

Abstract
P/Q-type Ca2+ currents through presynaptic CaV2.1 channels initiate neurotransmitter release, and differential modulation of these channels by neuronal calcium-binding proteins (nCaBPs) may contribute to synaptic plasticity. The nCaBPs calcium-binding protein 1 (CaBP1) and visinin-like protein-2 (VILIP-2) differ from calmodulin (CaM) in that they have an N-terminal myristoyl moiety and one EF-hand that is inactive in binding Ca2+. To determine whether myristoylation contributes to their distinctive modulatory properties, we studied the regulation of CaV2.1 channels by the myristoyl-deficient mutants CaBP1/G2A and VILIP-2/G2A. CaBP1 positively shifts the voltage dependence of CaV2.1 activation, accelerates inactivation, and prevents paired-pulse facilitation in a Ca2+-independent manner. Block of myristoylation abolished these effects, leaving regulation that is similar to endogenous CaM. CaBP1/G2A binds to CaV2.1 with reduced stability, but in situ protein cross-linking and immunocytochemical studies revealed that it binds CaV2.1 in situ and is localized to the plasma membrane by coexpression with CaV2.1, indicating that it binds effectively in intact cells. In contrast to CaBP1, coexpression of VILIP-2 slows inactivation in a Ca2+-independent manner, but this effect also requires myristoylation. These results suggest a model in which nonmyristoylated CaBP1 and VILIP-2 bind to CaV2.1 channels and regulate them like CaM, whereas myristoylation allows differential, Ca2+-independent regulation by the inactive EF-hands of CaBP1 and VILIP-2, which differ in their positions in the protein structure. Differential, myristoylation-dependent regulation of presynaptic Ca2+ channels by nCaBPs may provide a flexible mechanism for diverse forms of short-term synaptic plasticity.
Introduction
Neurotransmitter release initiated by Ca2+ entering through presynaptic Ca2+ channels increases steeply with the concentration of extracellular Ca2+ (Dodge and Rahamimoff, 1967; Mintz et al., 1995). CaV2.1 channels, which mediate P/Q-type currents, are the primary initiators of neurotransmitter release in glutamatergic nerve terminals (Takahashi and Momiyama, 1993; Dunlap et al., 1995; Mintz et al., 1995; Westenbroek et al., 1995; Sakurai et al., 1996). Therefore, regulation of CaV2.1 channels is expected to have profound effects on neurotransmitter release and synaptic plasticity (Forsythe et al., 1998).
Binding of Ca2+/calmodulin (CaM) to a site in the C-terminal domain of CaV2.1 channels causes facilitation followed by enhanced inactivation (Lee et al., 1999, 2000, 2003; DeMaria et al., 2001; Liang et al., 2003). Several CaM-related neuronal Ca2+-binding proteins (nCaBPs), including Ca2+-binding protein 1 (CaBP1), visinin-like protein-2 (VILIP-2), and neuronal Ca2+ sensor-1 (NCS-1/frequenin) regulate CaV2.1 channels in a manner that is distinct from CaM (Wang et al., 2001; Weiss and Burgoyne, 2001; Lee et al., 2002; Tsujimoto et al., 2002; Lautermilch et al., 2005). These nCaBPs contain four potential Ca2+-binding EF-hand motifs like CaM. However, one or two of the EF-hands in nCaBPs are inactive in binding Ca2+, and the nCaBPs contain a saturated 14-carbon fatty acyl (i.e., myristoyl) moiety covalently attached at their N termini, unlike CaM. For recoverin and some other nCaBPs, the myristoyl moiety is thought to form a Ca2+-myristoyl switch that regulates membrane anchoring in response to changes in intracellular Ca2+ (Ames et al., 1996, 1997; Braunewell and Gundelfinger, 1999; Burgoyne and Weiss, 2001; Haeseleer et al., 2002; Haeseleer and Palczewski, 2002). In the experiments reported here, we examined the regulation of CaV2.1 channels by the myristoyl-deficient mutants CaBP1/G2A and VILIP-2/G2A. Our results show that CaBP1/G2A and VILIP-2/G2A have lost their distinctive regulatory properties and instead modulate CaV2.1 channels in manner that is similar to regulation by endogenous CaM. Because myristoylation of CaBP1 is not required for binding to CaV2.1 channels in intact cells, these results show that myristoylation is required for differential regulation of CaV2.1 channels by CaBP1 and VILIP-2 that is distinct from their regulation by CaM. Myristoylation of nCaBPs is therefore an important determinant of the regulatory properties of presynaptic Ca2+ channels and of nerve terminal function.
Materials and Methods
Cell culture and transfection. Before transfection, tsA-201 cells were grown to ∼70% confluency in DMEM/Ham's F-12 with 10% fetal bovine serum (Invitrogen, Rockville, MD) and 100 U/ml penicillin and streptomycin at 37°C in 10% CO2. For electrophysiology and immunocytochemical experiments, cells in 35 mm dishes were transfected with cDNA encoding Ca2+ channel subunits α1A (2 or 1.2 μg), β2a (1.5 or 1 μg), α2δ (1 μg) with or without VILIP-2, VILIP-2/G2A, the long splice variant of CaBP1 (Haeseleer et al., 2000), or CaBP1/G2A (1 μg), using the Ca2+ phosphate method. cDNA encoding CD8 (0.3 μg) was included to identify transfected cells. For coimmunoprecipitation studies, cDNA encoding Ca2+ channel subunits α1A (20 μg), β2a (10 μg), and α2δ (15 μg) were transfected into 150 mm dishes with or without cDNA encoding CaBP1, CaBP1/G2A, or VILIP-2 (20 μg). Throughout the experiments, transfection of CaV2.1 channels will indicate cotransfection of α1A, β2a, and α2δ.
