

Abstract
The effects of kainic acid (KA) on neurogenesis in the developing rat hippocampus were investigated. Neonatal [postnatal day (P) 7] rats received a single bilateral intracerebroventricular infusion of KA (50 nmol in 1.0 μl) or vehicle. At P14, P25, P40, and P60, the spatial and temporal relationships between the neurodegeneration and neurogenesis induced by KA were explored using terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) to detect the dying cells and 5-bromodeoxyuridine (BrdU) to label newly generated cells.
There was progressive loss of neurons in the cornu ammonis (CA) 1 and CA3 subfields of the hippocampus at all time points in KA-treated rats. TUNEL staining identified dying cells at P14 through P60, mainly in the CA3 subfield. The number of TUNEL-positive cells decreased with age. Neurogenesis also was observed in the KA-treated hippocampus. The number of BrdU-positive cells in the dentate gyrus was significantly decreased at P14, when the number of TUNEL-positive cells is highest. However, at later time points (P40 and P60) the number of BrdU-positive cells in the dentate gyrus was significantly increased. In addition, the number of BrdU-positive cells was increased in the CA3 subfield at P40 and P60 in KA-treated rats. A substantial proportion (40%) of the newly generated cells in CA3 also expressed markers of immature and mature neurons (class III β-tubulin and neuronal nuclei). Newly generated cells in the CA3 subfield only rarely expressed glial markers (8%).
These results suggest that a single exposure to KA at P7 has both immediate (inhibition) and delayed (stimulation) effects on neurogenesis within the dentate gyrus of developing rats. KA administration resulted in both neuronal apoptosis and neurogenesis within the CA3 subfield, suggesting that the purpose of neurogenesis in the CA3 is to replace neurons lost to apoptosis.
Introduction
Regions of the vertebrate CNS, including the dentate gyrus of the hippocampus, retain the capacity for neurogenesis throughout adulthood (Altman, 1962, 1969; Kaplan and Hinds, 1977; Lois and Alvarez-Buylla, 1993; Luskin, 1993; Cameron et al., 1995; Gould et al., 1999; Alvarez-Buylla and Garcia-Verdugo, 2002). The study of this phenomenon offers the opportunity to identify and characterize circumstances that activate progenitor cells to form new neurons and the conditions required for these neurons to mature and establish functional connections. Neurogenesis in the dentate gyrus is modulated by hormones, stress, pharmacological actions (i.e., NMDA receptor activation), levels of physical activity, and the complexity of the environment (Gould et al., 1994, 1999, 2000; Cameron et al., 1995; van Praag et al., 1999; Eisch et al., 2000; Malberg et al., 2000). Also, neurogenesis in this region of the hippocampus may be required for normal formation of memories (Shors et al., 2001). However, the functionality of neurons newly generated under these circumstances has not yet been elucidated completely (Kempermann, 2002;Nottebohm, 2002).
Kainic acid (KA), an agonist for kainate and AMPA receptors, is an excitotoxin in the hippocampus (Campochiaro and Coyle, 1978; Nadler, 1978; Cook and Crutcher 1986). KA administration in rats is widely used as a model of epilepsy because it induces ongoing convulsions, degeneration of cornu ammonis (CA) neurons, and hyperexcitability of surviving CA neurons (Nadler, 1981; Dudek et al., 1994; Ben-Ari, 1985,2001). However, with slow administration and concomitant anticonvulsants, KA also can induce neuronal loss not associated with seizures (Olney, 1990). KA administration in adult rats increases the rate of neurogenesis within the dentate gyrus of the hippocampus (Bengzon et al., 1997; Parent et al., 1997, 1998; Gray and Sundstrom, 1998; Nakagawa et al., 2000). Granule cells newly formed after KA-induced seizures can migrate from the subgranular zone (SGZ) of the dentate gyrus into the hilus (Parent et al., 1997). Finally, Scharfman et al. (2000) demonstrated that newly formed granule-like neurons at the border of the hilus and CA3 subfield develop synchronous activity with neighboring pyramidal neurons. Although in adult rats KA induces both ongoing neuronal degeneration and adaptive neurogenesis, the relationship between these processes is not well understood.
