

Abstract
Cerebral amyloid angiopathy (CAA) is a common cause of brain hemorrhage in the elderly. It is found in the majority of patients with Alzheimer's disease (AD). The most common form of CAA is characterized by the deposition of the amyloid-β (Aβ) peptide in the walls of cerebral vessels, and this deposition can lead to hemorrhage and infarction. As in AD, the ϵ4 allele of apolipoprotein E (APOE) is a risk factor for CAA. To determine the effect of apoE on CAA and associated hemorrhage in vivo, we used two amyloid precursor protein (APP) transgenic mouse models that develop age-dependent Aβ deposition: PDAPP and APPsw mice. We found that both models developed an age-dependent increase in CAA and associated microhemorrhage, with the APPsw model having an earlier and more severe phenotype; however, when APPsw and PDAPP mice were bred onto an Apoe-/- background, no CAA was detected through 24 months of age, and there was little to no evidence of microhemorrhage. Biochemical analysis of isolated cerebral vessels from both PDAPP and APPsw mice with CAA revealed that, as in human CAA, the ratio of Aβ 40:42 was elevated relative to brain parenchyma. In contrast, the ratio of Aβ 40:42 from cerebral vessels isolated from old PDAPP, Apoe-/- mice was extremely low. These findings demonstrate that murine apoE markedly promotes the formation of CAA and associated vessel damage and that the effect of apoE combined with the level of Aβ40 or the ratio of Aβ 40:42 facilitates this process.
- Alzheimer's disease
- apolipoprotein E
- cerebral amyloid angiopathy
- amyloid β
- ratio
- hemorrhage
- transgenic models
Introduction
Cerebral amyloid angiopathy (CAA) consists of deposition of amyloid in brain arterioles, capillaries, and leptomeningeal vessels. The most common form of CAA results from deposition of the amyloid-β (Aβ) peptide in the walls of cerebral vessels, gradually replacing the smooth muscle cell layer (Vinters, 1987). The vast majority of patients diagnosed with Alzheimer's disease (AD) also have CAA. A major consequence of CAA is fatal lobar cerebral hemorrhage, and it also appears to play a role in ischemic brain lesions and leukoariaosis (Greenberg, 1998; Revesz et al., 2002).
The Aβ peptide is 38-43 amino acids in length and is derived from proteolytic processing of a longer precursor protein termed the amyloid-β precursor protein (APP). The predominant Aβ peptide present in CAA is Aβ40, whereas in brain parenchymal plaques it is Aβ42 (Joachim et al., 1988; Prelli et al., 1988; Suzuki et al., 1994; Alonzo et al., 1998; McCarron et al., 2000). Several transgenic mice have been created using APP constructs containing familial AD mutations that recapitulate many aspects of the amyloid-related pathology of AD (Hock and Lamb, 2001). Many of these models have been shown to have both diffuse and neuritic plaques in brain parenchyma, and a few have been shown to develop CAA (Calhoun et al., 1999; Holtzman et al., 2000a; Van Dorpe et al., 2000).
The role of apolipoprotein E (apoE) in the genetics and pathogenesis of AD has been well established (Strittmatter and Roses, 1995; Wisniewski et al., 1997). As with AD, the ϵ4 allele of APOE is a risk factor for developing CAA (Schmechel et al., 1993; Greenberg et al., 1995; Nicoll et al., 1997), whereas the ϵ2 allele is a risk factor for developing hemorrhage associated with CAA (Nicoll et al., 1996, 1997; Greenberg et al., 1998). Previous studies using APP transgenic mouse models of AD (in particular the PDAPP mouse) have shown that the absence of murine apoE does not result in a delay in the onset of Aβ deposition (Holtzman et al., 1999), but it does result in a decrease in the level of Aβ deposition and a marked delay in the onset of fibrillar Aβ deposits (amyloid) (Bales et al., 1997) as well as CAA up to 12 months of age (Holtzman et al., 2000a).
To date, the effects of apoE on CAA and its consequences have not been well studied. Herein, we examine the extent and effects of apoE on CAA in both APPsw (Tg2576) and PDAPP mice through 24 months of age, two different transgenic models with AD pathology. We found that CAA occurs earlier and to a much greater extent in APPsw than PDAPP mice but that both develop CAA-associated microhemorrhages. In the absence of apoE, however, CAA and CAA-associated microhemorrhages are markedly reduced even when assessed at a very old age. Finally, as in human CAA, the ratio of Aβ 40:42 is elevated relative to brain parenchyma and also reduced in the absence of apoE. Our findings demonstrate a critically important contribution of apoE to CAA pathogenesis.