Electrophysiological recording and data analysis. Whole-cell voltage-clamp recordings were obtained at room temperature 2-3 d after transfection. tsA-201 cells were incubated in an extracellular solution containing (in mm) 10 CaCl2, 150 Tris, 1 MgCl2, or 10 BaCl2, 120 Tris, 4 MgCl2, and CD8 beads (Dynal, Oslo, Norway) to allow visualization of transfected cells. The intracellular solution consisted of (in mm) 120 N-methyl-d-glucamine, 60 HEPES, 1 MgCl2, 2 Mg-ATP, and 0.5 EGTA. The pH of intracellular and extracellular solutions was adjusted to 7.3 with methanesulfonic acid. Recordings were made using an Axopatch 1D or 200B patch-clamp amplifier (Molecular Devices, Union City, CA) with PULSE software (HEKA Elektronik, Lambrecht, Germany) and filtered at 5 kHz. Leak and capacitive transients were subtracted using a P/-4 protocol. Because extracellular Ba2+ causes shifts in the voltage dependence of activation of -10 mV, voltage protocols were adjusted to compensate for this difference. Data analysis was performed using IGOR (Wavemetrics, Lake Oswego, OR). Normalized, individual traces of the data in Figures 2 and 7 were smoothed before measuring I200 or Ires using a binomial algorithm smoothing within 20 points. The measured data in Figure 3G were smoothed with the same algorithm. Curves were fit to the data from each cell included in the results of Figure 1 to determine values for the voltage of half-activation (V1/2) and the slope (k) using the following Boltzmann equation: y = (ymax - ymin)/(1 + exp(V1/2 - V/k)) + ymin. All averaged data represent the mean ± SEM. Statistical significance was determined using one-way ANOVA tests.

Myristoylation of CaBP1 is required to shift the voltage dependence of CaV2.1 activation. Normalized tail current-voltage curves were obtained by depolarizing from a holding potential of -80 mV to a variety of potentials and then repolarizing to -40 mV to elicit a tail current before returning to the holding potential. A, Normalized Ca2+ tail current-voltage curves from cells cotransfected with CaV2.1, β2a, α2δ (•, n = 15), and CaBP1 (○, n = 13), or CaBP1/G2A (□, n = 18). Inset, Current traces for every other voltage beginning at -40 mV. B, Voltage of half-activation (left) and slope (right) of the data shown in A. Inset, Current traces for every other voltage beginning at -50 mV. C, Normalized Ba2+ tail current-voltage curves from cells cotransfected with CaV2.1, β2a, α2δ (•, n = 12), and CaBP1 (○, n = 15), or CaBP1/G2A (□, n = 15). D, Voltage of half-activation (left) and slope (right) of the data shown in C. The data shown are averages ± SEM (*p < 0.05; **p < 0.01). Error bars represent SEM.
Coimmunoprecipitation assays. Two to three days after transfection, tsA-201 cells were washed twice with PBS and collected in ice-cold PBS containing protease inhibitors. Cells were then resuspended in lysis buffer and lysed by repeatedly passing cells through a syringe. For coimmunoprecipitation with Ca2+, cells were pretreated with 5 μm A23187 or ionomycin for 30 min at 37°C (Sigma, St. Louis, MO); the lysis buffer contained 2 mm CaCl2, 25 mm Tris, 500 mm NaCl, 0.1% Triton X-100, 1 mg/ml BSA, 0.05% NaN3, and protease inhibitors. For coimmunoprecipitation without Ca2+, the lysis buffer contained 10 mm EGTA, 25 mm Tris, 300 mm NaCl, 0.1% Triton X-100, 1 mg/ml BSA, 0.05% NaN3, and protease inhibitors. Cells were solubilized by rotating at 4°C for 30 min. Unsolubilized material was removed by centrifugation. The remaining solubilized lysates were then precleared with 5 mg of protein A-Sepharose. Subsequently, precleared lysates were incubated with 10 μg CNA5 (anti-CaV2.1) or 10 μg rabbit IgG for 1.5 h at 4°C, followed by the addition of 2.5 mg protein A-Sepharose for an additional 30 min. Immune complexes were then washed with lysis buffer and eluted from protein A-Sepharose by incubation in 2× SDS sample buffer containing 0.1 m DTT. Eluted proteins were then resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by Western blotting. CaV2.1 channels were detected with the CNA5 antibody (1:50). CaBP1 was detected with CaBP1 antiserum UW72, a generous gift from F. Haeseleer and K. Palczewski (1:1000) (Haeseleer et al., 2000). Primary antibodies were detected using horseradish peroxidase-linked protein A (Amersham Biosciences, Piscataway, NJ). ECL (Amersham Biosciences) was used to detect proteins by chemiluminescence.
