

Abstract
Regulation of the process of neuronal death plays a central role both during development of the CNS and in adult brain. The transcription factor myocyte enhancer factor 2 (MEF2) plays a critical role in neuronal survival. Cyclin-dependent kinase 5 (Cdk5) mediates neurotoxic effects by phosphorylating and inhibiting MEF2. How Cdk5-dependent phosphorylation reduces MEF2 transactivation activity remained unknown. Here, we demonstrate a novel mechanism by which Cdk5, in conjunction with caspase, inhibits MEF2. Using primary cerebellar granule neuron as a model, our investigation reveals that neurotoxicity induces destabilization of MEF2s in neurons. Destabilization of MEF2 is caused by an increase in caspase-dependent cleavage of MEF2. This cleavage event requires nuclear activation of Cdk5 activity. Phosphorylation by Cdk5 alone is sufficient to promote degradation of MEF2A and MEF2D by caspase-3. In contrast to MEF2A and MEF2D, MEF2C is not phosphorylated by Cdk5 after glutamate exposure and, therefore, resistant to neurotoxin-induced caspase-dependent degradation. Consistently, blocking Cdk5 or enhancing MEF2 reduced toxin-induced apoptosis. These findings define an important regulatory mechanism that for the first time links prodeath activities of Cdk5 and caspase. The convergence of Cdk5 phosphorylation-dependent caspase-mediated degradation of nuclear survival factors exemplified by MEF2 may represent a general process applicable to the regulation of other survival factors under diverse neurotoxic conditions.
Introduction
Survival is central to the normal function of both developing and mature neurons (Lewin and Barde, 1996; Mattson, 2000). Insults resulting from exposure to a variety of neurotoxins, both exogenous and endogenous, constitute the main triggering event of pathological apoptosis in the adult nervous system (Yuan and Yankner, 2000). Understanding the molecular mechanisms that regulate this process remains the focus of investigations by many laboratories.
Recent studies have revealed a key role of myocyte enhancer factor 2 (MEF2) in neuronal survival (Mao and Wiedmann, 1999; Mao et al., 1999; Okamoto et al., 2000; Li et al., 2001; Liu et al., 2003). MEF2, isoforms A-D, belongs to a family of MADS (MCM1, agamous, deficiens, and serum response factor) box transcription factors (Black and Olson, 1998). Structurally, they share a highly homologous N terminus that mediates MEF2 homodimerization and heterodimerization and DNA binding, whereas the C terminus of MEF2 is more divergent and comprises the transactivation domain (Molkentin et al., 1996). Studies show that MEF2 is a focal point of regulation; various signaling pathways target MEF2 directly, including the cyclin-dependent kinase 5 (Cdk5) pathway (Gong et al., 2003).
Cdk5 is a proline-directed serine/threonine kinase not active in regulating the cell cycle (Grant et al., 2001). Its activity is primarily restricted to postmitotic neurons as a result of the elevated expression of its positive regulators, p35 and p39 (Dhavan and Tsai, 2001). In addition to its role in the normal development of the CNS (Ko et al., 2001; Gupta and Tsai, 2003), many lines of evidence indicate that both abnormally high and low levels of Cdk5 activity are detrimental to the survival of neurons (Patrick et al., 1999; Ko et al., 2001; Tanaka et al., 2001; Li et al., 2002; Cruz et al., 2003). Various neurotoxic signals regulate Cdk5 activity by controlling the stability of p35 (Kusakawa et al., 2000; Lee et al., 2000). Consistent with this, deregulation of Cdk5 has been reported in models of amyotrophic lateral sclerosis (ALS), Neimann-Pick type C disease (NPTC), and Parkinson's disease (PD), as well as in surveys of tissues from Alzheimer's patients (Brion and Couck, 1995; Bajaj et al., 1998; Nguyen et al., 2001; Bu et al., 2002; Tseng et al., 2002; Smith et al., 2003).
Our recent data demonstrate a novel role of Cdk5 in the nucleus. Specifically, neurotoxins activate nuclear Cdk5, lead to the Cdk5-dependent MEF2 phosphorylation, and inhibit MEF2 function (Gong et al., 2003). This pathway underlies neurotoxin-induced neuronal apoptosis. How phosphorylation by Cdk5 inhibits MEF2 function was not clear. We report here a process by which Cdk5 and caspase regulate MEF2 in a concerted manner to execute neuronal apoptosis. Neurotoxins inhibit MEF2-dependent survival via caspase-mediated degradation of MEF2. Phosphorylation by Cdk5 is both necessary and sufficient to facilitate this degradation process. These findings demonstrate Cdk5 phosphorylation-dependent destabilization of specific MEF2 isoforms as an important regulatory mechanism in mediating neurotoxin-induced death. They reveal a novel cross talk between Cdk5 and caspase in regulating neuronal survival and apoptosis, which may represent a general mechanism applicable to other key regulatory factors.
Materials and Methods
Plasmids, recombinant adenoviruses, chemicals, and antibodies. Wild-type and mutated MEF2-dependent luciferase reporter constructs, dominant-negative Cdk5 (dnCdk5), and pcDNA3-MEF2DS444A were described previously (Gong et al., 2003). Recombinant adenoviruses expressing green fluorescent protein (GFP), MEF2D, MEF2DS444A, and dnCdk5 were generated according to He et al. (1998)
Polyclonal rabbit antibody specific for a phosphoserine-containing peptide corresponding to a sequence containing SPXR motif found in MEF2s was generated as described previously (Sharma et al., 1999; Gong et al., 2003). Antibodies were purchased from the following vendors: anti-MEF2C and cleaved caspase-3 (Asp175) antibodies from Cell Signaling Technology (Beverly, MA); anti-histone H1, anti-MEF2A, anti-Cdk5, and anti-p35/p25 polyclonal antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); anti-β-actin monoclonal antibody from Sigma (St. Louis, MO); and anti-MEF2D antibody from BD Transduction Laboratories (Lexington, KY). Propidium iodide (PI), poly-l-ornithine, N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (z-VAD.fmk), and glutamate were purchased from Sigma, roscovitine and active caspase-3 from Calbiochem (La Jolla, CA), Lipofectamine 2000 from Invitrogen (San Diego, CA), mammalian transfection system calcium phosphate from Promega (Madison, WI), a Poly Caspase Detection Kit from Biocarta (San Diego, CA), and restore Western blot stripping buffer from Pierce (Rockford, IL).
Culture of rat primary cerebellar granule neurons. Cultures of primary neurons were established as described by Mao and Wiedmann (1999). Briefly, cerebellum from postnatal day 6 rat pups was dissected and subjected to enzymatic dissociation. Dissociated cells were cultured on poly-l-ornithine-coated plates in basal medium Eagle's media. Neurons were treated with glutamate at 7 or 8 d in vitro (DIV) unless indicated otherwise.
