of serum, neurons injected with the fluorescent red dye marker DTR, the empty pHSVPrPUC vector (vector), or the p75S construct remain resistant to extracellular Aβ (Fig. 2D). Unexpectedly, in the presence of serum, microinjection of p75AS sensitizes human neurons to 100 nm extracellular Aβ1-42 toxicity, resulting in 75% cell death within 24 hr of treatment. Unlike the p75AS construct, another known neuroprotective protein antisense construct, prion protein (PrPAS) (Bounhar et al., 2001), did not alter the resistance of neurons to extracellular Aβ1-42 toxicity. Similar results were obtained in the absence of serum (data not shown).
A higher concentration of 10 μm Aβ1-42 is not neurotoxic to p75S-microinjected neurons (Fig. 3A), in contrast to the observed toxicity of Aβ in p75NTR-transfected neuroblastoma cells lines (Rabizadeh et al., 1994; Yaar et al., 1997, 2002; Kuner et al., 1998; Perini et al., 2002). Similarly, 10 μm Aβ1-42 does not exert a higher toxicity than the 100 nm Aβ1-42 concentration on the p75AS-microinjected neurons. Fibrillized Aβ1-42 (Aβ1-42f) and nonfibrillized Aβ1-42 (Aβ1-42nf) peptides are equally cytotoxic to the p75AS-microinjected neurons, whereas Aβ42-1f or Aβ42-1nf peptides are not toxic (Fig. 3B). The oligomeric nature of the nonfibrillized peptides was demonstrated previously (Zhang et al., 2002). Together, these results indicate that, in contrast to increased toxicity of extracellular Aβ in p75NTR-transfected neuronal cell lines (Rabizadeh et al., 1994; Perini et al., 2002), the p75NTR protects primary human neurons against Aβ toxicity.

Impact of Aβ concentration, fibrillar state, and pro-NGF on neuronal cell death. A, TUNEL-positive cell death of 100 nm versus 10 μm Aβ1-42 in vector-, p75S-, or p75AS-microinjected neurons. B, Extracellular toxicity of fibrillized and nonfibrillized Aβ1-42 and Aβ42-1 on p75S- or p75AS-microinjected neurons. One-way ANOVA followed by Scheffé's test compares p75S and p75AS with control (Ctl); *p < 0.0001. C, Western blot analysis of NGF with anti-NGF H20 in protein extracts from 24 hr serum-deprived (-S) or serum-treated (+S) neurons (50 μg/lane), adult or fetal brain (100 μg/lane), and conditioned media (CM) from serum-deprived neurons and serum. Mature recombinant NGF (R-NGF) was added as a control (100 ng/lane). Data in A and B represent the mean and SEM of three independent experiments.
Because pro-NGF binds with high affinity to p75NTR and can induce the cell death pathway in smooth muscle cells and oligodendrocytes (Lee et al., 2001; Beattie et al., 2002), we assessed levels of NGF in neuronal extracts, conditioned media, and the serum added to complete neuronal media (Fig. 3C). Western blot analysis fails to detect mature NGF in any of these samples but detects 34 kDa pre-pro-NGF in neuronal extracts, adult brains, and fetal brains. Pro-NGF (28 kDa) is present in the neuron-conditioned medium and in adult brain, confirming previous studies (Fahnestock et al., 2001). Therefore, these human neurons survive despite the presence of pro-NGF, Aβ, and p75NTR.
To further confirm the role of p75NTR against Aβ toxicity, neurons were incubated with the HB8737 monoclonal antibody to the extracellular domain of primate p75NTR (anti-p75ec). This p75ec antibody in the absence of Aβ has no effect on neuronal cell death in untreated, p75S-microinjected, or p75AS-microinjected neurons (Fig. 4A). However, anti-p75ec, but not antibodies to the intracellular domain of p75 (anti-p75ic), sensitizes neurons to extracellular Aβ1-42 (Fig. 4B). Addition of recombinant protein from the extracellular domain of human p75NTR to the anti-p75ec effectively competes out Aβ1-42 toxicity (Fig. 4C). The anti-p75ec effect is specific to the Aβ1-42-mediated toxicity, because neither sublethal doses of H2O2 nor etoposide (Paradis et al., 1996) induce toxicity in the presence of the anti-p75ec (Fig. 4D). These results confirm that p75NTR protects human neurons against Aβ1-42 toxicity.