For coimmunoprecipitations in which protein complexes were cross-linked before cell lysis, cells were washed twice with PBS at room temperature and then incubated with 2 mm dithio-bis(succinimidyl propionate) (DSP; Pierce, Rockford, IL) at room temperature for 30 min. The cross-linking reaction was quenched by washing the cells with Tris-buffered saline (TBS). Lysates were collected, and coimmunoprecipitations were performed as above.
Immunocytochemistry. Two to three days after transfection, tsA-201 cells were incubated in fresh media with or without 5 μm A23187 or ionomycin (Sigma) for 15 min. Cells were then fixed with 4% paraformaldehyde (PFA), rinsed with 0.1 m TBS, blocked sequentially with 2% avidin and 2% biotin, and then blocked again with 10% goat serum. For double-labeling experiments, we used the Zenon rabbit IgG labeling kit number 1 (Molecular Probes, Eugene, OR). Anti-CaBP1 (UW72) was then incubated for 5 min with 5 μl of Zenon rabbit IgG labeling reagent (goat anti-rabbit Fab fragments conjugated to Alexa Fluor 488). Labeled anti-CaBP1 was then blocked with 10 μl of the Zenon blocking reagent (nonspecific IgG) and incubated with the cells (1:150) for 60 min. Cells were postfixed with 4% PFA and then incubated with CNA5 (1:40) overnight at 4°C in 0.1 m TBS with 0.1% Triton X-100 and 10% goat serum, to label CaV2.1 channels. After extensive washing, cells were incubated with biotinylated anti-rabbit IgG (1:300; Vector Laboratories, Burlingame, CA) for 60 min at room temperature, rinsed, and then incubated with Texas Red avidin D (1:300; Vector Laboratories) for 60 min at room temperature. After final washes, coverslips were mounted on cells using Vectashield (Vector Laboratories). Cells were imaged using a Bio-Rad (Hercules, CA) MRC 600 confocal microscope in the W. M. Keck Imaging Facility at the University of Washington. Careful controls were performed in double-labeling experiments as both primary antibodies were made in rabbit. In addition to the lack of immunolabeling seen in untransfected cells and in controls with no primary antibody, we identified cells on each slide where only CaV2.1 or CaBP1 (or CaBP1/G2A) was immunolabeled. We also observed individual cells in which the subcellular localization of CaV2.1 was dramatically different from the subcellular localization of CaBP1 or CaBP1/G2A, indicating that the Zenon blocking reagent was able to block all of the available Fc sites of the anti-CaBP1 antibodies.

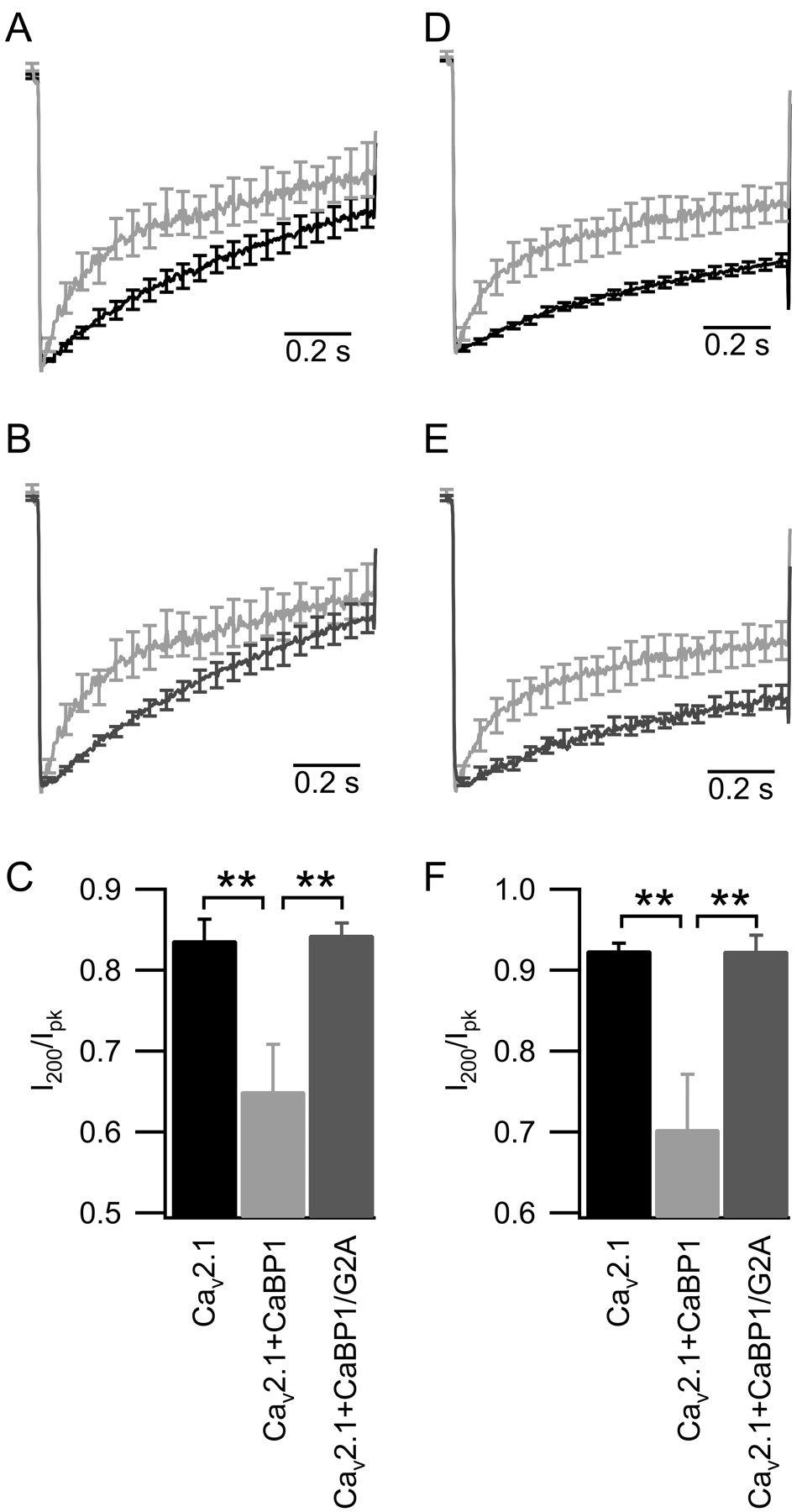
Myristoylation of CaBP1 is required to speed the inactivation of CaV2.1 channels. A, B, Mean time course of ICa elicited by a 1 s depolarization from -80 to +30 mV in cells transfected with CaV2.1, β2a, α2δ (A, black trace, n = 13), and CaBP1 (A, B, light gray trace, n = 11), or CaBP1/G2A (B, dark gray trace, n = 14). C, Ca2+ current amplitudes at 200 ms (I200) normalized to the peak (Ipk) and plotted for the data in A and B. D, E, IBa elicited by a 1 s depolarization from -80 to +20 mV in cells transfected with CaV2.1, β2a, α2δ (D, black trace, n = 13), and CaBP1 (D, E, light gray trace, n = 13), or CaBP1/G2A (E, dark gray trace, n = 11). F, Ba2+ current amplitudes at 200 ms (I200) normalized to the peak (Ipk) and plotted for the data in D and E. The data shown are smoothed averages ± SEM, with every 100th error bar plotted for clarity (*p < 0.05; **p < 0.01). Error bars represent SEM.