Materials and Methods
Animals and tissue preparation. The production, genotyping, and background strain (B6/SJL) of APPsw (Tg2576) and APPsw, Apoe-/- mice ages 12-18 months used in this study have been described previously (Hsiao et al., 1996; Holtzman et al., 2000a). APPsw mice overexpress human APP695 with the familial Swedish AD mutations at positions 670/671 under control of the prion promoter and were a generous gift from Dr. K. Ashe (University of Minnesota, Minneapolis, MN). The production, genotyping, and background strains of PDAPP and PDAPP, Apoe-/- mice ages 15-24 months used in this study have been described previously (Games et al., 1995; Bales et al., 1997; Holtzman et al., 1999). PDAPP mice overexpress human APP751 with the familial AD mutation at position 717 (APPV717F) under control of the neuronal-specific platelet-derived growth factor promoter. Animals were anesthetized with pentobarbital (150 mg/kg, i.p.) and perfused transcardially with 0.1 m PBS containing heparin (3 U/ml), pH 7.4. One hemibrain was immersion-fixed in PBS containing 4% paraformaldehyde overnight at 4°C. After fixation, the brain was cryoprotected in PBS containing 30% sucrose at 4°C. All experimental protocols were approved by the animal studies committee at Washington University.
Histological analysis. Coronal sections (50 μm) were cut on a freezing-sliding microtome and mounted on Superfrost Plus slides (Fisher Scientific, Houston, TX) and permeabilized with PBS containing 0.25% Triton X-100 (PBS-X) for 30 min at room temperature (RT). Every sixth section from the genu of the corpus callosum to the caudal end of the hippocampus (20-25 sections per animal) was examined. To assess for evidence of previous microhemorrhage, the Prussian Blue stain was performed as described (Gomori, 1936; Winkler et al., 2001). The Prussian Blue reagent stains microglia that have engulfed ferric iron-containing hemosiderin from red blood cells, indicating a previous hemorrhage. Briefly, sections were washed twice quickly in deionized water and incubated in 2% HCl containing 2% potassium ferrocyanide for 20 min. Slides were rinsed three times in PBS, coverslipped with 70% glycerol, and examined with a 10× objective for blue puncta. Microhemorrhage was defined as having at least two blue puncta surrounding a cerebral vessel. To confirm that microhemorrhage was associated with CAA (as shown in Fig. 1), the sections were costained with thioflavine-S as described previously (Bales et al., 1997). Prussian blue stain was always imaged first because of extremely rapid dissolution of the blue precipitate after UV excitation required for thioflavine-S imaging. For triple-label immunohistochemistry, sections were blocked with 2% dry milk-PBS-X for 1 hr at RT. Sections were then incubated with rabbit anti-mouse apoE sera (generous gift from Dr. R. Pitas, Gladstone Institute, University of California San Francisco) at 1:500 dilution in 1% dry milk-PBS-X overnight at 4°C. Sections were washed three times with PBS-X and incubated with goat anti-rabbit conjugated with Alexa-568 (Molecular Probes, Eugene, OR) in 1% milk-PBS for 1 hr at RT. Sections were washed three times with PBS-X and then incubated with a monoclonal antibody, m3D6 (Johnson-Wood et al., 1997) directed against the Aβ peptide (amino acid residues 1-5) conjugated with Alexa-488 in 1% dry milk-PBS. Sections were washed three times in PBS-X and then stained for fibrillar amyloid with the Congo red derivative X-34 dye (generous gift from Dr. W. Klunk, University of Pittsburgh, Pittsburgh, PA). This dye has the advantage of narrow emission spectra compared with thioflavine-S, which has fluorescent emission well into the 488 range (Styren et al., 2000). Fluorescein-conjugated monoclonal antibody against smooth muscle actin (Accurate Chemical, Westbury, NY) was used with Alexa 488 tyramide signal amplification using anti-fluorescein-peroxidase (Molecular Probes) and imaged on a Zeiss LSM 510 META laser scanning confocal microscope (Carl Zeiss, Jena, Germany).

CAA is present in both the APPsw and PDAPP mouse models of AD. Thioflavine-S staining of sections from a 12-month-old APPsw mouse (A) and a 24-month-old PDAPP mouse (B) demonstrating amyloid in cerebral vessels (arrows). Although the PDAPP model does develop CAA, the pathology is less extensive at equivalent ages when compared with the APPsw model. Also shown are parenchymal plaques (arrowheads). C, Two-photon image of CAA vessel from an 18-month-old APPsw mouse immunostained for smooth muscle actin (green, arrowhead) surrounded by amyloid (blue, arrow). D, Isolated cerebral vessels from a 27-month-old PDAPP mouse stained with thioflavine-S demonstrating presence of amyloid (arrows) in vessels. Scale bar: A, B, 100 μm.