Transfection of cells. Primary neurons in DMEM were transfected by the calcium phosphate method at 4-6 DIV as described by Mao and Wiedmann (1999). SH-SY5Y cells were transfected by the lipofectamine method. In general, neurons were returned to conditioned full media after transfection and then treated if needed. A 3:1 DNA ratio of effector versus β-galactosidase (β-Gal) or GFP vector was used for survival and luciferase assays.
Infection of SH-SY5Y cells. SH-SY5Y cells were split at 80% confluence and seeded into uncoated plates. When the cells grew to 60% confluence, recombinant adenoviruses were added individually or in combination to the cells. After 24 h, the cells were treated with H2O2 as indicated.
Cytoplasmic and nuclear fractionation. Cytoplasmic and nuclear fractionation was performed using an EZ nuclei isolation kit (NUC-101; Sigma) according to the manufacturer's protocol, which involves three cycles of thorough cell lysis and washing.
In vivo metabolic labeling. Primary neurons were washed and cultured in phosphate-free media for 3-4 h with or without roscovitine pretreatment and then metabolically labeled with 1 mCi/ml [32P]orthophosphate for an additional 3 or 4 h. For neurotoxic treatment, toxic insults were added to cells at the time of metabolic labeling with or without 10 μm roscovitine.
In vitro kinase assays. Purified Cdk5/p25 complex or Cdk5 complex immunoprecipitated from cell lysates with anti-Cdk5 polyclonal antibody was incubated with MEF2 substrates. The Cdk5 kinase reaction was performed as described by Gong et al. (2003).
Luciferase reporter gene assays. Primary neurons were transiently transfected with various constructs using the calcium phosphate transfection procedure as described by Mao and Wiedmann (1999). A β-galactosidase expression plasmid was used to determine the efficiency in each transfection. The total amount of DNA for each transfection was kept constant by using control vectors. Cell lysates were analyzed for luciferase and β-Gal activity according to the manufacturer's instruction (BD Bioscience, Franklin Lake, NJ).
Cellular dehydrogenase activity by cell counting kit-8. Water-soluble tetrazolium salt (WST-8; Dojindo Molecular Technologies, Tokyo, Japan) was added directly to the cells and bioreduced by the cellular dehydrogenases to give a colored formazan product, which is soluble in the tissue culture medium. The intensity of the color reaction was measured by a Packard (Meridian, CT) Spectracount.
Survival assays. The survival assays were performed as described previously (Blair et al., 1999; Mao et al., 1999). Neurons were stained with propidium iodide without permeabilization. GFP-positive cells with or without apoptotic nuclei (pyknotic or fragmented) were counted using a fluorescence microscope in a blind manner. Three hundred or more transfected cells were counted for each treatment. Apoptotic rates were calculated as the apoptotic cells in the total number of GFP-positive cells counted.
Reverse transcription-PCR amplification. Total RNA was isolated with a Trizol total RNA preparation kit (Invitrogen). One microgram of RNA extracted from primary CGNs was used to generate first-strand cDNA using an oligo-dT 15 primer. PCR amplification of rat MEF2C was performed using the following specific oligonucleotide primers (P1, nucleotides 1636-1656, AGACCCAAATTGTGGCGGTCG; P2, nucleotides 1837-1860, GTATCTCGAAGGGGTGGTGGTACG); the conditions were 94°C for 30 s, 60°C for 30 s, and 72°C for 45 s (35 cycles).
Statistics method. The results were analyzed using one-way ANOVA for independent or correlated samples.
Results
Glutamate-induced neuronal death correlates with inhibition of MEF2 activity and degradation of MEF2A and MEF2D
To investigate how neurotoxins regulate MEF2 function, we assessed the effects of the excitotoxin glutamate on MEF2 activity in primary CGNs. Cerebellar granule neurons cultured at 7 DIV were treated with glutamate, and nuclear morphology was assessed by Hoechst 33258 staining. Exposure to glutamate caused nuclear chromatin condensation, suggesting increased apoptosis (Fig. 1A, top). Consistent with this, exposure to glutamate reduced neuronal viability determined by cellular dehydrogenase activity in a time-dependent manner (Fig. 1A, bottom). These data indicate that excitotoxin glutamate triggers CGN apoptosis. To correlate reduced neuronal survival and MEF2 activity, CGNs were transiently transfected with a MEF2-dependent luciferase reporter construct and exposed to glutamate. Exposure to glutamate caused a time-dependent decrease of MEF2-dependent reporter activity, indicating reduced transactivation potential of MEF2 after glutamate treatment (Fig. 1B). Western blot analysis using isoform-specific MEF2 antibodies showed that the levels of MEF2A and MEF2D decreased over time after glutamate treatment (Fig. 1C). This reduction of MEF2A and MEF2D level is not a result of a general decrease in protein but rather a result of specific degradation, because the level of transcription factor c-Jun was not altered under the same condition (Fig. 1D). Together, these data provide direct, correlative evidence linking reduced levels of MEF2 protein with inhibition of MEF2 function and decreased neuronal survival. This suggests MEF2 destabilization as an important mechanism regulating its function in glutamate toxicity.
Phosphorylation of MEF2 by glutamate-induced activation of nuclear Cdk5 accompanies its degradation
To assess the protein degradation mechanisms that may be involved in regulation of MEF2 stability, we tested the effects of inhibiting caspases and proteasome on MEF2 level. Treating CGNs with either caspase inhibitor z-VAD.fmk or proteasome inhibitor I greatly reduced glutamate-induced degradation of MEF2D, with inhibition of proteasome pathway increasing MEF2D level above that of CGNs control (Fig. 2A, left). Consistent with this, inhibition of proteasome also increased MEF2D level in the absence of glutamate toxicity, whereas under the same condition, z-VAD.fmk did not enhance MEF2 level (Fig. 2A, right). Together, these results suggest that proteasome pathway is also involved in the normal turnover of MEF2D, whereas caspase pathway plays a more specific role in regulation of MEF2 stability under neurotoxic conditions. Therefore, we focused our subsequent studies on caspase-mediated regulation of MEF2.
Because previous studies have shown that the sensitivity of cerebellar granule neurons to glutamate changes overtime in culture (Frandsen and Schousboe, 1990; Chuang et al., 1992; Atlante et al., 2003), we correlated the degradation of MEF2 with cellular sensitivity to glutamate toxicity in CGNs cultured for 4, 8 and 12 d. Glutamate exposure had little effect on the level of MEF2D in CGNs at day 4 in culture but reduced MEF2D at day 8 and 12 in a time-dependent manner (Fig. 2B). Moreover, glutamate treatment led to a significant activation of caspase (indicated by red fluorescence via poly caspase activation kit) and increased conversion of Cdk5 activator p35 to its more potent cleavage product p25, a marker for Cdk5 hyperactivation, in day 8 and 12 CGNs compared with that of day 4 CGNs. Similarly, day 4 CGNs were more resistant to glutamate-induced death measured by cellular dehydrogenase activity, whereas glutamate caused a time-dependent death of day 8 and 12 CGNs. Collectively, these results indicate that glutamate-induced degradation of MEF2 correlates closely with activation of Cdk5, enhanced caspase activity, and increased cell death. Given that the effects of glutamate measured in Figure 2B are qualitatively similar for day 8 and 12 CGNs, our subsequent experiments were performed using day 8 CGN as a model.