In experiments in which only CaBP1 was labeled, after the initial blocking steps were performed, cells were incubated with anti-CaBP1 (1:300) overnight at 4°C in 0.1 m TBS with 0.1% Triton X-100 and 10% goat serum, and subsequent steps were performed as above. Line-scan analysis was performed using IGOR (Wavemetrics).
Results
Effect of myristoylation of CaBP1 on voltage-dependent activation of CaV2.1 channels
To investigate the functional role of myristoylation of CaBP1, we used a mutant, CaBP1/G2A, in which the glycine that is normally myristoylated has been mutated to alanine, a mutation that is known to prevent N-terminal myristoylation of proteins (Rocque et al., 1993; Olshevskaya et al., 1997). We began by characterizing the voltage dependence of CaV2.1 activation in cells cotransfected with wild-type or myristoyl-deficient CaBP1. Cotransfection of CaBP1 with CaV2.1 causes a positive shift and a reduction in the slope of the voltage dependence of activation (Lee et al., 2002). The voltage of half-activation (V1/2) of Ca2+ currents (ICa; V1/2 = 18.6 ± 2.2 mV), measured from the normalized tail current-voltage curve in cells cotransfected with CaV2.1 and CaBP1, was shifted by +8.0 mV compared with cells transfected with CaV2.1 alone (V1/2 = 10.6 ± 1.8 mV). The V1/2 for activation in cells cotransfected with CaV2.1 and CaBP1/G2A (V1/2 = 7.7 ± 1.0 mV) was indistinguishable from that of cells transfected with CaV2.1 alone. (Fig. 1A,B, left). The slope of the activation curve was reduced in cells cotransfected with CaV2.1 and CaBP1 (slope factor k = 8.1 ± 0.4 mV per e-fold change in current) compared with cells transfected with CaV2.1 alone (k = 6.0 ± 0.4 mV). In contrast, there was no significant difference between the slope of the voltage dependence of activation between cells cotransfected with CaV2.1 plus CaBP1/G2A (k = 5.7 ± 0.2 mV) versus cells transfected with CaV2.1 alone (Fig. 1A,B, right). These data indicate that myristoylation of CaBP1 is required for the positive shift and the reduction of the slope of voltage-dependent activation of CaV2.1 channels.
Regulation of CaV2.1 by CaBP1 is Ca2+ independent (Lee et al., 2002). To test the importance of myristoylation in the absence of Ca2+, we characterized the voltage dependence of activation of CaV2.1 channels with the replacement of Ca2+ with Ba2+ as the charge carrier. In cells cotransfected with CaV2.1 and CaBP1, the activation curve for Ba2+ current (IBa) was shifted to more positive potentials (V1/2 = 10.6 ± 2.7 mV) compared with cells transfected with CaV2.1 alone (V1/2 = 4.6 ± 2.2 mV). When CaBP1/G2A was cotransfected with CaV2.1, the voltage dependence of activation (V1/2 = 0.1 ± 1.8 mV) was indistinguishable from that for cells transfected with only CaV2.1 and was significantly different from cells cotransfected with CaV2.1 and wild-type CaBP1 (Fig. 1C,D, left). The slopes of the activation curves for IBa are not significantly different in any of the three conditions (Fig. 1C,D, right). These results demonstrate a requirement for myristoylation of CaBP1 to shift the voltage dependence of activation. Because CaBP1/G2A retains the ability to bind to CaV2.1 channels in transfected tsA-201 cells (Figs. 4C, 5), these results indicate that myristoylation is required for this functional effect of CaBP1.