Quantitation of CAA and Aβ load. The percentage of cross-sectional area covered by CAA vessels (percentage CAA load) as defined by thioflavine-S-positive vessels was quantified using unbiased stereological principles. CAA load was determined in the cortex and overlying leptomeningeal vessels immediately dorsal to the hippocampus in three sections, each separated by 300 μm. StereoInvestigator image analysis software (MicroBrightField, Williston, VT) was used to quantify percentage CAA load using the Cavalieri point counting method (Cavalieri, 1966). Percentage Aβ load in hippocampus was quantified using stereological techniques as described previously (DeMattos et al., 2002).
Isolation of cerebral vessels and parenchymal tissue. The isolation of cerebral vessels for biochemical analysis was performed essentially as described (Zlokovic et al., 1993) with minor modifications. Briefly, animals were anesthetized and perfused transcardially with 0.1 m PBS with heparin (3 U/ml). The brain was removed gently and placed in ice-cold vessel buffer consisting of HBSS (Invitrogen, Carlsbad, CA) containing 15 mm HEPES, 1 mm pyruvate, 25 mm glucose, 25 mm NaHCO3, 0.1% BSA, and 1% dextran (∼64,000 mol weight). After the cerebellum was removed, the brain was homogenized in a loose-fitting glass Dounce homogenizer in fivefold excess of vessel buffer. An equal volume of 26% dextran was added for a final concentration of 13.5%, and the tissue was immediately centrifuged at 6200 × g in a Beckman SW40 Ti ultracentrifuge swinging bucket rotor for 30 min at 4°C. Using this protocol, the vessels form a pellet, whereas the parenchymal “vessel-free” material forms a solid, compact disc at the top of the solution with little protein in the clear interface between the vessel pellet and the parenchymal disc. This parenchymal material was gently aspirated with a transfer pipette, collected in a conical tube with 50 ml of ice-cold PBS, and centrifuged at 2000 × g to pellet the material. The parenchymal pellet was washed once with vessel buffer (minus dextran and BSA). The vessel pellet was resuspended in 10 ml of vessel buffer (minus dextran) and passed over a 40 μm nylon mesh (Fisher Scientific) to capture vessels. Vessels were washed extensively with vessel buffer (minus dextran) and were recovered by inverting the mesh and collecting into a 50 ml conical tube with a stream of vessel buffer (minus dextran). Vessels were spun in a tabletop centrifuge at 2000 × g and washed once with 30 ml of vessel buffer (minus dextran). Purity of similarly prepared samples was verified by light microscopy. Vessel and parenchymal material were lysed in 5 m guanidine, 50 mm Tris, pH 8, with protease inhibitor mixture (Roche, Indianapolis, IN) and 1 mm PMSF (Sigma, St. Louis, MO) rotating for 3 hr at RT.
Acid-urea gel and ELISA. Denaturing acid-urea PAGE followed by immunoblotting was used to identify forms of Aβ in tissue lysates as described previously (DeMattos et al., 2002). Aβ40 and Aβ42 were quantified by ELISA as described previously (DeMattos et al., 2001).
Statistical analysis. Because CAA load and microhemorrhage data were not distributed normally, Mann-Whitney two-tailed t test was used to compare APPsw, Apoe+/+ with APPsw, Apoe -/- mice or PDAPP, Apoe+/+ with PDAPP, Apoe -/- mice at the same age in regard to percentage CAA load or microhemorrhage. To compare Aβ levels by ELISA and Aβ deposition between PDAPP and APPsw mice of the same age, a two-tailed t test was used. For statistical analyses, Prism version 3.00 software was used (GraphPad, San Diego, CA).