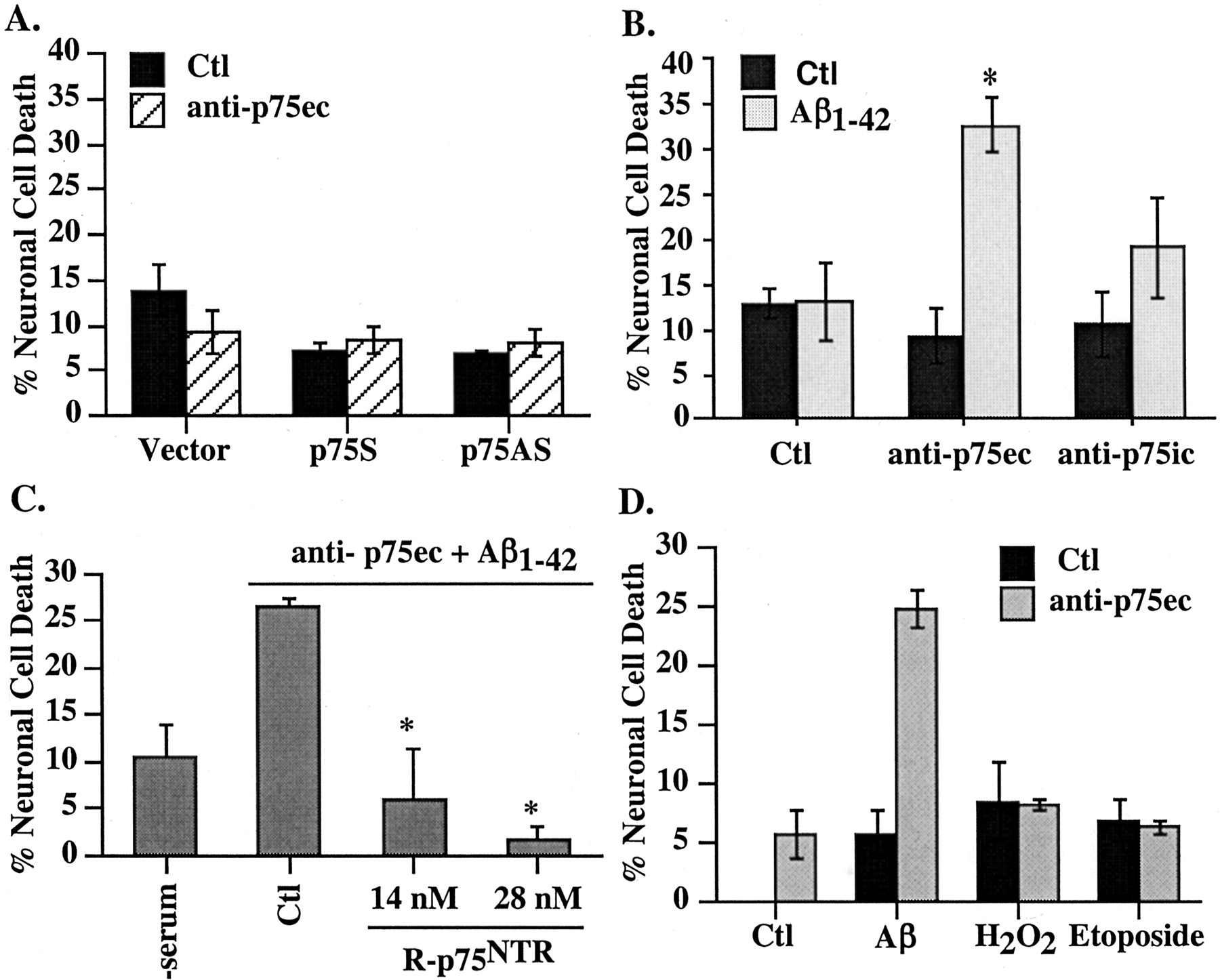
Anti-extracellular p75 antibody specifically induces neuronal cell death in the presence of Aβ. A, TUNEL-positive cell death in vector-, p75S-, or p75AS-microinjected neurons treated for 24 hr with anti-p75ec. B, TUNEL-positive cell death in serum-deprived neurons treated with anti-p75ec or with anti-p75ic in the absence [control (Ctl)] or presence of 100 nm extracellular Aβ1-42. The difference between Aβ1-42 alone versus p75 antibodies plus Aβ1-42 was assessed by one-way ANOVA followed by Scheffé's test; *p < 0.003. The increase in cell death with anti-p75ic is not significantly different from control. C, Competition of anti-p75ec-induced Aβ toxicity by R-p75NTR; *p < 0.02 compared with Ctl (one-way ANOVA). D, TUNEL-positive cell death in neurons treated for 24 hr with 100 nm Aβ1-42, 0.1 μm H2O2, or 20 μm etoposide in the absence or presence of 1 μg/ml anti-p75ec. Data represent mean and SD.