Glutamate-induced neuronal apoptosis correlates with the degradation of MEF2A and MEF2D. A, Cerebellar granule neurons were treated with glutamate (100 μm). Chromatin condensation was revealed by Hoechst 33258 staining; arrows indicate apoptotic cells (top). Loss of neuronal viability was quantified by dehydrogenase activity at various times after glutamate exposure (bottom). B, Glutamate inhibited MEF2-dependent luciferase activity in a time-dependent manner. CGNs were transfected with a MEF2-dependent luciferase reporter construct and then exposed to glutamate. Error bars represent SD. mt, Mutant; wt, wild type. C, Glutamate promoted degradation of MEF2A and MEF2D proteins. CGNs were treated with glutamate; MEF2A and MEF2D protein levels were determined by Western blotting using anti-isoform-specific antibodies. Histone H1 was used as a loading control after reprobing the same membrane. D, The level of c-Jun was not altered by glutamate. CGNs were treated with glutamate for the times indicated. The levels of c-Jun, MEF2D, and actin were determined by Western blotting.
Our above data (Fig. 2B) suggest that conversion of p35 to p25, a key step in deregulation of Cdk5, is associated closely with glutamate toxicity and MEF2 degradation. Because our previous studies have shown that neurotoxins deregulate nuclear Cdk5 activity in primary cortical neurons, we tested the possibility that deregulation of nuclear Cdk5 by glutamate accompanies MEF2 degradation in cerebellar granule neurons by direct kinase assay. First, the effect of glutamate on CGN nuclear Cdk5 was determined. Cerebellar granule neurons were treated with glutamate. Nuclear Cdk5 kinase activity was determined in vitro using MEF2 as a substrate after anti-Cdk5 immunoprecipitation. Glutamate toxicity significantly enhanced nuclear Cdk5 activity, whereas nuclear Cdk5 protein level remained unchanged (Fig. 3A). Consistent with this finding, glutamate significantly increased the incorporation of 32P into MEF2D when CGNs were metabolically labeled with [32P]orthophosphate, and this increase was blocked by coincubation with a selective Cdk5 inhibitor, roscovitine (Fig. 3B). Cdk5 phosphorylates MEF2D at Ser444 (or its equivalents in MEF2A and MEF2C) and inhibits MEF2 transactivation activity (Gong et al., 2003). The phosphorylation of MEF2D at Ser444 in CGNs was studied using a phospho (p)-specific anti-MEF2 antibody described previously (Gong et al., 2003). Glutamate treatment induced MEF2D phosphorylation at Ser444, which was essentially blocked by cotreating cells with roscovitine (Fig. 3C). Several caspases cleave in vitro transcribed and translated MEF2 (Li et al., 2001; Okamoto et al., 2002), generating various fragments dependent on the specific caspases tested. Therefore, activation of specific caspases was investigated. Glutamate treatment led to cleavage and activation of procaspase 3 in CGNs (Fig. 3D). Moreover, inhibition of caspase activity by z-VAD.fmk mostly prevented glutamate-induced degradation of both MEF2A and MEF2D (Fig. 3E). In addition, the restored MEF2 was not recognized by phospho-MEF2 antibody, suggesting that the restored MEF2 is functional. Similarly, z-VAD.fmk restored MEF2 activity measured by MEF2-dependent reporter assay (Fig. 3F). Consistent with these, z-VAD.fmk added after significant MEF2 degradation had taken place was less effective in preventing neuronal death (Fig. 3G). Together, these findings suggest that phosphorylation of MEF2A and MEF2D at a site recognized by Cdk5 accompanies or parallels the caspase-mediated cleavage of MEF2 and neuronal death after neurotoxic insults.

Caspase-dependent degradation of MEF2 after glutamate treatment. A, Degradation of MEF2 was mediated by both caspase and proteasome-dependent processes. CGNs were pretreated with z-VAD.fmk or proteasome inhibitor I and then treated with (left) or without (right) glutamate. Levels of MEF2D and actin were determined by Western blotting. B, Glutamate-induced MEF2D degradation correlated with the generation of Cdk5 activator p25, activation of caspases, and loss of CGN viability. CGNs were treated with glutamate on different days in culture for the time indicated. MEF2D, p25, and actin levels were determined by Western blotting (top). Activation of caspase was determined by the Poly Caspase Detection kit (middle), and cell viability was determined by dehydrogenase level (bottom graph). Error bars represent SD.
Glutamate-induced cleavage of MEF2 requires Cdk5
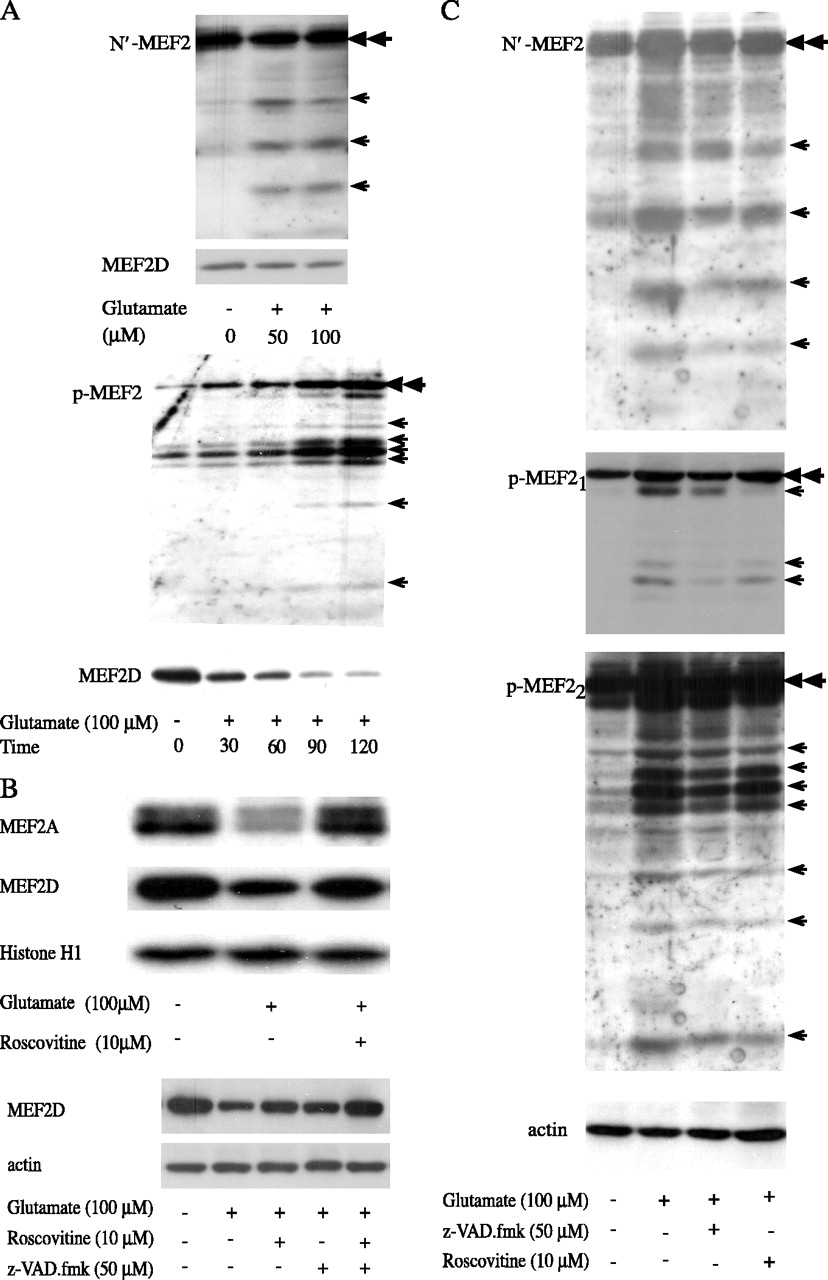