The effects of excitotoxins on the developing CNS are not as well studied, perhaps because of reports that the neonatal hippocampus is resistant to their acute toxicity (Albala et al., 1984; Wolf and Keilhoff, 1984; Ribak and Navetta, 1994). However, because of similarities between the effects of excitotoxins and hypoxia on the vertebrate CNS (Olney, 1990), such studies may be relevant for understanding the pathogenesis of neuropsychiatric disorders like schizophrenia that appear in adults but are associated with neurodevelopmental injury (Csernansky and Bardgett, 1998).
Intracerebroventricular administration of a low dose of KA to neonatal rats produces a progressive loss of CA3 and CA1 hippocampal neurons without generating seizures (Montgomery et al., 1999). The delayed death of CA neurons is apoptotic and coincides with the onset of pubescence (Humphrey et al., 2002). In the present study, we further investigated the effects of low-dose KA on neurodevelopment by examining the dynamic processes driving both neurodegeneration and neurogenesis within the developing hippocampus. In particular, we tested the hypothesis that neurogenesis is a mechanism by which CA neurons lost to KA-induced apoptosis might be replaced.
Materials and Methods
Female Sprague Dawley dams with litters were obtained from Harlan Bioproducts (Indianapolis, IN). The animals were maintained in an environmentally controlled facility with a fixed 12 hr light/dark cycle (lights on at 7:00 A.M.), 60% relative humidity, and food and water provided ad libitum. At P7, all litters were culled to six to eight pups of similar weight. The pups were then randomly assigned to the different experimental groups. Procedures involving animals were conducted in accordance with institutional guidelines that are in compliance with national laws and policies (National Institutes of Health Guide for the Care and Use of Laboratory Animals, NIH publication number 85–23, 1985).
KA administration. All solutions were mixed fresh daily. Neonatal rats (P7) were anesthetized with a 4% halothane/oxygen mixture and maintained with a 2% halothane/oxygen mixture throughout the remainder of the procedure. They were affixed to a surgical platform formed from dental acrylic and attached to a David Kopf Instruments (Tujunga, CA) stereotaxic apparatus. The scalp was washed with a 10% betadine solution, and a 1 mm incision was made to expose bregma. After noting the initial coordinates of bregma (in millimeters), the coordinates of −1.0 anteroposterior, ±1.1 mediolateral and −3.3 dorsoventral were used to perform bilateral infusions of KA into the lateral ventricles.
A 1.0 μl Hamilton (Reno, NV) syringe with a beveled needle was attached to the stereotaxic apparatus and slowly lowered (−3.3 mm) into one of the lateral ventricles over a period of 1 min. The gentle downward motion of the needle easily penetrated the soft skull. After 2 min, infusion of either 25 nmol KA in 0.5 μl (Sigma, St. Louis, MO) or vehicle (artificial CSF) into the ventricle began, at the rate of 0.1 μl every 2 min. After 0.5 μl had been infused into the ventricle, a 2 min rest was allowed for diffusion of the solution before the needle was slowly withdrawn. Using initial coordinates from bregma, the location of the opposite lateral ventricle was then determined, and an equal amount of KA (25 nmol KA in 0.5 μl) was infused into the opposite ventricle.
Incisions were closed with veterinary tissue adhesive, and the animals were placed in a warmed recovery cage. When all surgeries on the litter were complete, the pups were returned to the dam in the home cage. All pups were monitored closely for seizures for 5 d. No experimental animals had seizures (n = 70 total).
5-Bromodeoxyuridine injection. 5-Bromodeoxyuridine (BrdU) (Sigma) was dissolved in 0.9% NaCl and sterile filtered. Beginning at P11, P21, P37, and P57, respectively, each group of animals received daily intraperitoneal injections of BrdU for 3 d (50 mg/kg body weight at a concentration of 15 mg/ml) (del Rio and Soriano, 1989; Soriano et al., 1991; van Praag et al., 1999). Four animals (two P14 and two P40) received one dose of BrdU (150 mg/kg) 30 min before they were killed.
Perfusion and tissue preparation. At P14, P25, P40, and P60 (i.e., 24 hr and 30 min after the last BrdU injection), animals were deeply anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and then transcardially perfused with 30–60 ml of heparinized saline flush, followed by 4% paraformaldehyde in PBS, pH 7.4, for 15 min. After perfusion, all brains were removed and postfixed in the same fixation solution overnight at 4°C and then transferred to 30% sucrose for 48 hr. Serial sections of the brains were cut (40 μm sections) to encompass the entire hippocampus (from bregma −2.30 to −5.20 mm) (Paxinos and Watson, 1986) using a freezing microtome. All sections were stored in PBS/sodium azide (0.06%) until needed for analysis.