Resistance of human neurons to extracellular Aβ is PI3K dependent but Akt independent
The PI3K-Akt survival pathway has been implicated in various neurotrophin-mediated neuroprotection including p75NTR (Roux et al., 2001). Preincubation of neurons with 200 nm or 10 μm the PI3K inhibitor wortmannin for 1 hr, followed by exposure to Aβ and wortmannin, sensitizes human neurons to 100 nm extracellular Aβ1-42 (Fig. 5A). Similar results were obtained with LY294002, another PI3K inhibitor (results not shown). Control reverse peptide Aβ42-1 is not toxic to wortmannin-treated cells. As expected, wortmannin decreases phospho-PI3K relative to total PI3K levels (Fig. 5B). In addition, wortmannin considerably inhibits the activity of PI3K (Fig. 5C). Aβ1-42 alone reduces both the levels and activity of PI3K.

Wortmannin sensitizes neurons to Aβ1-42 toxicity. A, TUNEL-positive neuronal cell death in neurons pretreated 1 hr with 200 nm or 10 μm wortmannin and exposed to 100 nm extracellular Aβ1-42 in the continued presence of wortmannin for 24 hr. Data represent the mean and SEM of three independent experiments. One-way ANOVA followed by Scheffé's compared wortmannin-treated cells with untreated cells; *p < 0.001. B, Western blot analysis of total PI3K in neurons treated with Aβ1-42 and wortmannin. C, Autoradiogram of phosphorylated phosphatidylinositol (PIP) from immunoprecipitated PI3K. The concentration of wortmannin was 200 nm. Wtm, Wortmannin; Ctl, control; ORI, origin.
Resistance of human neurons to extracellular Aβ is Akt independent
The requirement of PI3K for neuronal survival in the presence of extracellular Aβ raises the possibility that Akt phosphorylation through PI3K activation is responsible for p75NTR-mediated neuroprotection against extracellular Aβ1-42. Western blot analysis of the phospho-Akt473 epitope shows the expected inhibition of active Akt phosphorylation at amino acid 473 (Fig. 6A). However, microinjection of either wild-type or constitutively active Akt (Akt-active) constructs in the neurons before treatment with wortmannin and Aβ1-42 does not protect against Aβ1-42 toxicity (Fig. 6B). An Akt DN form also has no effect. However, the Akt constructs are functional in these neurons, because the Akt WT and the Akt active partially or completely inhibit the neuronal cell death induced by a 96 hr serum deprivation (Fig. 6B). The Akt DN enhances cell death by serum deprivation, although this is more detectable 48 hr after serum deprivation (32% cell death in Akt-DN-injected neurons compared with 12% in control cells). Furthermore, anti-p75ec highly stimulates PI3K-dependent Akt phosphorylation at amino acids 308 and 473 despite rendering neurons susceptible to Aβ1-42 toxicity (Fig. 6C,D). These results indicate that the p75NTR protection against extracellular Aβ1-42 does not involve the Akt survival pathway. In addition, the results show that activation of Akt is not sufficient to protect against extracellular Aβ toxicity.

p75NTR does not protect human neurons through Akt activation. A, Western blot analyses with phospho-Akt (pAkt) 473, total Akt, or β-actin in neurons treated with 10 μm wortmannin for 6 hr. B, TUNEL-positive cell death in neurons pretreated 1 hr with 10 μm wortmannin and microinjected with wild-type Akt (Akt WT), constitutively active Akt (Akt active), or dominant-negative Akt (Akt DN) before a 24 hr treatment with extracellular Aβ1-42 and wortmannin. Controls are serum deprived [-serum (-S)] for 96 hr after microinjection with the Akt constructs. Data represent the mean and SEM of three independent experiments. C, Western blot analyses of phospho-Akt (P-Akt) 473 or 308, total Akt, and β-actin in proteins from neurons treated with p75ic or p75ec antibodies in the absence or presence of 100 nm Aβ1-42. D, Western blot of phospho-Akt (P-Akt) 308 in proteins from neurons treated with anti-p75ec in the presence of 200 nm or 10 μm wortmannin. Wtm, Wortmannin; Ctl, control.