Results
Age-dependent CAA and associated microhemorrhage occur in APPsw and PDAPP mice
Of the numerous APP transgenic mice that have been described that develop Aβ deposits in the brain, spontaneous intracerebral microhemorrhage in association with CAA has been reported previously in only one type of these APP transgenic models (Winkler et al., 2001, 2002; Pfeifer et al., 2002). We first set out to determine whether other mouse models with Aβ deposition also have evidence of CAA and spontaneous microhemorrhage in association with CAA. The APPsw model shows a more severe CAA phenotype than does the PDAPP model at equivalent ages, despite higher Aβ parenchymal plaque load and total Aβ levels in PDAPP mice at 12 and 15 months of age (Table 1). Aβ deposition in the form of both diffuse and fibrillar parenchymal plaques begins at 7-10 months of age in both PDAPP and APPsw mice. CAA occurs concurrently in APPsw mice. By 12 months of age, CAA is more prominent in APPsw mice with comparable levels not seen in PDAPP mice until 24 months of age (Fig. 1A,B). The difference in CAA severity between the two animal models may be attributable to the fact that at both young and older ages, APPsw mice have higher brain tissue levels of Aβ40 and an increased ratio of Aβ 40:42 (Table 1). CAA from both models is typical of that seen in other mouse models and in human CAA cases, with a ring of amyloid surrounding the vessel wall (Fig. 1C). The presence of CAA can also be seen in isolated cerebral vessels from APP transgenic mice (Fig. 1D). We found that once CAA was demonstrable, microhemorrhages occurred in association with CAA in both APPsw and PDAPP mice (Fig. 2). Microhemorrhages were associated almost exclusively with amyloid-containing vessels (identified as thioflavine-S positive). Although more rare, occasional macrohemorrhages associated with CAA were also seen in APPsw mice 15 months of age and older.
Comparison of Aβ levels and parenchymal plaque load between APPsw and PDAPP models

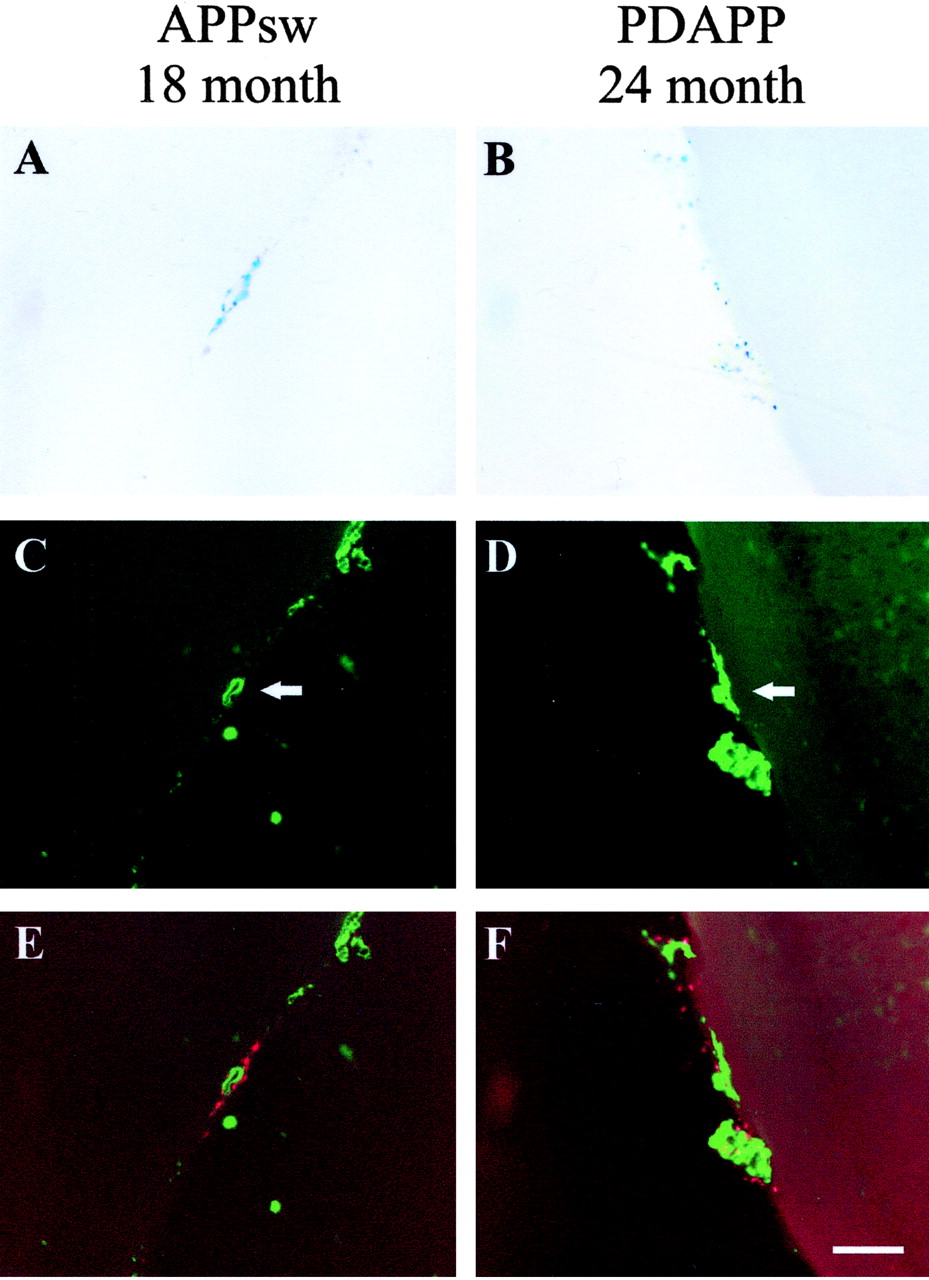
CAA and associated microhemorrhage occur in both the aged APPsw and PDAPP mice. A, B, Prussian Blue staining indicates microhemorrhage in an 18-month-old APPsw mouse and 24-month-old PDAPP mouse, respectively. C, D, Thioflavine-S-positive vessels (arrows) in an 18-month-old APPsw mouse and 24-month-old PDAPP mouse, respectively. E, F, Merged images of A and C and B and D showing colocalization of microhemorrhage (red) with CAA (green). Prussian Blue images were digitally converted to red using Adobe Photoshop. Scale bar, 100 μm.