Our above data demonstrated that the level of full-length MEF2A and MEF2D is reduced after glutamate treatment. To show that degradation of MEF2 involves specific cleavage, we sought to demonstrate the generation of discrete MEF2 fragments by glutamate toxicity. Commercially available isoform-specific anti-MEF2A and MEF2D antibodies are raised against very limited number of epitopes, which seem to be essentially lost after cleavage of MEF2, making these antibodies poor reagents in detecting MEF2 fragments. Therefore, we characterized MEF2 fragmentation using an antibody that recognizes the conserved N terminus of MEF2 and the phospho-MEF2 antibody. Cerebellar granule neurons were treated with glutamate, and samples adjusted for equal amounts of MEF2 loading were tested. Exposure to glutamate led to generation of several discrete fragments recognized by anti-N′ MEF2 antibody (Fig. 4A, top, N′MEF2 blot, arrows). Comparable amount of full-length MEF2 was loaded as indicated by the double arrowhead and by MEF2D level when the membrane was reprobed (Fig. 4A, top, MEF2 blot). Similarly, glutamate treatment also generated bands specifically recognized by anti-phospho-MEF2 antibody in a time-dependent manner (Fig. 4A, bottom, p-MEF2 blot, arrows). The increase in phospho-MEF2 fragments was accompanied by a decline of full-length MEF2D (Fig. 4A, bottom, MEF2D blot) (equal histone H1 loading not shown). Together, these findings strongly support the notion that at least part of the process in glutamate-induced degradation of MEF2 involves specific cleavage of MEF2. The close correlation between MEF2 phosphorylation and degradation suggests that phosphorylation of MEF2 by Cdk5 facilitates its degradation. To test this possibility, Cdk5 activity was blocked using roscovitine. Roscovitine reduced glutamate-induced degradation of the full length of MEF2A and MEF2D (Fig. 4B, top). Together with findings in Figures 2 and 3, our data indicate that both roscovitine and z-VAD.fmk partially protected MEF2 from degradation. Combination of the two inhibitors resulted in a greater extent of protection (Fig. 4B, bottom), suggesting the possible presence of both overlapping and nonconvergent mechanisms. To show that inhibition of Cdk5 and caspases also attenuated glutamate-induced fragmentation of MEF2, MEF2 cleavage products were revealed using either N′ terminal or phospho-MEF2 antibodies as described in Figure 4A. Inhibition of either Cdk5 or caspase resulted in a clear reduction of glutamate-induced fragmentation of MEF2 (Fig. 4C, arrows; the same membrane was probed with N′ and p-MEF2 antibodies; the bottom p-MEF2 is the overexposure of the middle p-MEF2 panel to show the smaller fragments). To show the isoform identity of the cleaved MEF2 fragments, a blot with the specific phospho fragments generated after glutamate treatment was reprobed successively with anti-MEF2A and MEF2D antibodies (Fig. 5A). The phospho fragments were recognized by either MEF2A or MEF2D antibodies (Fig. 5A, middle two panels), confirming that the phospho bands originate from cleavage of MEF2A or D. Addition of either z-VAD.fmk or roscovitine simultaneously reduced glutamate-induced accumulation of phospho -as well as isoform-specific MEF2 fragments. Likewise, roscovitine restored MEF2 activity in reporter assay (Fig. 5B).

Correlation and activation of nuclear Cdk5 and caspase by glutamate. A, Glutamate activated nuclear Cdk5 kinase activity. An in vitro Cdk5 kinase assay was performed using purified C′ fragment of MEF2D as a substrate after anti-Cdk5 immunoprecipitation from nuclear lysates (top). Levels of Cdk5 and histone H1 protein were determined by Western blotting (middle and bottom, respectively). The Cdk5 inhibitor roscovitine was included with glutamate where indicated. B, Glutamate stimulated incorporation of [32P]orthophosphate into MEF2. CGNs were treated with glutamate and metabolically labeled with [32P]orthophosphate. 32P incorporation in MEF2D was determined by autoradiography after anti-MEF2D immunoprecipitation (top). The bottom panel shows the MEF2D level after immunoprecipitation. C, MEF2D was phosphorylated at the Cdk5 recognition site, Ser444. The level of MEF2 phosphorylated at Ser444, and total MEF2 were determined by Western blotting using phospho-specific anti-MEF2 antibody and anti-MEF2D antibody (reprobing the same membrane), respectively. Quantitation is shown at the bottom (n = 3). D, Glutamate activated caspase 3 in CGNs. Activated caspase 3 was determined by Western blotting (top). Z-VAD.fmk was added with glutamate where indicated. The bottom panel shows histone H1 protein level. E, Increased phosphorylation and degradation of MEF2 correlated. Equal amounts of protein were loaded under each condition based on histone H1. Western blotting analyses were performed using antibodies to MEF2A, MEF2D, phospho-MEF2, and histone H1 after reprobing the same membrane. F, Inhibition of caspase rescued MEF2-dependent reporter activity. mt, Mutant; wt, wild type. G, Inhibition of caspase reduced glutamate-induced death of CGNs. CGNs were treated with glutamate for 6 h, and cell viability was measured by WST assay. Pre and Post indicate that Z-VAD.fmk was added 30 min before or 4 h after glutamate exposure, respectively. The total length of Z-VAD.fmk treatment was 2 h. n = 3. Error bars represent SD.