CaBP1/G2A allows Ca2+-dependent facilitation of CaV2.1 channels. A, Voltage protocol used to measure paired-pulse facilitation. B-D, Voltage dependence of CaV2.1 tail currents (ICa) evoked before (p1, •) or after (p2, ○) a 50 ms depolarization to +10 mV to allow Ca2+ entry.B, Cells transfected with CaV2.1, β2a, and α2δ (n = 10). C, Cells cotransfected with CaV2.1, β2a, α2δ and CaBP1 (n = 9). D, Cells cotransfected with CaV2.1, β2a, α2δ, and CaBP1/G2A (n = 14). E, Paired-pulse ratios of tail currents from data in B-D elicited by an initial depolarization from -80 to +80 mV (*p < 0.05; **p < 0.01). F, Repetitive depolarizations (2 ms)from -80 to +20 mV in Ca2+ were given at a frequency of 166 Hz. The first current elicited in the train was normalized to 1.0, with each subsequent current expressed as a fraction of the first and plotted against time. ICa from cells transfected with CaV2.1, β2a, α2δ (•, n = 8), and CaBP1 ( , n = 5) or CaBP1/G2A (□, n = 9). The data shown are averages ± SEM, with every 10th data point and every 20th error bar plotted for clarity (*p < 0.05; **p < 0.01). The peak amplitude of ICa was substantially reduced by coexpression of CaBP1 (Fig. 6). Therefore, in F, we compared results from cells with peak ICa < 1.6 nA to avoid artifacts caused by greater Ca2+ entry in the control cells.
, n = 5) or CaBP1/G2A (□, n = 9). The data shown are averages ± SEM, with every 10th data point and every 20th error bar plotted for clarity (*p < 0.05; **p < 0.01). The peak amplitude of ICa was substantially reduced by coexpression of CaBP1 (Fig. 6). Therefore, in F, we compared results from cells with peak ICa < 1.6 nA to avoid artifacts caused by greater Ca2+ entry in the control cells.
Effect of myristoylation of CaBP1 on inactivation of CaV2.1 channels
To further examine whether myristoylation of CaBP1 was required for acceleration of inactivation of CaV2.1 channels, we compared the effects of wild-type and CaBP1/G2A on inactivation of ICa (Fig. 2A,B) and quantitated the results as the ratio of the current remaining 200 ms after the initiation of a 1 s depolarization to the peak current during the depolarization (I200/Ipk) (Fig. 2C). The rate of inactivation of ICa is faster in cells in which CaV2.1 is coexpressed with CaBP1 (I200/Ipk = 0.65 ± 0.06) (Fig. 2A, light gray trace) compared with cells in which CaV2.1 is modulated by endogenous CaM (I200/Ipk = 0.83 ± 0.03) (Fig. 2A, black trace; C), as described previously (Lee et al., 2002). ICa inactivates more slowly in cells coexpressing CaV2.1 and CaBP1/G2A (I200/Ipk = 0.84 ± 0.02) (Fig. 2B, dark gray trace) than in cells coexpressing CaV2.1 channels and wild-type CaBP1 (Fig. 2B, light gray trace; C) and is not different from inactivation of CaV2.1 when channels are modulated by endogenous CaM (Fig. 2C). These data indicate that myristoylation of CaBP1 is required to accelerate the rate of inactivation of ICa conducted by CaV2.1 channels.
To examine the effect of Ca2+, we performed experiments in which Ba2+ was used as the charge carrier instead of Ca2+. Wild-type CaBP1 accelerates the inactivation of IBa, (Fig, 2D, light gray trace). In contrast, CaBP1/G2A does not accelerate inactivation of CaV2.1 compared with wild-type CaBP1 (Fig. 2E, dark gray trace) [I200/Ipk = 0.92 ± 0.02 for CaBP1/G2A compared with 0.7 ± 0.07 for CaBP1 (Fig. 2F)]. Similar to experiments measuring the rate of ICa inactivation, the rate of CaV2.1 inactivation of IBa is not significantly different when channels are expressed alone (I200/Ipk = 0.92 ± 0.01) or coexpressed with CaBP1/G2A (I200/Ipk = 0.92 ± 0.02) (Fig. 2F).
Ca2+-dependent facilitation of CaV2.1 channels coexpressed with CaBP1/G2A
Ca2+/CaM binding to the IQ-like domain in the C terminus of CaV2.1 channels produces Ca2+-dependent facilitation in response to paired pulses or trains of action potential-like stimuli (Lee et al., 2000, 2003), whereas coexpression of CaBP1 blocks facilitation (Lee et al., 2002). We assessed whether CaBP1/G2A could also block Ca2+-dependent facilitation of CaV2.1 channels. In cells expressing CaV2.1 alone, where channels are modulated by endogenous CaM, Ca2+ influx during a short prepulse produced a significant increase in the tail current elicited by a subsequent test pulse across a wide range of test pulse potentials (Fig. 3A,B,E). In cells expressing both CaV2.1 and CaBP1, the amplitude of the tail current elicited after the prepulse was not increased (Fig. 3C,E). In contrast, facilitation was observed with the same voltage protocol when CaBP1/G2A was coexpressed with CaV2.1 channels (Fig. 3D,E), indicating that myristoylation of CaBP1 is required to block Ca2+/CaM-induced facilitation of CaV2.1 channels. These data support the conclusion that myristoylation of CaBP1 is required to produce functional modulation of CaV2.1 channels that is distinct from modulation by CaM.