CAA load increased with age in both the APPsw and PDAPP models (Fig. 3A,C); however, the absolute amount of CAA in the APPsw model is far greater than in the PDAPP model at the same age. In 15-month-old mice, the percentage cortical CAA load (percentage area of the cortex and overlying leptomeninges covered by CAA) in APPsw mice is 1.46 versus 0.38% in PDAPP mice (Fig. 3A,C). Cerebral microhemorrhage also increased with age in the APPsw and PDAPP models (Fig. 3B,D). Although there was a substantial increase in microhemorrhages between 12 and 15 months of age in APPsw mice, the number of hemorrhages at 15 and 18 months was not statistically different.

Age-dependent CAA and microhemorrhage in APP transgenic mice is prevented in the absence of apoE. A, CAA load increases with age in the cortex of 12-, 15-, and 18-month-old APPsw, Apoe+/+ mice. APPsw, Apoe-/- mice have a significant reduction (p < 0.05) in CAA at 15 and 18 months. B, The frequency of microhemorrhage also increases in 12-, 15-, and 18-month-old APPsw, Apoe+/+ mice. APPsw, Apoe-/- mice have a significant reduction (p < 0.05) in microhemorrhage at 15 and 18 months. C, CAA load increases in the cortex between 15 and 24 months of age in PDAPP, Apoe+/+ mice. PDAPP, Apoe-/- mice have a significant reduction (p < 0.05) in CAA at 15 and 24 months of age as compared with PDAPP, Apoe+/+ mice. D, The frequency of microhemorrhage increases in 15- and 24-month-old mice. PDAPP, Apoe-/- mice have a significant reduction (p < 0.05) in microhemorrhage at 15 and 18 months as compared with PDAPP, Apoe+/+ mice.
ApoE promotes CAA and associated microhemorrhage in APPsw and PDAPP mice
We sought to determine whether apoE, forms of which are associated with altered risk of CAA and hemorrhage, is directly involved in the pathogenesis of CAA. Previous results have shown that murine apoE promotes parenchymal amyloid deposition as well as CAA up to 12 months of age in the APPsw model (Holtzman et al., 2000a). The effect of apoE on CAA at older ages and whether it influences CAA-associated hemorrhage is unknown. We first asked whether apoE directly associates with CAA in APPsw mice. ApoE immunoreactivity colocalized to many parenchymal and cerebrovascular deposits of Aβ in the form of amyloid in APPsw mice (Fig. 4). Similar results were obtained in PDAPP mice (data not shown). We next examined the development of CAA in APPsw and PDAPP mice lacking apoE between 12 and 24 months of age. Strikingly, even up to 18 months of age, a time when there is otherwise substantial CAA in APPsw mice, none of the APPsw, Apoe-/- mice (n = 13) developed any detectable CAA (Fig. 3A). The absence of CAA was also associated with a dramatically reduced number of microhemorrhages up to 18 months of age in APPsw, Apoe-/- mice (Fig. 3B). Similar effects of apoE were also seen in the PDAPP model. None of the PDAPP, Apoe-/- mice studied (n = 8) had evidence of CAA in any brain region up to 24 months of age (minimum of 20 sections sampled per brain) (Fig. 3C). There was also a decrease in the number of microhemorrhages in PDAPP, Apoe-/- mice up to 24 months of age (Fig. 3D). This dramatic effect of apoE on CAA is remarkable given that although there is a delay in thioflavine-S-positive amyloid deposition in the absence of apoE, Aβ still deposits as amyloid in the parenchyma of aged APPsw, Apoe-/- mice (Fig. 4) as has been reported previously in very old (18-24 months) PDAPP mice (Fagan et al., 2002). PDAPP, Apoe-/- mice, up to 24 months of age, had no CAA despite the presence of thioflavine-S-positive amyloid in the brain parenchyma of most of these animals at old ages (Fagan et al., 2002).