To establish the direct role of Cdk5 in MEF2 degradation, Cdk5 was blocked using a more specific dominant-negative approach. To achieve higher efficiency of gene delivery for this assay, a neuroblastoma cell line, SH-SY5Y, which expresses endogenous Cdk5, was used. Western blot analysis showed that SH-SY5Y cells express endogenous MEF2D (Fig. 6A) and other MEF2 isoforms (data not shown). Oxidative stress induced a dose-dependent degradation of MEF2D (Fig. 6A). This was accompanied by an increase in its phosphorylation at Ser444 (Fig. 6B), suggesting that a mechanism(s) in SH-SY5Y cells regulates MEF2 stability similar to that observed in CGNs. To show that oxidative stress-induced degradation of MEF2 requires phosphorylation of MEF2 at its Cdk5 site (Ser444 for MEF2D), MEF2D was introduced into SH-SY5Y cells by recombinant adenoviruses expressing wild-type MEF2D (Ad-MEF2D) or Ser444 to Ala mutant (Ad-MEF2DS444A). Infection of SH-SY5Y cells with Ad-MEF2D or Ad-MEF2DS444A led to significant and comparable increases in MEF2D expression (Fig. 6C, top left). Oxidative stress significantly decreased the level of wild-type MEF2D in cells compared with untreated control (Fig. 6C, middle left). In contrast, MEF2DS444A was much more resistant to H2O2-induced degradation, suggesting that the degradation process requires phosphorylation of MEF2D at Ser444. To corroborate with this result, Cdk5 was blocked by overexpressing a dominant-negative Cdk5 (Ad-dnCdk5). dnCdk5 greatly protected MEF2D from H2O2-induced degradation compared with Ad-GFP control (Fig. 6C, right).
Phosphorylation by Cdk5 alone is sufficient to promote caspase-mediated degradation of MEF2
Our above data suggest that Cdk5 is required for neurotoxin-induced MEF2 degradation. To establish directly whether phosphorylation by Cdk5 is sufficient to mediate MEF2 degradation, the effects of phosphorylation of MEF2 on degradation was tested by caspase-3 cleavage assay. Purified glutathione S-transferase MEF2D (GSTMEF2D) was phosphorylated in the presence of [γ-32P]ATP by Cdk5/p25 in vitro. After phosphorylation, GSTMEF2D was incubated with caspase-3, and the level of GSTMEF2D was determined by anti-MEF2D Western blotting. Coincubation of GSTMEF2D with Cdk5/p25 significantly increased 32P incorporation in MEF2D compared with controls (Fig. 7A, left top). Incubation of phosphorylated GSTMEF2D with purified active caspase-3 resulted in MEF2D degradation evidenced by both the loss of 32P-MEF2D signal and the reduction of MEF2D (Fig. 7A, left bottom). On the contrary, unphosphorylated GSTMEF2D was much more resistant to caspase-3-mediated degradation under the same experimental condition (the near-complete loss of 32P signal compared with the moderate loss of GSTMEF2 on immunoblot reflects the efficiency of phosphorylation of MEF2 by Cdk5 in vitro). To show that degradation of full-length GSTMEF2D involves specific cleavage, GSTMEF2D was phosphorylated in vitro by Cdk5 and treated with or without caspase-3. The effect on GSTMEF2D was assessed using an anti-GST antibody. Caspase-3 led to a reduction of full-length GSTMEF2D undergone Cdk5 phosphorylation and simultaneous generation of a band that was not detectable under other conditions (Fig. 7A, right, arrow). These findings suggest that phosphorylated MEF2D is preferentially degraded by caspase-3. To extend this observation to CGNs in the context of neurotoxicity, we treated CGNs with glutamate, immunoprecipitated MEF2D, and incubated it with caspase-3. Consistent with our previous findings, caspase-3 did not alter the level of MEF2D immunoprecipitated from control CGNs (Fig. 7B). However, caspase-3 significantly reduced the level of MEF2D in CGNs exposed to glutamate. This decrease in MEF2D was not caused by variations in loading, because the amounts of MEF2D before and after immunoprecipitation was about the same as determined by anti-MEF2D Western blotting (Fig. 7B). To strengthen the role of Cdk5 in caspase-3-mediated degradation of MEF2, we tested the effect of increased expression of Cdk5/p35 on MEF2 degradation by caspase-3. MEF2D either alone or in combination with p35/Cdk5 was transiently expressed in SH-SY5Y cells (Fig. 7C, top). The effect of caspase-3 on overexpressed MEF2D was assessed as described in Figure 7B. Coexpression of p35/Cdk5 rendered MEF2D more susceptible to caspase-3-mediated degradation compared with MEF2D alone (Fig. 7C, bottom). Together with finings in Figure 6, these results indicate strongly that phosphorylation by Cdk5 is both necessary and sufficient for neurotoxin-induced caspase-mediated degradation of MEF2.

Cdk5 inhibitor roscovitine prevented glutamate-induced MEF2 degradation. A, Glutamate induced fragmentation of MEF2. CGNs were treated with glutamate as indicated. For the top panels, Western blotting with an antibody anti-N′ MEF2 shows fragmentation of MEF2 (top blot). The same membrane was reprobed for MEF2D for equal loading (bottom blot). For the bottom panels, anti-phospho-MEF2 blotting was performed (top blot). The same membrane was probed for MEF2D (bottom blot) and H1 (equal H1 loading data not shown). The double arrowhead and single arrow indicate the positions of the full length of MEF2A/D and MEF2 fragments, respectively. B, Cdk5 and caspase inhibitors reduced glutamate-induced degradation of MEF2. CGNs were incubated with glutamate with or without pretreatment of Cdk5 and/or caspase inhibitors. The same membranes were probed successively with anti-MEF2A, MEF2D, and histone H1 antibodies (top 3 panels) or with anti-MEF2D and actin antibodies (bottom 2 panels). C, Caspase and Cdk5 inhibitors reduced MEF2 fragmentation. CGNs were treated as described in B. Western blotting with anti-N′ and anti-phospho-MEF2 antibodies were performed. p-MEF22 is the overexposure of p-MEF21 to show fragmentation. The double arrowhead and single arrow indicate the positions of the full length of MEF2A/D and MEF2 fragments, respectively.
Neurotoxin-induced, Cdk5-dependent degradation is MEF2 isoform specific
CGNs express multiple isoforms of MEF2 including MEF2C. We studied whether MEF2C was regulated by neurotoxins in the same manner as MEF2A and MEF2D. Surprisingly, exposure of CGN to glutamate did not alter the level of MEF2C, whereas the same treatment induced significant degradation of MEF2A and MEF2D (Fig. 8A). Because the data showed that neurotoxin-induced degradation of MEF2 is Cdk5 dependent (Fig. 6), the phosphorylation of MEF2C was determined after glutamate treatment. Consistent with its resistant to degradation, MEF2C was not phosphorylated at Ser388 (its equivalent to Ser444 in MEF2D) after glutamate exposure as determined by phospho-MEF2 Western blotting (Fig. 8B). The apparent lack of MEF2 phosphorylation was not attributable to inability of the phospho-MEF2 antibody to detect MEF2C phosphorylated at Ser388; it detected phospho-MEF2C phosphorylated in cells by Cdk5 (Fig. 8 D). Ser388 of MEF2C resides in an alternatively spliced exon B (Fig. 8C, top), raising the possibility that the lack of MEF2C degradation may be because of the absence of this exon in a MEF2C splicing variant expressed in CGNs. This was tested with PCR primers to generate fragments with different lengths based on the presence or absence of the alternatively spliced exon B. Reverse transcription (RT)-PCR of total RNA isolated from CGN generated two fragments with the lengths corresponding to the two MEF2C variants with and without the alternatively spliced exon B (Fig. 8C, middle). These data suggest that CGNs express MEF2C splicing variants with and without exon B. To confirm that MEF2C protein carrying exon B is expressed in CGN and can be phosphorylated by Cdk5, MEF2C was immunoprecipitated from CGN and incubated with Cdk5/p25 as described above. Incubation with Cdk5 resulted in the robust phosphorylation of MEF2C (Fig. 8C, bottom).