The affinity of CaBP1/G2A for CaV2.1 channels is reduced. Lysates from cells (transfected as indicated) were collected in medium with 10 mm EGTA or 2 mm Ca2+ and were immunoprecipitated with an antibody recognizing the α1 subunit of CaV2.1 or IgG. The immunoprecipitate was analyzed by immunoblotting for the presence of CaBP1 or CaBP1/G2A. Top panels show immunoblots of immunoprecipitates, and bottom panels show immunoblots of lysates. A, Cells were analyzed without cross-linking. B, Cells were treated with the bifunctional cross-linker DSP, as described in Materials and Methods, before lysis. C, The extent of coimmunoprecipitation in B was estimated from densitometric scans of CaBP1 and CaBP1/G2A in the coimmunoprecipitate and normalized for expression of the corresponding bands in the lysate. The value for wild-type CaBP1 was set to 1, and the fraction of CaBP1/G2A binding was normalized by the same value for side-by-side experiments. The data shown are mean ± SEM (n = 8 in EGTA; n = 7 in 2 mm Ca2+).
To further test whether myristoylation of CaBP1/G2A is required for modulation of CaV2.1 channels, we measured facilitation in response to trains of 2 ms depolarizations from -80 to +20 mV. These brief repetitive depolarizations maximize Ca2+/CaM-dependent facilitation by preventing Ca2+-dependent inactivation (Lee et al., 2000) and are similar to trains of action potentials and therefore represent more physiologically relevant stimuli. ICa modulated by endogenous CaM in cells expressing CaV2.1 alone showed increasing facilitation as the number of stimuli increased (Fig. 3F, black circles). Cells coexpressing CaBP1 and CaV2.1 had much reduced facilitation, which became insignificant by 800 ms of stimulation (Fig. 3F, gray circles). ICa in cells coexpressing CaBP1/G2A and CaV2.1 channels exhibited an intermediate effect (Fig. 3F, open squares). Initially, ICa showed facilitation that was not different from the facilitation induced by CaM. However, as the number of stimuli increased, facilitation of ICa decreased, and, by the end of the train of 250 stimuli, facilitation of ICa remaining in cells coexpressing CaBP1/G2A was significantly less than in cells expressing only endogenous CaM. Our working model for the effects of nCaBPs on CaV2.1 channels (see Discussion) is that CaBP1 blocks facilitation by inducing such rapid, voltage-dependent inactivation during each depolarizing stimulus that facilitation is occluded. Our results with CaBP1/G2A show that this effect is not detectable in paired-pulse experiments and in the initial few depolarizations during trains of stimuli. In contrast, CaBP1/G2A does accelerate pulse-wise inactivation and thereby reduces facilitation later in trains of stimuli, as illustrated in Figure 3F. These data suggest that CaBP1/G2A retains some ability to enhance inactivation, which builds up from pulse to pulse in repetitive pulse experiments. A <1% increase in cumulative inactivation of ICa in each depolarization/repolarization cycle in the 250-pulse train would be sufficient to reduce the degree of facilitation by the extent observed in Figure 3F.
Binding of CaBP1/G2A to CaV2.1 channels
Modulation of CaV2.1 channels by CaBP1/G2A was indistinguishable from modulation of channels by CaM in every functional test we performed except one, the modulation in response to trains of action potential-like stimuli. Because myristoylation is known to be important for membrane targeting of many nCaBPs, it was possible that CaBP1/G2A was not localized at the plasma membrane, where it could exert functional effects on the channel. Alternatively, CaBP1/G2A may bind to CaV2.1 channels but may not be capable of modulating their function normally. To distinguish between these possibilities, we assessed the ability of CaBP1/G2A to bind to CaV2.1 channels in transfected cells. CaBP1 coimmunoprecipitated with CaV2.1 in cotransfected tsA-201 cells when cells were treated with the Ca2+ ionophore ionomycin to increase intracellular Ca2+ or when Ca2+ was buffered with EGTA (Fig. 4A), consistent with previous work (Lee et al., 2002). In coimmunoprecipitations performed side by side with wild-type CaBP1, CaBP1/G2A did not coimmunoprecipitate with the channel as effectively as wild type in the presence or absence of Ca2+ (Fig. 4A), indicating that the CaBP1/G2A does not bind to the channel as stably as CaBP1. However, the appearance of a faint band corresponding in size to CaBP1/G2A in the coimmunoprecipitate without Ca2+ suggests that CaBP1/G2A can bind to CaV2.1 channels stably enough to be detected after extraction and immunoprecipitation.