Nissl (thionin) staining. Every eighth section throughout the hippocampus was mounted on a gelatin-coated slide and allowed to air-dry overnight. The slides were then rehydrated in double-distilled water, submerged in a standard thionin solution for 15–45 sec until the desired depth of staining was achieved, and then gradually dehydrated for 5 min in successive baths of ethanol (i.e., 50, 75, 90, 95, and 100%). Each slide was then given two 15 min baths in 100% xylenes and coverslipped for light microscopy.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. The methods of Gavrieli et al. (1992) were used. Tissue sections were subjected to proteolytic pretreatment with proteinase K (2 μg/ml) in Tris-EDTA buffer (TE, pH 7.4) for 15 min at room temperature to increase labeled nucleotide binding. Sections were rinsed (three times at 5 min each) in TE before undergoing a quick wash with terminal deoxynucleotidyl transferase (TdT) buffer (25 mm Tris-HCl, 200 mm sodium cacodylate, and 5 mm cobalt chloride). Sections were then incubated in the labeling solution [20 U terminal transferase, 0.5 mm digoxigenin-11-dUTP (Boehringer Mannheim, Indianapolis, IN) in TdT buffer at 37°C for 2 hr]. The reaction was terminated by incubating the sections in a 1:100 solution of 300 mm NaCl in 30 mm sodium citrate for 30 min at room temperature. Sections were rinsed (three times at 5 min each) in Tris-buffered saline (TBS, 0.05 m) and then placed in 2% bovine serum albumin, 0.3 Triton X-100 in TBS for 30 min at room temperature, and then overnight at 4°C in anti-digoxigenin Fab fragments conjugated with alkaline phosphatase (Boehringer Mannheim) diluted 1:500 in TBS with 0.3% Triton X-100. Afterward, sections were again rinsed in TBS (three times at 5 min each), and then placed in 10 ml of a color buffer solution of 100 mm Tris-HCl, 100 mm NaCl, 50 mm MgCl, pH 9.5, containing 50 μl 4-nitroblue tetrazolium salt and 37.5 μl 5-bromo-4-chloro-3-indolyl-phosphate for 5–10 min to produce a dark blue reaction. The sections were then rinsed for a final time in TBS (three times at 5 min each), dehydrated, and coverslipped before being examined via light microscopy.
BrdU labeling. Tissue sections were first denatured in 2N HCl for 60 min at 37°C and then neutralized in 0.1m borate buffer, pH 8.5, for 10 min. After washing in PBS, the sections were incubated for 1 hr in 5% normal horse serum and 0.2% TritonX-100, then with anti-mouse BrdU (1:800; Boehringer Mannheim) for 48 hr at 4°C, and then with secondary antibody (biotinylated horse anti-mouse; Vector Laboratories, Burlingame, CA) for 2 hr, followed by amplification with an avidin–biotin complex (Vector Laboratories). BrdU-positive cells were visualized with DAB (Vector Laboratories) or nickel intensification (50 mm NiCl2).
Double-labeling immunohistochemistry. Sections containing BrdU-positive cells were double-labeled using neuron-specific [class III β-tubulin (TuJ1) and neuronal nuclei (NeuN)] or glia-specific (GFAP) markers. Selected tissue sections were first incubated with an avidin–biotin blocking solution, and then with primary antibody for TuJ1 (1:500; Babco, Richmond, CA) and NeuN (1:500; Chemicon, Temecula, CA) or GFAP (1:4; Incstar, Stillwater, MN). Visualization of the second marker was accomplished using species-specific secondary antibodies conjugated with cyamine dye (Cy3), fluorescein isothiocyanate (1:200, Jackson ImmunoResearch, West Grove, PA), or Alexa 488 (1:100; Molecular Probes, Eugene, OR) for confocal microscope (Olympus Fluoview 500, equipped with Coherent UV channel). The sections were optically sliced in the Z plane using 1.0 μm intervals, and cells were rotated in orthogonal planes to confirm the colocalization of labeling for BrdU and TuJ1, NeuN, or GFAP.