Toxicity of extracellular Aβ is p53-Bax independent but inhibited by caspase inhibitors and lithium chloride
We subsequently investigated the involvement of p53 in extracellular Aβ1-42 cytotoxicity, because intracellular Aβ1-42 toxicity is mediated through the p53-Bax cell death pathway in the human primary cultures (Zhang et al., 2002). Microinjection of a cDNA expressing either wild-type p53 (p53WT) or a p53 dominant negative (p53DN) with either p75AS or followed by wortmannin treatment does not alter the toxicity of extracellular Aβ1-42 (Fig. 7A) despite confirmation that the p53DN can inhibit intracellular Aβ1-42 toxicity (intracellular Aβ1-42 yields 51% cell death vs 9.9% cell death in the presence of p53DN). Similarly, three neutralizing antibodies to Bax, N-20, 6A7, and 2D2, are unable to prevent extracellular Aβ1-42-mediated toxicity in the presence of wortmannin, although they completely inhibit toxicity mediated by microinjected Bax cDNA expression (Fig. 7B). Therefore, Bax is unlikely to be involved in extracellular Aβ1-42 toxicity in the human neurons. However, cell death is caspase dependent, as shown by the inhibition of toxicity with the pan-caspase inhibitor Boc-D-fmk (Fig. 7C). Furthermore, glycogen synthase kinase 3β (GSK3β) activation may be involved for cell death, because lithium chloride (LiCl2), which can inhibit GSK3β, prevents extracellular Aβ1-42 toxicity (Fig. 7D). These results suggest that extracellular Aβ1-42 toxicity is mediated through a p53-Bax-independent but caspase-dependent pathway that may require GSK3β activation.

Pathways of extracellular Aβ1-42-induced toxicity. A, Cell death in neurons microinjected with p53WT or p53DN comicroinjected with p75AS or treated with wortmannin in addition to a 100 nm extracellular Aβ1-42 treatment. There was no significant difference with p53 microinjections compared with the absence of p53. B, Cell death in cells microinjected with Bax-neutralizing antibodies N20, 2D2, or 6A7 comicroinjected with Bax cDNA or treated with 100 nm extracellular Aβ1-42 and 200 nm wortmannin. The APP or N20 treatments were compared with the rabbit sera (Rb-sera), and 2D2 and 6A7 were compared with mouse IgG (Mo-IgG) by one-way ANOVA followed by Scheffé's test; *p < 0.05; **p < 0.005. C, Neuronal cell death in neurons microinjected with p75AS and treated with 100 nm Aβ1-42 or treated with wortmannin and Aβ1-42 in the absence or presence of 5 μm Boc-D-fmk caspase inhibitor. Recombinant active caspase-6 (R-Csp-6) was microinjected as a positive control for the Boc-D activity. The difference between the absence or presence of Boc-D-fmk was assessed by one-way ANOVA followed by Scheffé's test; *p < 0.0001. Data represent the means and SEMs of three independent experiments. D, Cell death from 100 nm extracellular Aβ1-42 with either p75AS or wortmannin in the absence or presence of 20 mm LiCl2. Data represent the mean and SEM of three independent experiments. The difference between the absence or presence of LiCl2 was assessed by one-way ANOVA and Scheffé's test; *p < 0.0001. Ctl, Control; Wtm, wortmannin.
Discussion
In the present study, we show that p75NTR protects human primary neurons in culture against extracellular Aβ1-42-mediated apoptosis. Human primary neurons naturally resist 0.1-10 μm extracellular Aβ1-42 (Mattson et al., 1992; Zhang et al., 2002). Here, we found that 100 nm Aβ treatments increase the levels of p75NTR. An increase in p75NTR has been observed in brain or neurons after seizures (Roux et al., 1999), zinc toxicity (Park et al., 2000), in axotomized corticospinal neurons (Giehl et al., 2001), and mechanical injury of the spinal cord (Widenfalk et al., 2001). In these situations, the increase of p75NTR is associated with apoptosis. Because Aβ1-42 does not induce cell death in human neurons but sensitizes neurons to cell death in the presence of low levels of oxidative stress (Paradis et al., 1996), we assumed that the increase of p75NTR would mediate cell death. Unexpectedly, two independent types of experiments suggest that p75NTR protects against rather than induces Aβ-mediated cell death. First, inhibiting the Aβ-mediated increase in p75NTR in human neurons with antisense constructs sensitizes the neurons to 100 nm Aβ. Second, an antibody to the extracellular domain of p75NTR also sensitizes neurons to Aβ-mediated toxicity. In contrast, neurons overexpressing p75NTR resist extracellular Aβ toxicity with fibrillized Aβ or soluble Aβ even with higher 10 μm concentrations that are 2500 times higher than the concentration of 4 nm found in the CSF of early-onset Alzheimer patients (Nakamura et al., 1994).