Triple-labeling demonstrates apoE colocalization to CAA in vessels (arrows) in APPsw mice. Eighteen-month-old APPsw mice (A-H) and 18-month-old APPsw, Apoe-/- mice (I-L) were immunostained for apoE (A, E, I) and Aβ (B, F, J) and with the Congo red derivative X-34 as a marker of fibrillar amyloid (C, G, K). Although fibrillar Aβ (arrowheads) begins to deposit in the parenchyma of old APPsw, Apoe-/- mice at later ages, no evidence for CAA was found in these same mice. Scale bars: (shown in D for A-D; shown in L for E-L), 100 μm.
Ratio of Aβ40 to Aβ42 is markedly increased in cerebral vessels of aged APPsw and PDAPP mice: dependence on apoE
Previous biochemical experiments from human brain as well as immunohistochemical staining in APP transgenic mouse brain suggest that, in contrast to parenchymal plaques in which there is an enrichment of Aβ42, there is a relative increase in Aβ40 associated with CAA (Joachim et al., 1988; Prelli et al., 1988; Suzuki et al., 1994; Alonzo et al., 1998; McCarron et al., 2000). Because apoE dramatically influenced both the deposition of amyloid in cerebral vessels as well as one of its consequences (i.e., microhemorrhage), we sought to determine whether the ratio of Aβ40 to Aβ42 (40:42) was altered in the cerebral blood vessels of aged PDAPP and APPsw mice in the presence and absence of apoE. Biochemical assessment of cerebral vessels isolated from 24-month-old PDAPP mice by acid-urea gel electrophoresis followed by Western blotting showed a marked increase in the ratio of Aβ 40:42 compared with whole brain or parenchymal lysate (Fig. 5, Table 2). Aβ40- and Aβ42-specific ELISA analysis confirmed this elevated ratio in vessel preparations (Table 2). Of the two aged 24-month-old PDAPP mice examined, PDAPP animal 5 showed a 20-fold increase in the ratio of Aβ 40:42 in cerebral vessels (40:42 ratio = 6.1) compared with whole brain lysate (40:42 ratio = 0.3), whereas animal 6 showed a more modest threefold increase (ratio of Aβ 40:42 of 1.1 for vessel vs 0.4 for whole brain) (Table 2). Of the two aged PDAPP, Apoe-/- mice examined, both animals had a very low ratio of Aβ 40:42 in cerebral vessels as well as in whole-brain lysate. Animal 7 had a ratio of Aβ 40:42 in cerebral vessels of 0.08, and animal 8 had a ratio of Aβ 40:42 of 0.16 (Table 2). In the one 18-month-old APPsw mouse examined biochemically, the vessels and brain parenchyma both had a high ratio of Aβ 40:42, although this ratio was much higher in vessels (19.4) versus brain parenchymal material (5.7). We also examined the ratio of Aβ 40:42 in young PDAPP mice in the presence and absence of apoE. The ratio of Aβ 40:42 was similar in both groups (Table 2). This suggests that the combination of apoE together with the levels of Aβ40 or ratio of Aβ 40:42 predispose to CAA.

Ratio of Aβ 40:42 is increased in cerebral blood vessels of APPsw and PDAPP mice shown in Table 2. Shown is immunoblot of cerebral vessels and parenchymal brain tissue lysates run on acid-urea polyacrylamide gels to differentiate Aβ 1-40 and 1-42. Both the 18-month-old APPsw and 24-month-old PDAPP mice tested revealed an elevated ratio of Aβ 40:42 in cerebral vessel lysates as compared with brain parenchyma. Immunoblots were probed with an anti-Aβ antibody mixture of 3D6 (N-terminal specific), 21F12 (42 specific), and 266 (central domain specific). WB, Whole brain; V, vessels; P, parenchyma.