Cell counting and data analysis. Coded slides were first examined for the presence TUNEL- and BrdU-positive cells in the dentate gyrus and in the CA3 and CA1 subfields of the hippocampus by an examiner blind to the group assignment of each animal. The number of TUNEL- and BrdU-positive cells was then quantified using an optical fractionators method in which every eighth section through the rostral–caudal extent of the hippocampus was examined. The code was not broken until the analysis was complete. All TUNEL-positive cells in the hippocampus CA1 and CA3 (no TUNEL-positive cells were observed within dentate gyrus) were counted regardless of size and shape. All BrdU-positive cells within the SGZ, the hilus of the dentate gyrus, and the CA1 and CA3 subfields were counted regardless of size or shape. To enable counting of individual neurons within cell clusters, quantification was performed using 400× and 1000× magnification. A BrdU-positive cell was counted as being in the SGZ of the dentate gyrus only if it was in direct contact with the SGZ. Cells located more than two cell diameters away from the SGZ were classified as hilar. The number of BrdU-positive cells in each anatomical region was then determined, the total number of these cells throughout the selected 10 sections of the hippocampus was calculated, and the mean number of cells per section was used for comparison.
Double-labeled sections were viewed with an Olympus BX-60 fluorescence microscope, and the percentage of a sample of 25 BrdU-labeled cells that were positive for each of the specific neuronal or glial markers was determined.
Data analysis. The data were analyzed using two-way ANOVA with experimental group and time as factors. When significant group effects, time effects, or group × time interactions were found,post hoc analyses were performed using a Fisher's protected least squares design (PLSD) test. The limit for statistical significance was maintained at p values <0.05; in cases where multiple comparisons were used, a Bonferroni correction was applied.
Results
KA injection at P7 was followed by visually apparent reductions in the numbers of hippocampal pyramidal neurons at both early (P14) and delayed (P25, P40, and P60) time points (Fig.1). The magnitude of KA-induced neuronal loss was smaller than that seen in adult animals (Shetty and Turner, 2001). Moreover, neuronal loss at P40 and P60 appeared to be relatively specific to the CA3 subfield. In some animals, CA3 pyramidal neurons were nearly absent at these time points (Fig.1C,D). These qualitative results were highly consistent with our previous quantitative findings (Montgomery et al., 1999; Humphrey et al., 2002).

Neuronal loss in KA-treated animals. Thionin-stained sections of the hippocampus are shown for KA-treated and control animals at P14, P25, P40, and P60. In KA-treated animals, loss of pyramidal neurons is initially apparent in the CA3 subfield at P14 and P25 (B, D,arrows). However, at P40 and P60, pyramidal neurons are nearly absent in the CA3 subfield of KA-treated animals (F, H, arrows). Scale bar, 100 μm.
TUNEL-positive cells appeared in the CA1 and CA3 subfields at P14 and P25, but only in the CA3 subfield at P40 and P60 (Fig.2). No TUNEL-positive cells were detected in the dentate gyrus at any time point. Furthermore, inspection of adjacent Nissl-stained sections showed no qualitative evidence of necrotic cells, cellular debris, or an inflammatory response that would be consistent with cell death. At P40 and P60, TUNEL-positive cells were especially notable within the pyramidal cell layer adjacent to the CA3 subfield, where pyramidal neurons were nearly absent (Fig. 2C,D). Quantitative analysis revealed the highest number of TUNEL-positive cells at P14, diminishing to very low levels by P40 and P60 (Fig.3). The presence of TUNEL-positive cells at later time points (P40 and P60) suggests that KA administration at P7 initiated an ongoing degenerative process (Humphrey et al., 2002).

TUNEL-positive cells in KA-treated animals. TUNEL-stained and Nissl (neutral red) counterstained sections of the hippocampus are shown for KA-treated and control animals at P14, P25, P40, and P60. In the KA-treated animals, TUNEL-positive pyramidal neurons undergoing apoptosis were observed in the CA3 subfield at all time points (arrows), adjacent to the areas where pyramidal neurons are notably absent (F, H). No TUNEL-positive cells were observed in the control animals at any time point. Scale bar, 100 μm.