These results contrast with strong evidence that p75NTR increases the susceptibility of PC12, SK-N-BE, NIH3T3, and SK-N-MC cell lines to extracellular Aβ toxicity (Rabizadeh et al., 1994; Yaar et al., 1997, 2002; Kuner et al., 1998; Perini et al., 2002). The reason for the difference in our results may be explained by differential activation of signal transduction pathways in primary neurons versus tumor cell lines, cell-type or species-specific effects of Aβ, or differential expression of the other neurotrophic receptors. It is not the first time that these human neurons react unexpectedly in cell survival or cell death. For example, these neurons undergo apoptosis through caspase-6, and not the usual caspase-3 (LeBlanc et al., 1999). We assume that signal transduction pathways relating to survival and cell death may be highly regulated to account for the extensive life span of human neurons. However, it is important to consider that, in several other situations, p75NTR is neuroprotective. In PC12 cells expressing both TrkA and p75NTR, p75NTR mediates NGF-dependent survival through Akt activation (Roux et al., 2001; Bui et al., 2002). Anti-p75 antibodies and antisense cDNA block this pathway and result in cell death (Bui et al., 2002). Here, it is unlikely that TrkA is involved, because the neuroprotection occurs independently of the Akt pathway known to be linked to TrkA activation (Miller and Kaplan, 2001). p75NTR also has been shown to promote survival through receptor-interacting protein 2 (Khursigara et al., 2001).
The neuroprotective role of p75NTR in these human neurons is supported by in vivo studies. p75NTR immunoreactivity is decreased in the vulnerable basal forebrain cholinergic neurons of the NBM of AD patients compared with normal individuals (Salehi et al., 2000; Mufson et al., 2002). Furthermore, cognitive impairment is inversely proportional to the level of p75NTR in mild cognitive impairment (MCI) and in early AD individuals suggesting that the lack of p75NTR predisposes to AD (Mufson et al., 2002). However, in the temporal cortex of late AD individuals, p75NTR immunoreactivity is increased (Mufson and Kordower, 1992). Similarly, p75NTR protein levels increase in APP Swedish or PS1 mutant transgenic mice with severe extracellular Aβ deposits, and these mice fail to display neuronal loss (Jaffar et al., 2001). The increase of p75NTR could be triggered by the accumulation of extracellular Aβ similar to the Aβ-mediated increase of p75NTR observed in human neuronal cultures. Therefore, it would be predicted on the basis of our results that these neurons would have enhanced protection against Aβ. Thus, one explanation for these observations is that the lack of p75NTR in NBM leads to MCI, early neurodegeneration, and AD, whereas neurons expressing p75NTR have an extended life span in normal individuals, and mildly or later affected regions of AD brains.
We show that neuroprotection against Aβ occurs through a PI3K-dependent pathway, because both wortmannin and LY294002 render the human neurons sensitive to extracellular Aβ. However, the well known PI3K-activated neuronal survival factor Akt is not involved in this neuroprotective function of p75NTR, because phosphorylation of the Akt 473 and 308 sites indicative of an active state of the Akt is induced by the HB8737 p75ec antibody that blocks the resistance of neurons to Aβ toxicity. Furthermore, constitutively active and wild-type Akt cannot prevent Aβ toxicity, and the Akt dominant-negative form does not enhance Aβ toxicity despite the ability of these wild-type and constitutively active Akt to prevent human neurons against serum deprivation. The lack of a role for Akt in p75NTR-mediated survival is unexpected, given that PI3K activity is required. However, the results are not entirely unexpected, because human neurons that are terminally differentiated early and have a long life span may be expected to have several other mechanisms to ensure long-term survival. Therefore, these results suggest a PI3K-dependent, but Akt-independent, neuroprotective pathway. Two other survival pathways have been linked to p75NTR: neurotrophin-mediated survival through ceramide in subplate neurons (DeFreitas et al., 2001) and NGF-activated survival through RIP2-TRAF (TNF-R-associated factor) in Schwann cells (Khursigara et al., 2001). Additional work will be needed to determine which PI3K downstream survival pathway is responsible for p75NTR-mediated neuroprotection against extracellular Aβ.