It is possible that the binding of CaBP1/G2A to the channel was reduced because of incorrect targeting of CaBP1/G2A to the plasma membrane or because of a reduction in the stability of the complex of CaBP1/G2A and CaV2.1, which allowed dissociation during extraction and immunoprecipitation. To address the latter possibility, we performed coimmunoprecipitations after treating intact cells with a bifunctional cross-linker (DSP) to covalently link complexes of CaV2.1 and CaBP1/G2A in situ in intact cells. Under these conditions, CaBP1/G2A did coimmunoprecipitate with CaV2.1 channels both when Ca2+ was elevated by treating cells with Ca2+ ionophore before cross-linking and when cell lysates were buffered with EGTA. However, the amount of CaBP1/G2A that coimmunoprecipitated with the channel appeared to be reduced compared with wild-type CaBP1 in some experiments (Fig. 4B). Densitometric scans of the immunoprecipitated bands and normalization to the level of expression in the corresponding lysate samples indicated a significant decrease in coimmunoprecipitation of CaBP1/G2A compared with wild type in the absence of Ca2+ (Fig. 4C) (0.47 ± 0.09, SEM; n = 8; p < 0.01) but no decrease in the coimmunoprecipitation of CaBP1/G2A compared with wild type in the presence of Ca2+ (0.84 ± 0.15, SEM; n = 7; p > 0.05). Thus, CaBP1/G2A does bind to CaV2.1 channels effectively in transfected tsA-201 cells, although its binding may be reduced compared with wild-type CaBP1 in the absence of Ca2+.
Colocalization of CaBP1/G2A with CaV2.1 channels
We performed immunocytochemical experiments to further assess the ability of CaBP1/G2A to bind to CaV2.1 in intact cells. First, we examined the subcellular localization of CaBP1 or CaBP1/G2A expressed alone in tsA-201 cells. In cells transfected with CaBP1 only, immunolabeling was seen at or near the plasma membrane with exclusion from the nucleus (Fig. 5A). A similar pattern of immunolabeling was observed in cells that were treated with ionomycin to increase intracellular Ca2+ (Fig. 5B), indicating that CaBP1 does not undergo a Ca2+-dependent membrane translocation, similar to other nCaBPs such as NCS-1 and K+ channel interacting protein 1 (KChip1) (O'Callaghan and Burgoyne, 2003, 2004). In contrast, in cells transfected with CaBP1/G2A, immunolabeling of CaBP1/G2A was more diffuse, without exclusion from the nucleus (Fig. 5C), consistent with an important role of myristoylation in targeting of CaBP1 (Haynes et al., 2004). The pattern of immunolabeling was similar when cells were treated with ionomycin (Fig. 5D). No labeling was detected in control experiments in which transfected cells were labeled with a secondary antibody only (data not shown). In addition, untransfected cells treated with both primary and secondary antibodies showed no labeling (data not shown). These data indicate that myristoylation of CaBP1 is necessary for targeting at or near the plasma membrane in tsA-201 cells without Ca2+ channels.
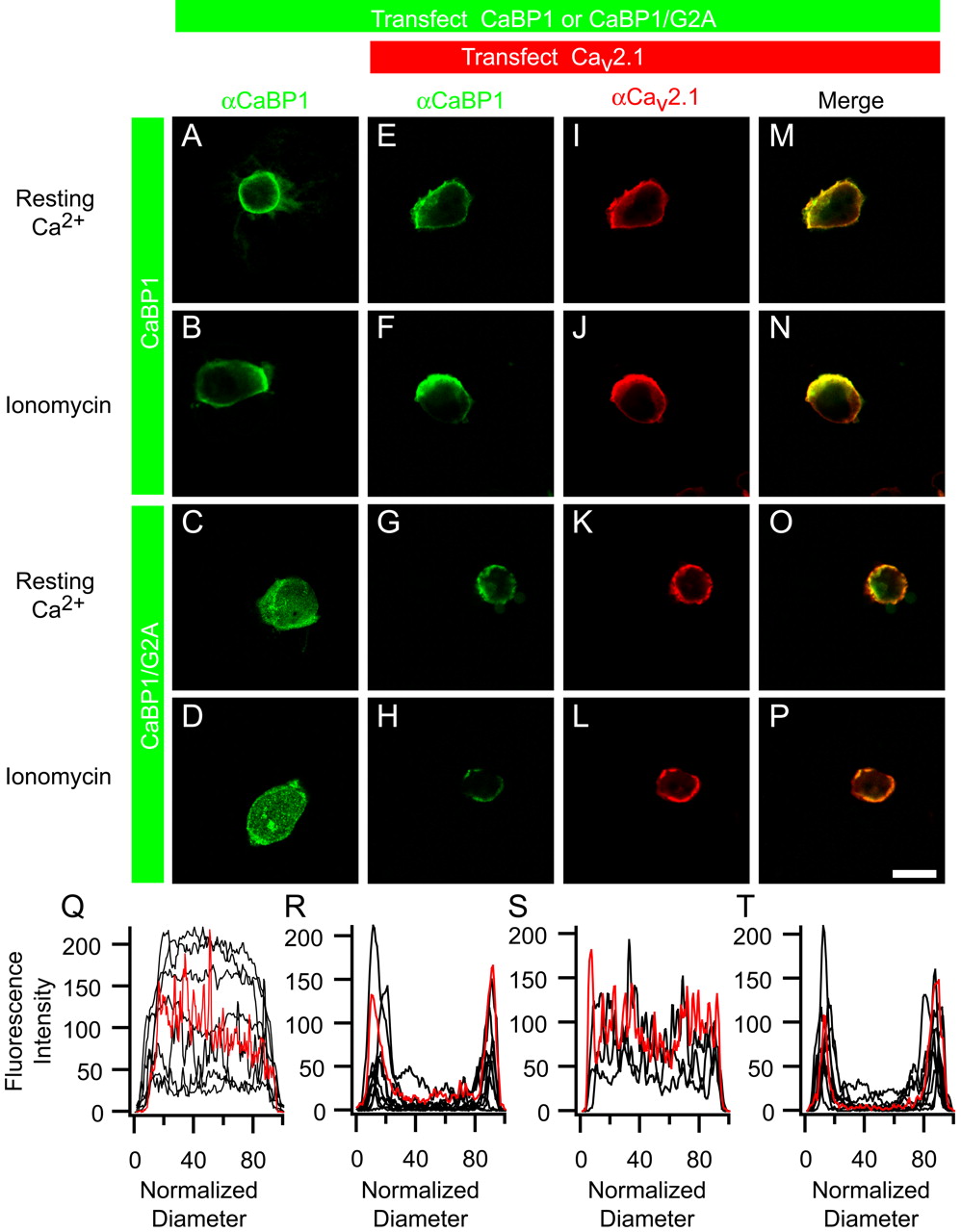