Quantitative analysis of TUNEL-positive cells in KA-treated groups in CA3 and CA1. Data represent the mean number of TUNEL-positive cells per section of hippocampus ± SEM. The number of TUNEL-positive cells was greatest at P14 and declined to very low levels by P60. In the CA3 subfield, a one-way ANOVA revealed a significant effect of time (F = 4.58; df = 3, 43; p < 0.007) (A). Forpost hoc analyses (Fisher's PLSD) performed at each time point, asterisks denote significant differences (p < 0.01) between the number of TUNEL-positive cells at P14 and all later time points (P25, P40, and P60). In the CA1 subfield, a one-way ANOVA revealed a similar trend (F = 1.49; df = 3, 43; p = 0.23) (B). No TUNEL-positive cells were found in control animals at any time point (n = 6 at each time point) (also see Fig. 2).
Visual inspection of hippocampal sections labeled with BrdU revealed clusters of BrdU-positive cells within the dentate gyrus SGZ in both KA-treated and control animals at all time points (Fig.4). Clusters of BrdU-positive cells with irregularly shaped nuclei extended from the hilar border into the inner granular cell layer. Also, at P14–25, BrdU-positive cells were observed in the hilus and within the CA3 and CA1 subfields of the hippocampus in both KA-treated and control animals (Fig.4A–D). In control animals, the number of BrdU-positive cells declined with increasing age (Fig.4A,C,E,G). This trend was less apparent in KA-treated animals (Fig.4B,D,F,H). Similar differences in the frequency of BrdU-positive cells were observed between KA-treated and control animals in anterior, middle, and posterior regions of the hippocampus. BrdU-positive cells also were observed within the neocortex and other brain regions (data not shown).
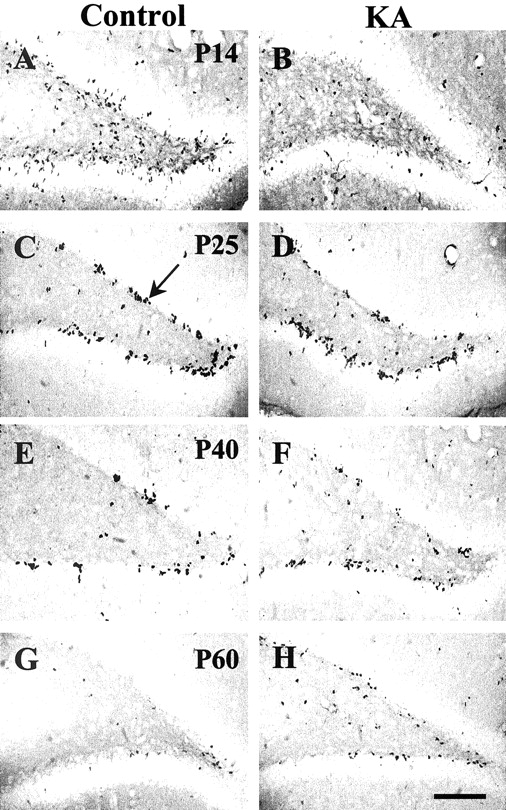
Representative sections showing BrdU-positive cells in the dentate gyrus in control and KA-treated animals at P14, P25, P40, and P60. At P14, BrdU-positive cells are dispersed throughout the dentate gyrus in control sections (A). After KA administration, the number of BrdU-positive cells was visibly diminished in P14 sections (B). At P25, P40, and P60, most of the BrdU-labeled cells were located in the SGZ of the dentate gyrus (C, arrow). The number of BrdU-positive cells appears to be increased at P40 and P60 in KA-treated animals compared with control animals (F,H). Scale bar, 100 μm.