Ratio of Aβ 40:42 is increased in vessels from aged mice with CAA and is dependent on apoE
Discussion
Although there is considerable data on factors that facilitate parenchymal Aβ deposition, less is known about mechanisms leading to CAA. We used two different mouse models with Aβ deposition, PDAPP and APPsw transgenic mice, to investigate the effects of apoE expression on CAA and associated microhemorrhage. We demonstrate that both of these models develop an age-dependent deposition of Aβ in the form of amyloid in cerebral vessels, with the APPsw model developing more severe pathology. We also show that a major consequence of CAA (i.e., hemorrhage) also occurs in an age-dependent manner in these models. Although the effect of apoE on parenchymal amyloid deposition has been well studied, much less is known about its role in CAA. We show here, in both the PDAPP and APPsw mice, that the age-dependent development of CAA is prevented completely in the absence of apoE in animals examined up to 24 months of age. The latter occurs despite deposition of Aβ in the parenchyma in all of these animals, some of which is fibrillar (thioflavine-S positive) at older ages. Of note, Aβ deposition in blood vessels of PDAPP and APPsw mice is all fibrillar, and there does not appear to be diffuse Aβ deposits in vessels. Thus, the effect of removing apoE is not to delay conversion of diffuse deposits to fibrillar deposits; it prevents the conversion directly from soluble to fibrillar Aβ. Importantly, and concomitant with the prevention of CAA, there is an almost complete absence of microhemorrhages in the Apoe-/- mice. Additionally, as is the case in human patients with CAA, cerebral vessels isolated from aged PDAPP and APPsw mice with CAA have an elevated amount of Aβ40 relative to Aβ42; however, cerebral vessels isolated from aged PDAPP, Apoe-/- mice have a dramatically reduced ratio of Aβ 40:42. Taken together, our results demonstrate that in addition to the levels of Aβ40 and the ratio of Aβ 40:42, apoE plays a critical role in CAA formation and its consequences.
CAA is a major cause of often fatal lobar cerebral hemorrhage (Vinters, 1987). Although mutations in APP outside of the Aβ coding region result in rare forms of familial AD (Goate et al., 1991; Tanzi and Bertram, 2001), it has also been found that familial mutations within the Aβ coding region itself give rise to severe forms of CAA, including hereditary cerebral hemorrhage with amyloidosis Dutch-type (Levy et al., 1990; Revesz et al., 2002) and Italian-type (Tagliavini et al., 1999). Other clinically relevant effects of CAA, such as ischemia, are just now beginning to be investigated (Greenberg, 2002). A recently described mutation within the Aβ region in an Iowa family is associated with dementia and severe CAA but not cerebral hemorrhage (Grabowski et al., 2001). Interestingly, many of the mutations that result in familial AD appear to result from an increase in production of Aβ42, a particularly amyloidogenic form of Aβ (Citron et al., 1992, 1997; Cai et al., 1993; Haass et al., 1994; Borchelt et al., 1996, 1997; Eckman et al., 1997). Mutations in APP that lead to CAA do not appear to increase Aβ production, but in some way alter its fibrillogenic properties, toxicity, and clearance (Levy et al., 1990; Davis and Van Nostrand, 1996; Miravalle et al., 2000; Grabowski et al., 2001; Nilsberth et al., 2001; Van Nostrand et al., 2001; Monro et al., 2002).
Characterization of suitable animal models is necessary to study the pathogenesis of and potential treatment strategies for CAA as well as AD. Recently, Jucker and colleagues (Winkler et al., 2001) demonstrated that the APP23 mouse model of AD, in which the neuron-specific thy-1 promoter is used to drive expression of APPsw, also develops extensive CAA and has an age-dependent increase in microhemorrhages. Our data show that hemorrhage associated with CAA occurs in two other commonly used APP transgenic mice and that the APPsw mutation favors the formation of CAA and hemorrhage as compared with the APPV717F mutation. Recently, Jucker and colleagues (Pfeifer et al., 2002) showed that a passively administered N-terminal Aβ antibody resulted in an increase in CAA-associated hemorrhage. The exact cause of this effect has yet to be delineated, although it may have been caused by the antibody recognizing Aβ in an amyloid conformation in the vessel wall. Our results presented here indicate that the PDAPP and APPsw models also display this hemorrhage phenotype and may be useful in examining and defining future immunization strategies.