Isoform-specific recognition of phospho-MEF2 fragments. A, Recognition of phospho-MEF2 fragments by isoform-specific MEF2A and MEF2D antibodies. CGNs were treated with glutamate as in Figure 4C, and MEF2 fragments were analyzed by Western blotting using phospho-MEF2 antibody. The same membrane was reprobed successively with antibodies to MEF2A, MEF2D, and histone H1. B, Inhibition of Cdk5 rescued MEF2-dependent reporter activity. MEF2 reporter assay was performed as described in Figure 1 B (n = 3). mt, Mutant; wt, wild type. Error bars represent SD.
To test whether the presence of exon B in MEF2C affects its degradation, human embryonic kidney 293 (HEK293) cells were transiently transfected with constructs expressing MEF2C with or without exon B (MEF2C + b or MEF2C - b), Cdk5, and p25. The phosphorylation level of MEF2C + b and MEF2C - b by Cdk5 was determined by phospho-MEF2 blotting after normalizing the loading for overexpressed MEF2C + b and MEF2C - b. Coexpression of Cdk5 and p25 led to a significant phosphorylation of MEF2C + b but not MEF2C - b (Fig. 8D, top). To assess whether Cdk5/p25 differentially affects the stability of MEF2C - b and MEF2C + b, the levels of MEF2C proteins were normalized with the general loading control β-actin. Overexpression of Cdk5 and p25 led to a modest reduction of MEF2C - b compared with control (Fig. 8D, middle). By comparison, Cdk5/p25 caused a much great reduction of MEF2C + b (Fig. 8D, bottom). Consistently, MEF2C - b was also more resistant to glutamate-induced inhibition after glutamate treatment of CGNs in reporter assay (Fig. 8E). Together with the data presented above, these studies demonstrate for the first time the differential regulation of MEF2C isoforms and splicing variants via selective phosphorylation by Cdk5.
Enhancing MEF2 protects neurons from neurotoxin-induced apoptosis
Whether inhibiting Cdk5 rescued MEF2 function and protected CGNs from glutamate-induced apoptosis was assessed by survival assays. CGNs were transfected with a construct expressing GFP with or without cotransfection of other vectors or treatment. Neurons were exposed to glutamate, stained with PI, which identifies apoptotic neurons, and scored for the incorporation of PI to GFP-positive neurons (Blair et al., 1999; Gong et al., 2003). An example of the assay is shown in the top panel of Figure 9A, which typifies glutamate-induced colocalization of GFP and PI signals. Quantitative analysis showed that glutamate significantly increased the number of apoptotic neurons, whereas treatment with either roscovitine or overexpression of dnCdk5 greatly reduced neuronal apoptosis (Fig. 9A, middle). CGN death assessed by PI staining correlated well with CGN viability measured by dehydrogenase activity (Fig. 9A, bottom) and with change of MEF2 function (Fig. 9B), suggesting that enhanced MEF2 activity may be responsible for increased survival of CGNs after roscovitine or dnCdk5 treatment. This conclusion was strengthened by the findings that overexpression of MEF2DS444A, which is expressed at a level comparable with that of MEF2D, offered CGNs greater protection from glutamate-induced apoptosis and inhibition of gene activation than MEF2D (Fig. 9C,D). This is consistent with the notion that resistance to Cdk5 phosphorylation enhances the neuroprotective effect of MEF2. To extend these observations to more mature neurons, the protective effect of blocking Cdk5 and enhancing MEF2 was tested in CGNs cultured 12 d in vitro. dnCdk5 and MEF2DS444A significantly reduced the number of apoptotic neurons after glutamate treatment (Fig. 9E). These results are consistent with our findings that glutamate toxicity induces Cdk5 activation and degradation of MEF2 in both 8 and 12 DIV CGNs (Fig. 2B), suggesting that glutamate-induced inhibition of MEF2 plays a role in the death of more mature cerebellar granule neurons.

Neurotoxicity induced Cdk5-dependent cleavage of MEF2. A, Expression and degradation of MEF2D in SH-SY5Y neuronal cells. Endogenous MEF2D was determined by Western blotting after hydrogen peroxide treatment (top). The membrane was reprobed for β-actin as a loading marker (bottom). B, Hydrogen peroxide induced phosphorylation of endogenous MEF2D in SH-SY5Y cells. The level and phosphorylation of MEF2D were analyzed as described by immunoblotting. C, Hydrogen peroxide-induced, Cdk5-dependent degradation of MEF2D. Expression of MEF2D and MEF2DS444A by recombinant adenoviruses is equal (top left). SH-SY5Y cells were infected with the indicated recombinant adenoviruses, and MEF2D level was determined by immunoblotting. MEF2DS444A was more resistant to hydrogen peroxide-induced degradation (middle left). SH-SY5Y cells were infected with recombinant adenoviruses as indicated and then treated with hydrogen peroxide. The levels of MEF2D, MEF2DS444A, and β-actin were analyzed by Western blotting. The bottom left graph indicates the relative level of MEF2 under various treatments (n = 3). Dominant-negative Cdk5 blocked MEF2 degradation (right). The experiment was performed as described for the middle left panel. Error bars represent SD.
Discussion
It is well established that caspases play a central role in initiation and execution of apoptotic process by cleaving a number of cytoplasmic and nuclear elements (Earnshaw et al., 1999). More recent findings implicate the abnormally high Cdk5 activity in the neuronal loss in animal models and human disease tissues of Alzheimer's disease, PD, NPTC, ALS, and acute neuronal injury (for review, see Maccioni et al., 2001; Nguyen et al., 2002; Shelton and Johnson, 2004). This study describes a novel mechanism by which Cdk5 and caspase pathways converge on a nuclear survival factor to induce neuronal death. We show that neurotoxin regulates neuronal apoptosis in part by reducing MEF2 protein via a Cdk5-dependent mechanism. Phosphorylation of MEF2 by nuclear Cdk5 targets MEF2 for caspase3-mediated degradation and promotes neurotoxin-induced apoptosis. These findings establish nuclear survival factor MEF2 as a cross-point at which Cdk5 and caspase concertedly mediate neurotoxic effects and provide a basis to test the intriguing possibility that degradation of MEF2 may play a role in the loss of neurons in neurodegenerative disorders.