Quantitative analysis of BrdU labeling within the dentate gyrus SGZ and in the CA3 and CA1 subfields of the hippocampus was then performed. Every eighth section through the rostral–caudal extent of the hippocampus was analyzed, so the data represent an unbiased sampling of BrdU-positive cells within the hippocampus. In the dentate gyrus SGZ at P14, the number of BrdU-positive cells was significantly lower in KA-treated animals than in controls (Fig.5A). However, at later time points, this trend was reversed with the number of BrdU-positive cells becoming greater in KA-treated animals than in controls. In the CA1 subfield of the hippocampus, the numbers of BrdU-positive cells were equivalent in KA-treated and control animals at P14 and P25 (Fig.5B). At later time points, BrdU-positive cells were rarely observed in the CA1 subfield in both groups of animals. The pattern of BrdU-positive cells in the CA3 subfield of the hippocampus was similar to that in the dentate gyrus SGZ. KA-treated animals again showed greater numbers of BrdU-positive cells than control animals at later time points (Fig. 5C). However, at P14 and P25, equivalent numbers of BrdU-positive cells were found within the CA3 subfield in KA-treated and control animals. BrdU-positive cells were distributed primarily within the stratum radiatum and stratum pyramidal of the CA3 subfield, consistent with areas where neuronal loss also was observed (Fig. 6A). No BrdU staining was observed in the CA3 subfield of control animals (Fig.6B).

Quantitative comparison of BrdU-positive cells in the dentate gyrus (A), the CA1 subfield (B), and the CA3 subfield (C) of the hippocampus of KA-treated and control animals. In both the dentate gyrus and CA3 subfield, BrdU-positive cells were more numerous in KA-treated animals than in control animals at the later time points (i.e., P40 and P60). BrdU-positive cells were more numerous in control animals than in KA-treated animals only in the dentate gyrus at P14. In the dentate gyrus, a two-way ANOVA revealed significant effects of time (F = 100.5; df = 3, 58; p< 0.0001) and a time × group interaction (F= 10.2; df = 3, 58; P = < 0.0001). In the CA1 subfield, a two-way ANOVA revealed a significant effect of time (F = 6.0; df = 3, 58; p = 0.0013) alone. In the CA3 subfield, a two-way ANOVA revealed significant effects of time (F = 19.0; df = 3, 58; p < 0.0001) and experimental group (F = 13.2; df = 1, 58; p = 0.0006), as well as a nearly significant trend for time × group interaction (F = 2.3; df = 3, 58;p = 0.087). For post hoc analyses performed at each time point, asterisks denote significant differences (p < 0.05) between KA-treated animals and control animals.

Distribution of BrdU-positive cells in a KA-treated animal at P40. BrdU-positive cells were distributed primarily within the stratum radiatum and stratum pyramidal of the CA3 subfield in a KA-treated animal at P40 (arrows).A, BrdU-stained section from a KA-treated animal;B, BrdU-stained section from a control animal. Scale bar, 100 μm.
Newly formed cells in the hippocampus could have resulted from in situ glial proliferation or from neurogenesis in response to KA-induced pyramidal cell death. To differentiate among these possibilities, the distribution of BrdU-immunostaining colocalized with neuronal (TuJ1 and NeuN) and glial (GFAP) markers was examined in KA-treated P40 animals using Cy3 and Alexa 488 staining. In the dentate gyrus, where the greatest number of BrdU-positive cells were observed, some new granule cells in the SGZ labeled with BrdU also were positive for TuJ1, indicating that their origin is neurogenesis rather than glial proliferation (Fig. 7).

Granule cells colabeled with BrdU and TuJ1 in the dentate gyrus of a KA-treated animal at P40. Confocal laser scanning microscopy verified that some new cells labeled by BrdU in the subventricular zone are TuJ1 positive (arrows).A, BrdU labeling; B, TuJ1 labeling;C, colocalization of BrdU and TuJ1 labeling in several granule cells. Scale bar, 50 μm.
In CA3, the region most damaged by KA administration, BrdU-positive cells were observed embedded in the pyramidal cell layer (Fig.8). Colabeling with NeuN revealed that some of these BrdU-positive cells also are NeuN positive, suggesting that they are new pyramidal neurons (Fig.8A,B). In addition, some of the new CA3 cells were labeled for both BrdU and TuJ1, a marker for both immature as well as mature neurons (Fig. 8C,D). Overlap of BrdU and GFAP staining was rare (data not shown).

Pyramidal cells in CA3 colabeled by BrdU and TuJ1 in KA-treated animals at P40. Confocal laser scanning microscopy shows BrdU labeling (A) and the overlap of NeuN labeling with BrdU labeling in a CA3 pyramidal cell (B) of a KA-treated animal at P40. BrdU labeling of a different section shows BrdU labeling (C) and the overlap of TuJ1 labeling with BrdU labeling in another CA3 pyramidal cell (D) of a KA-treated animal. Scale bar, 20 μm.