In 1993 the ϵ4 allele of APOE was shown to be a genetic risk factor for developing AD (Corder et al., 1993; Rebeck et al., 1993; Strittmatter et al., 1993) and subsequently also found to be a risk factor for CAA (Schmechel et al., 1993; Greenberg et al., 1995; Nicoll et al., 1997). Additionally, in patients who have sustained brain hemorrhage attributable to CAA, there is an overrepresentation of ϵ2 allele of APOE (Greenberg et al., 1996, 1998; Nicoll et al., 1996). Several lines of evidence demonstrate that apoE interacts with Aβ both in vitro (Strittmatter et al., 1993; Wisniewski et al., 1993; LaDu et al., 1994; Munson et al., 2000) and in vivo (Naslund et al., 1995; Wisniewski et al., 1995; Permanne et al., 1997; Russo et al., 1998). Previously it has been shown that murine apoE markedly facilitates the fibrillogenesis of Aβ in brain parenchyma in vivo in the PDAPP model (Bales et al., 1997, 1999; Holtzman et al., 1999, 2000b) as well as parenchymal and cerebrovascular amyloid in the APPsw model up to 12 months of age (Holtzman et al., 2000a).
Murine apoE has a profound effect on the development of CAA and its consequences. Furthermore, the effect of apoE is even more profound on the deposition of Aβ in cerebral vessels than it is on deposition of Aβ in brain parenchyma. The reason for this difference is unknown. Clearance of Aβ40 from the brain has been suggested to occur via active transport across the blood-brain barrier (Ghersi-Egea et al., 1996; Shibata et al., 2000; Ji et al., 2001) as well as along peri-arterial interstitial fluid drainage pathways into cervical lymph nodes and then into peripheral circulation (Weller et al., 1998). The basement membrane along peri-arterial drainage pathways contains an abundance of molecules known to bind apoE, such as heparin sulfate proteoglycans (Strittmatter and Bova Hill, 2002). In addition, endothelial cells that make up the blood-brain barrier express apoE receptors such as low-density lipoprotein receptor family members (Zlokovic et al., 1996; Shibata et al., 2000). If apoE-Aβ complexes remain bound to these molecules, it could provide a different environment attributable to factors such as charge and surface structure that facilitates Aβ fibrillogenesis. Whether apoE derived from different species (i.e., rodent versus human) and different human apoE isoforms will have similar effects on Aβ accumulation in the form of CAA is not known. Previous studies suggest that human apoE, in addition to playing a role in Aβ fibrillogenesis, may also play an important role in Aβ clearance from brain parenchyma (Holtzman et al., 1999; Fagan et al., 2002). Whether it also plays a role in influencing Aβ clearance in relation to CAA remains to be defined.
Our findings also support the hypothesis that alterations in Aβ metabolism, such as the ratio of Aβ 40:42, may be a key pathologic event in the development of CAA. The increase in ratio of Aβ 40:42 in cerebral vessels with CAA is in close agreement with previous observations in humans with CAA (Joachim et al., 1988; Prelli et al., 1988; Suzuki et al., 1994; Alonzo et al., 1998; McCarron et al., 2000). A biochemical analysis of one mouse model of AD with a significant CAA component, the APP/Ld model, found that the vessels isolated from these aged mice had an eightfold increase in the ratio of Aβ 40:42 as compared with brain parenchyma (Van Dorpe et al., 2000). The APPV717F mutation results in a low ratio of Aβ 40:42 compared with the APP670/671 Swedish mutation (Hsiao et al., 1996; Johnson-Wood et al., 1997). Humans and mice with the APPV717F mutation have little CAA (Murrell et al., 1991), whereas humans and mice with the APP670/671 Swedish mutation have substantial CAA (Lannfelt et al., 1994). Despite developing greater overall levels of parenchymal Aβ load, PDAPP mice have less CAA than APPsw mice. This may be attributable to the fact that APPsw mice have higher levels of Aβ40 and an increase in the ratio of Aβ 40:42 compared with PDAPP mice at young and older ages. Although the ratio of Aβ 40:42 does not appear to be altered by apoE at young ages, there is a further reduction in this ratio in older PDAPP mice lacking apoE. Taken together, this suggests that levels of Aβ40 and the ratio of Aβ 40:42 combined with apoE influence the probability of CAA formation. Determining how apoE influences the development of CAA and its consequences such as cerebral hemorrhage may lead to new insights into the pathogenesis and treatment of CAA.
Footnotes
This work was supported by National Institutes of Health Grants AG13956, AG05681, AG11355, and NS034467. Special thanks to Malca Kierson, Robert Brendza, and Margaret Racke for technical support.
Correspondence should be addressed to Dr. David M. Holtzman, Washington University School of Medicine, Department of Neurology, 660 South Euclid Avenue, Box 8111, St. Louis, MO 63110. E-mail: holtzman{at}neuro.wustl.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237889-08$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}