Increasing evidence suggests that transcription-dependent mechanisms play a critical role in the survival and death of neurons (for review, see Brunet et al., 2001). Our data suggest that Cdk5 regulates nuclear survival machinery. In addition to MEF2, Cdk5 is shown to target other survival/death-related factors such as p53 and possibly retinoblastoma protein (Lee et al., 1997; Zhang et al., 2002). It would be interesting to test whether Cdk5 and caspases regulate other factors relevant to neuronal survival and/or apoptosis in a similarly coordinated manner. It is widely known that aberrant activation of cell-cycle proteins (various Cdks) is associated with apoptosis in several important tissues. The mechanism for this is not entirely clear. Given that Cdk5 and other Cdks share their substrate recognition site, our findings suggest that a mechanism by which aberrant activation of Cdks induces death is to target essential proteins for caspase-dependent degradation.
Regulation of MEF2 is implicated in neuronal survival and in caspase-mediated apoptosis (Mao and Wiedmann, 1999; Mao et al., 1999; Li et al., 2001; Gaudilliere et al., 2002; Okamoto et al., 2002; Liu et al., 2003). The results presented here provide a mechanism linking the proapoptotic effects of Cdk5 and caspase to MEF2. In support of the importance of this regulation, MEF2 mutants resistant to Cdk5 phosphorylation retain better transactivation potential and protect neurons from toxin-induced death. Although it remains possible that part of the protective effect is attributable to sequestration of Cdk5 or even caspases by overexpressed MEF2 mutants, this is unlikely to be a major mechanism, because MEF2AS408A mutant did not significantly affect phosphorylation at non-S408 sites by Cdk5 (Gong et al., 2003). As to whether phosphorylation by Cdk5 is sufficient to suppress MEF2 activity, our previous studies showed that overexpression of Cdk5 in neurons significantly inhibits transactivation activity of GAL4-MEF2A before causing significant change in GAL4-MEF2A protein, suggesting that MEF2 phosphorylated by Cdk5 is transcriptionally inactive possibly before its degradation. In support of this, MEF2A expressed in unstressed HEK293 cells is phosphorylated at its Cdk5 site but fail to activate MEF2-dependent reporter gene expression (data not shown). Together, these data suggest a two-step process by which phosphorylation by Cdk5 first reduces the transactivation potential of MEF2 while at the same time serves as a signal to target it specifically for caspase-dependent degradation. This mode of regulation provides cells an option to fine turn MEF2 in situations not accompanied by gross caspase activation. On the other hand, Cdk5 and caspase can operate fully in succession to ensure a permanent shutdown of the survival factor MEF2 when cells are under severe stress. Consistent with the notion that phosphorylation-mediated inhibition of MEF2 activity and its degradation are distinct biochemical steps, our previous studies found that Cdk5 inhibits MEF2 function without causing significant decline in MEF2 level in primary cortical neurons under similar conditions (Gong et al., 2003). The reason for this difference is not clear. One possible explanation is that the kinetics of caspase activation in cortical neurons may be different from that in cerebellar granule neurons, and therefore it takes a longer time point than tested to observe a significant decline of MEF2 level in cortical neurons.

Cdk5-mediated phosphorylation of MEF2 facilitated caspase-3-dependent degradation of MEF2. A, Caspase-3 degraded GSTMEF2D phosphorylated by Cdk5 in vitro. Purified GSTMEF2D was incubated with or without purified Cdk5/p25 in the presence of [γ-32P]ATP and then incubated with or without activated caspase-3 for 90 min and autoradiographed after Cdk5 kinase assay (top left). The same membrane was probed with anti-MEF2D antibody (bottom left). The right panel represents similar experiments as described in the left panel using cold ATP in kinase assay. The GSTMEF2D level after caspase-3 treatment was determined with an anti-GST antibody. The arrow indicates a fragment present only in caspase-3-treated phosphorylated GSTMEF2D. B, Endogenous MEF2D phosphorylated by Cdk5 in glutamate-treated CGNs was targeted for degradation by caspase-3. Glutamate treatment reduced MEF2D and increased phosphorylated MEF2D in CGNs. The same amounts of MEF2D from the lysates were subjected to anti-MEF2D immunoprecipitation (middle). The precipitated MEF2D was incubated with or without caspase-3 as described in A (bottom). C, Increasing Cdk5 activity was sufficient to enhance caspase-3-mediated degradation of MEF2D. SH-SY5Y cells were transiently transfected with control, MEF2D, or MEF2D plus p35/Cdk5 vectors. The susceptibility of overexpressed MEF2D to caspase-3-mediated degradation was determined as described in B. The top panel shows the same amounts of MEF2D and enhanced levels of p35 present in the lysates before caspase-3 treatment. The bottom panel shows that coexpression of p35/Cdk5 enhances degradation of MEF2D by caspase-3.
The data showed that MEF2A and MEF2D are cleaved to generate discrete fragments after exposure to neurotoxins. Neurotoxin activates caspase 3, which preferentially degrades phosphorylated MEF2. These data strongly support a caspase-dependent degradation process of MEF2, consistent with a previous report (Okamoto et al., 2002). Interestingly, our data suggest that proteasome may also participate in degradation of MEF2. However, different from the role of caspase in mediating MEF2 degradation after toxic stress, proteasome pathway seems to be involved in regulating MEF2 level with or without neurotoxicity. Whether these two processes are linked is unknown. It is possible that under stress, caspase mediates the initial cleavage of MEF2, the fragments of which in turn are further degraded through a proteasome-dependent pathway.
How phosphorylation by Cdk5 enhances degradation of MEF2 by caspase remains to be determined. One possibility is that phosphorylation causes a conformation change in MEF2, thus allowing access to caspase cleavage sites within MEF2. Another possibility is that phosphorylation by Cdk5 facilitates or stabilizes a direct interaction between MEF2 and caspase, which in turn allows efficient cleavage of MEF2 by caspase. Caspase 3 cleaves MEF2 at multiple sites present. Whether degradation of MEF2 by caspase is an ordered manner is not clear. Both conformation change and binding models would be consistent with an ordered cleavage of MEF2 by caspase. Although our finding suggests that phosphorylation by Cdk5 alone is sufficient to enhance MEF2 degradation by caspase, it does not rule out the possibility that there may be parallel and nonconvergent effects of Cdk5 and caspase on MEF2 degradation. Consistent with this, cotreatment of neurons with Cdk5 and caspase inhibitors can result in a somewhat greater protection of MEF2 than single treatment (Fig. 4B). Together with our findings that the relative protective effect of roscovitine and z-VAD.fmk is a dynamic process (data not shown), these data suggest that there may exist nonoverlapping mechanism of Cdk5 and caspase on MEF2 degradation dependent on the stage of neuronal stress.