We quantified a random sample of new CA3 cells that were double-labeled in the KA-treated group. Of 25 BrdU-labeled cells, 28% were positive for TuJ1, 12% were positive for NeuN, and 8% were positive for GFAP. Notably, in both the dentate gyrus SGZ and CA3 subfield of the hippocampus, many BrdU-positive cells were observed that were unlabeled by TuJ1, NeuN, or GFAP. Presumably, these new cells had not yet differentiated into either neurons or glia.
To rule out nonspecific incorporation of BrdU into CA3 cells, BrdU (150 mg/kg, i.p.) was injected into P14 and P40 KA-treated rats that were killed 30 min later. No BrdU labeling was observed at either time point (data not shown). Moreover, a random sample of sections from P40 KA-treated rats was double-labeled with BrdU and TUNEL, and no cells were found in the CA3 subfield or elsewhere that were positive for both BrdU and TUNEL.
Discussion
In this study, the effect of low-dose KA administration on neurogenesis in the hippocampus of developing rats was evaluated systematically. To avoid the confound of neuronal changes secondary to ongoing seizures, all animals were closely monitored for seizure activity, and no animals used in this study had seizures. We confirmed our previous findings that intracerebroventricular KA administration at P7 induced a progressive neuronal loss associated with apoptosis (Montgomery et al., 1999; Humphrey et al., 2002). However, for the first time we report that the delayed loss of neurons at P40 and P60 was accompanied by an increase in neurogenesis in the CA3 subfield, which is close to the site of injury, as well as in the dentate gyrus, a brain area normally capable of this phenomenon in adult animals. A significant number of these newly formed cells were labeled by the neuron-specific markers, TuJ1 and NeuN. This was especially true in the dentate gyrus; however, 40% of newly formed cells within the CA3 subfield also were labeled with TuJ1 or NeuN. These findings suggest that neurogenesis may be a mechanism by which the developing hippocampus responds to apoptosis-induced loss of CA neurons.
The results of some previous studies suggest that the neonatal rat hippocampus is resistant to acute excitotoxicity and that this resistance is attributable to the immaturity of glutamatergic afferents (Albala et al., 1984; Wolf and Keilhoff, 1984; Ribak and Navetta, 1994). In the newborn rat brain, the expression of some subtypes of glutamate receptors is not complete at early stages of postnatal life (Snyder-Keller and Costantini, 1996; Wullner et al., 1997; Scherer and Gallo, 1998; Nansen et al., 2000). However, other investigators have demonstrated that KA administration can induce loss of hippocampal neurons in rats as early as P7 (Cook and Crutcher, 1986; Leite et al., 1996). Moreover, KA-induced loss of hippocampal neurons progresses long after the initial administration of KA, via an apoptosis-related mechanism (Montgomery et al., 1999; Humphrey et al., 2002). The present report significantly extends these findings by demonstrating that KA-induced delayed neuronal loss in developing rats is accompanied by an increase in neurogenesis, perhaps as an adaptation to replace lost neurons.
Several investigators have reported that an increase in neurogenesis occurs in the dentate gyrus after KA-induced seizures in adult rodents (Bengzon et al., 1997; Parent et al., 1997, 1998; Gray and Sundstrom, 1998; Scott et al., 1998a,b). Although some studies suggest that the newly generated cells migrate to the CA as replacements for lost neurons, these neurons also may become involved in aberrant neuronal circuits that can then become the substrate for ongoing seizures (Parent et al., 1997; Scharfman et al., 2000). Conversely, some studies suggest that seizures in neonatal rodents result in a reduction rather than an increase in neurogenesis (Wasterlain and Plum, 1973;Wasterlain, 1976, 1978; Wasterlain and Suga, 1980; McCabe et al., 2001).
In the present study, seizures were not observed in P7 rats treated with a low dose of KA. In previous experiments, we also showed that electrographic evidence of seizure activity was present in <5% of such animals (Montgomery et al., 1999). Therefore, we believe that the changes in neurogenesis observed in this study were the result of adaptation to disruptions in hippocampal architecture induced by KA rather than from its capacity to induce seizures. However, there is a similarity between the changes in neurogenesis associated with experimentally induced seizures (see above) and with single administration of an excitotoxin such as KA, which suggests that there may be an underlying relationship between the two phenomena. In regard to this possibility, hyperexcitability and increases in the release of endogenous excitatory neurotransmitters have been associated with both experimentally induced seizure foci and excitotoxin administration (Ben-Ari, 1985; Cook and Crutcher, 1986; Leite et al., 1996; Bengzon et al., 1997).