The pathways mediating Aβ toxicity were also examined. In contrast to the toxicity of intracellular Aβ toxicity (Zhang et al., 2002), neither p53 dominant-negative forms nor Bax-neutralizing antibodies inhibit extracellular Aβ-mediated toxicity. These results indicate that extracellular Aβ-mediated toxicity in human neurons is independent of the p53-Bax proapoptotic pathway. However, inhibition of cell death with LiCl2, an inhibitor of GSK3β (Klein and Melton, 1996), suggests that GSK3β could be involved in the extracellular Aβ-mediated prodeath pathway. Lithium protects PC12 cells, cerebellar granule neurons, and rat cortical neuronal cultures against extracellular Aβ toxicity (Alvarez et al., 1999; Wei et al., 2000). GSK3β also mediates Aβ toxicity in primary rodent neuronal cultures (Takashima et al., 1993). Given that Akt is a strong inhibitor of GSK3β (Pap et al., 1998), it is surprising that, in our cultures treated with the p75NTR antibody, the active Akt is unable to prevent Aβ-mediated toxicity (Pap et al., 1998). The regulation of GSK3β is quite complex (for review, see Grimes and Jope, 2001), and it is presently unclear how it is activated by extracellular Aβ in human neurons. Akt is a well known inactivator of GSK3β through Ser9 phosphorylation. However, after initial dephosphorylation by phosphatase 2A, GSK3β can be activated by phosphorylation at Tyr216 via calcium or Fyn signal transduction. Both calcium and Fyn have been implicated in extracellular Aβ toxicity in other systems (Mattson et al., 1992; Zhang et al., 1994; Williamson et al., 2002). Interestingly, GSK3β phosphorylates tau and has been implicated in amyloid- and non-amyloid-induced neurodegeneration in vivo and in vitro, thereby linking two important AD pathological hallmarks (Takashima et al., 1993; Lucas et al., 2001). GSK3β activation also induces caspase activation in other systems, consistent with our result that extracellular Aβ toxicity is dependent on caspase activity (Mora et al., 2002). However, more work is needed to fully identify a role for GSK3β in extracellular Aβ toxicity of human neurons.
In summary, we showed that p75NTR protects human neurons against extracellular Aβ toxicity. These results contrast with observations that show that p75NTR promotes Aβ toxicity in neuroblastoma cell lines. However, the results agree with in vivo observations that indicate a neuroprotective role for p75NTR against cognitive impairment. On the basis of our results, we propose the alternate hypothesis that p75NTR expression can be neuroprotective against extracellular Aβ-mediated toxicity in human neurons. The neuroprotective role of p75NTR may be exploited to develop therapies that would protect neurons in AD patients against extracellular Aβ-mediated toxicity.
Footnotes
This work was supported by the Alzheimer Society of Canada, National Institutes of Health National Institute of Neurological Disorders and Stroke (RO1 NS31700), Canadian Institute for Health Research (MOP-15118), the Fonds de Recherche en Santé du Québec (A.L.B.), and the Rochester Nathan Shock Center for Excellence (H.J.F.). We gratefully acknowledge assistance on statistical evaluations from the Consultation Service (Center for Clinical Epidemiology and Community Studies, Lady Davis Institute, Jewish General Hospital). We thank Beverly Akerman, Jennifer Hammond, Hala Lahlou, and Ann Casey for technical assistance. We thank Dr. Uri Saragovi for helpful discussion and Neurochem, Inc., for the preparation of the amyloid peptides.
Correspondence should be addressed to Dr. Andréa LeBlanc, The Bloomfield Center for Research in Aging, Lady Davis Institute for Medical Research, Sir Mortimer B. Davis Jewish General Hospital, 3755 ch. Côte Sainte-Catherine, Montreal, Quebec, Canada H3T 1E2. E-mail: andrea.leblanc{at}mcgill.ca.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237385-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}