Findings presented here show that neurotoxin-nuclear Cdk5 pathway differentially regulates MEF2 isoforms in CGNs. Whereas MEF2A and MEF2D are phosphorylated and degraded in a Cdk5-dependent manner after exposure to neurotoxin, MEF2C is not phosphorylated and, therefore, appears completely resistant to degradation under the same experimental conditions. Because the Cdk5 phosphorylation site resides in the alternatively spliced exon B in MEF2C, one possibility is that MEF2C expressed in CGNs may lack exon B and, thus, is resistant to phosphorylation and caspase recognition. However, semiquantitative RT-PCR data show that a significant portion of MEF2C in CGNs contains exon B, and when immunoprecipitated from CGN, it can be phosphorylated by Cdk5 in vitro. Therefore, alternative splicing cannot explain entirely the absence of MEF2C phosphorylation in CGNs. Alternatively, MEF2C may complex with a factor(s) that prevents its access by Cdk5 or be actively dephosphorylated by a phosphatase(s). Consistent with the latter possibility, phosphatase calcineurin activity maintains MEF2 in a hypophosphorylated active state in neurons (Mao and Wiedmann, 1999).

MEF2C resisted glutamate-induced phosphorylation and degradation in CGNs. A, Glutamate exposure did not alter MEF2C but reduced MEF2A and MEF2D levels in CGNs. B, Glutamate exposure did not increase MEF2C phosphorylation. CGNs were treated with glutamate as described in Figure 3A. Levels of phospho-MEF2D and phospho-MEF2C were determined by phospho immunoblotting (top). The same membrane was reprobed successively for MEF2D and MEF2C. C, Expression of alternatively spliced variants of MEF2C in CGNs. The alternatively spliced exons A and B in MEF2C are diagramed (top). RT-PCR analysis of MEF2C mRNA shows the expression of the splicing variants expressed by CGN (MEF2C - b and + b are vector controls) (middle). Cdk5 kinase assay of MEF2C immunoprecipitated from CGNs (anti-MEF2C IP) was performed as described in Figure 7A. MEF2C C′ is a bacterially expressed C′ MEF2C fragment used as a positive substrate control (bottom). D, Cdk5/p25 induced phosphorylation and degradation of MEF2C + b but not MEF2C - b in HEK293 cells. Cdk5 induced phosphorylation of MEF2C + b but not MEF2C - b (top). HEK293 cells were transfected with various constructs indicated. The amounts of MEF2C and its level of phosphorylation were determined by successive immunoblotting using anti-MEF2C (indicated by MEF2C) and phospho-MEF2 antibody (indicated by p-MEF2C). The same lysates were used to analyze MEF2C levels. The amounts of protein loaded were quantified using β-actin as a control. The levels of MEF2C - b (middle) and MEF2C + b (bottom) were determined by anti-MEF2C immunoblotting. E, MEF2C - b was more resistant to glutamate-induced inhibition than MEF2C + b. CGNs were transfected with MEF2 reporter and the constructs indicated and treated with glutamate. The change of luciferase activity relative to untreated controls is presented. Error bars represent SD.
Structurally, genes for MEF2A, C, and D all contain alternatively spliced exons A and B. Because all the alternatively spliced exons for MEF2s are within the large C′ transactivation domain (Yu et al., 1992; Breitbart et al., 1993; Leifer et al., 1993; McDermott et al., 1993; Martin et al., 1994), it raises the possibility that their presence or absence may bestow MEF2 variants with distinct gene regulation potential. Despite of this important implication in its biology, to our knowledge, the role of the alternatively spliced exons in MEF2 has not been illustrated. Therefore, the finding that the Cdk5 phosphorylation site in MEF2C resides in the alternatively spliced exon B ascribes a functional role to the alternatively spliced exons of MEF2. Our data show that MEF2C with or without exon B exhibits markedly different stability in response to Cdk5/p25-dependent phosphorylation and subsequent degradation by caspase. The differential regulation of MEF2C splicing variants raises an interesting possibility that cells may display different degrees of sensitivity to Cdk5-mediated toxicity dependent on the ratio and pattern of MEF2C splicing variants expressed. MEF2C is the major isoform expressed in various brain regions including hippocampus during certain stages of development (Ikeshima et al., 1995; Lyons et al., 1995). Our findings offer a model to correlate MEF2C splicing variants and susceptibility to toxicity in specific brain region.
The finding that glutamate reduces MEF2 activity, whereas MEF2C remains unphosphorylated at its Cdk5 site, suggests that MEF2C activity is by a Cdk5-independent mechanism. It is possible that MEF2C needs to dimerize with MEF2A or D to function. Therefore, reduced MEF2C activity can be viewed as a consequence of glutamate-induced degradation of MEF2A and MEF2D. Consistent with this, studies in non-neuronal cells showed that various MEF2 heterodimers and homodimers may have distinct functions in MEF2-dependent gene activation (Ornatsky and McDermott, 1996). The findings presented here provide a basis for testing these possibilities.

Enhanced MEF2 function rescued CGNs from glutamate-induced apoptosis. A, Blocking Cdk5 rescued CGN from glutamate-induced apoptosis. CGNs were transfected with a construct encoding GFP (green), treated with glutamate, and stained with PI (red). The top panel shows exemplary image of transfected neurons unexposed (top row) or exposed (bottom row) to glutamate. The middle graph quantifies neuronal apoptosis by PI staining (n = 4). The bottom graph shows the quantitative correlation of glutamate-induced neuronal death determined by dehydrogenase activity. B, Blocking Cdk5 restored MEF2 function after glutamate treatment. CGNs were transfected and treated as indicated. MEF2 reporter assay was performed (n = 3). C, D, MEF2DS444A rescued CGN from glutamate-induced apoptosis to a greater degree than did wild-type MEF2D (C; n = 4) and enhanced MEF2 activity in reporter assay (D; n = 3). The experiments were performed as described for A and B, respectively. E, Blocking Cdk5 rescued 12 DIV CGNs from glutamate-induced apoptosis. The experiments were performed as described for the middle panel in A using CGNs after 12 d in culture (n = 3). mt, Mutant; wt, wild type. Error bars represent SD.
Footnotes
This work was supported by National Institutes of Health Grants HD39446 and AG023695 to Z.M. and Grant AG19193 to Z.X. We thank L.-H. Tsai and J. H. Wang for Cdk5 constructs, H. H. Shin and T. H. Han for the MEF2-luciferase plasmid, and S. Gregory for reading this manuscript.
Correspondence should be addressed to Zixu Mao, Departments of Pharmacology and Neurology, Center for Neurodegenerative Disease, Emory University School of Medicine, Whitehead Building, Room 505, 615 Michael Street, Atlanta, GA 30322. E-mail: zmao{at}pharm.emory.edu.
X. Gong's present address: Department of Pediatrics, Women and Infants Hospital, Providence, RI 02905.
X. Wang's present address: Departments of Pharmacology and Neurology, Center for Neurodegenerative Disease, Emory University School of Medicine, Atlanta, GA 30322.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254823-12$15.00/0
↵* X.T. and X.W. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}