Neurogenesis in the adult CNS has been observed most commonly in those brain regions where progenitor cells continue to exist (Kuhn et al., 1996; Jin et al., 2001; Gage, 2002). The dentate gyrus of the hippocampus is among these areas, and granule cells continue to differentiate from progenitor cells throughout adulthood (Gould and Cameron, 1996). Experimentally induced seizures in adult animals may increase the rate of neurogenesis in the dentate gyrus (Parent et al., 1997; Scharfman et al., 2000). Moreover, newly formed cells in the dentate gyrus have been observed to migrate into the hilus and CA3 subfield of the hippocampus, while at the same time retaining at least some of the properties of granule cells (Scharfman et al., 2000). Of particular relevance to our findings, Magavi et al. (2000) recently induced apoptotic degeneration of neurons in layer VI of anterior neocortex in adult mice and observed that neural precursors in the cortical subventricular zone were induced to differentiate in situ into mature neurons, possibly to replace the lost neurons.
In this study, none of the BrdU-positive cells were also labeled with TUNEL, which suggests that cellular injury was not the explanation for the observed increases in BrdU labeling. Also, we observed that some of the BrdU-positive cells in both the dentate gyrus and CA3 were colabeled with neuron-specific markers. Although these findings suggest that the BrdU-positive cells in these areas were nascent neurons, their origin remains somewhat unclear. It is possible that the apparent increase of such cells in KA-treated animals may be attributable to improved survival of newly formed cells that were normally appearing via neurogenesis. This possibility is plausible in the dentate gyrus where neurogenesis during adulthood occurs but is much less likely in the CA3 subfield where neurogenesis does not usually occur (Gould and Cameron, 1996). Also, none of the BrdU-positive cells were labeled with the glia-specific marker, GFAP, which suggests that the newly formed cells in the dentate gyrus or CA3 subfield did not arise from glial elements.
Some cells colabeled by BrdU and TuJ1 or NeuN were observed in positions intermediate between the dentate gyrus SGZ and the CA3 subfield of the hippocampus. The presence of newly formed cells colabeled with neuron-specific markers intermediate between these brain regions suggests that newly formed neurons may have been migrating from the dentate gyrus or the subventricular zone to the CA3 subfield, perhaps to replace neurons in the CA3 subfield lost to apoptosis. Future studies colabeling the BrdU-positive cells with markers specific for immature neurons, such as Hu and TOAD-64 (turned on after division) or Doublecortin, a protein found exclusively in migrating neurons, could help to elucidate the origin of newly formed neurons in the CA3 subfield and in positions intermediate between the dentate gyrus SGZ and the CA3 subfield. Physiological studies of these neurons would then be needed to determine whether they are functional and, more specifically, whether they have been integrated into circuits of CA pyramidal neurons damaged by KA administration. These future studies should include behavioral measures to assess the relationships between the functionality of such circuits and changes in cognitive behaviors (i.e., memory) that depend on the integrity of hippocampal circuits.
In conclusion, increased neurogenesis appears to occur in concert with ongoing neurodegeneration after the administration of a low dose of KA to neonatal rats. Thus, excitotoxicity in neonatal rats initiates a complex set of events that have long-term consequences for hippocampal structure and function. Our results support the general hypothesis that the developing CNS is capable of regeneration in response to neuronal injury or degeneration under specific conditions (Horner and Gage, 2000; Magavi et al., 2000). It is not yet known whether the newly generated neurons are functionally normal. Furthermore, it remains to be investigated whether their presence could contribute to either the pathogenesis of neuropsychiatric diseases or the processes that support functional recovery.
Footnotes
This research was supported by Public Health Service Grants MH62130 (Conte Center) and MH/AG 60883.
Correspondence should be addressed to Dr. John G. Csernansky, Department of Psychiatry, Washington University School of Medicine, Campus Box 8134, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail:csernanj{at}medicine.wustl.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}