Myristoyl-deficient CaBP1 colocalizes with CaV2.1 channels in tsA-201 cells. A, B, Confocal images of tsA-201 cells transfected with CaBP1 alone and fixed at resting Ca2+ levels (A) or after treatment with ionomycin (B). C, D, Confocal images of cells transfected with CaBP1/G2A alone and fixed at resting Ca2+ levels (C) or after treatment with ionomycin (D). E, F, I, J, M, N, Confocal images of cells transfected with CaV2.1, β2a, α2δ, and CaBP1 fixed at resting Ca2+ levels (E, I, M) or after treatment with ionomycin (F, J, N). G, H, K, L, O, P, Confocal images of cells transfected with CaV2.1, β2a, α2δ, and CaBP1/G2A fixed at resting Ca2+ levels (G, K, O) or after treatment with ionomycin (H, L, P). Scale bar, 50 μm. Q, Line scans of cells transfected with CaBP1/G2A alone, showing fluorescence intensity as a function of position across the cell diameter after fixation at resting Ca2+ levels (n = 8). R, Line scans of cells transfected with CaV2.1, β2a, α2δ, and CaBP1/G2A, fixed at resting Ca2+ levels (n = 10). S, Line scans of cells transfected with CaBP1/G2A alone, fixed after treatment with ionomycin (n = 4). T, Line scans of cells transfected with CaV2.1, β2a, α2δ, and CaBP1/G2A, fixed after treatment with ionomycin (n = 7). Red traces in Q-T correspond to line scans of the cells in images in C, G, D, and H, respectively, whereas black traces were obtained from other similarly treated cells.
Subsequently, we examined the ability of CaBP1 and CaBP1/G2A to colocalize with CaV2.1 channels in intact cells. When tsA-201 cells were cotransfected with CaBP1 and CaV2.1 and fixed at resting Ca2+ levels, immunolabeling showed colocalization of CaBP1 and CaV2.1 at or near the plasma membrane (Fig. 5E,I,M), consistent with the binding of CaBP1 to CaV2.1 observed in coimmunoprecipitation studies. When cells were treated with ionomycin to increase intracellular Ca2+ levels, the pattern of immunolabeling (Fig. 5F,J,N) was not different from labeling of cells at resting Ca2+ levels, consistent with the finding that regulation of CaV2.1 channels by CaBP1 is Ca2+ independent (Lee et al., 2002). Cells cotransfected with CaBP1/G2A and CaV2.1 also showed colocalization of CaBP1/G2A and CaV2.1 at or near the plasma membrane at both resting Ca2+ levels (Fig. 5G,K,O) and elevated intracellular Ca2+ levels (Fig. 5H,L,P). Thus, coexpression of CaV2.1 channels alters the distribution of CaBP1/G2A from general distribution throughout the cell when expressed alone (Fig. 5C,D) to colocalization with CaV2.1 channels at or near the plasma membrane when expressed together (Fig. 5G,H,O,P). These results indicate that CaBP1/G2A binds to CaV2.1 channels in intact tsA-201 cells, consistent with our coimmunoprecipitation results on cross-linked complexes of CaBP1/G2A and CaV2.1 channels.

CaBP1 and VILIP-2 differentially modulate CaV2.1 expression. A, Ca2+ tail current-voltage curves from cells cotransfected with CaV2.1, β2a, α2δ (•, n = 15), and CaBP1 (○, n = 13), or CaBP1/G2A (□, n = 18) show myristoylation of CaBP1 is required to decrease CaV2.1 current density. The data shown are averages ± SEM (*p < 0.05; **p < 0.01). B, Top, Immunoblots for the α12.1 subunit or heat shock protein 90 (Hsp90) from lysates of cells transfected with CaV2.1, β2a, α2δ (left), plus CaBP1 (middle), or VILIP-2 (right). Bottom, Lysates from cells transfected with CaV2.1, β2a, α2δ (n = 9), and CaBP1 (n = 6), or VILIP-2 (n = 5) were analyzed by immunoblotting for α12.1, Hsp90, and CaBP1 or VILIP-2. Both isoforms of α12.1 (Sakurai et al., 1996) were included in the densitometric scan. α12.1 expression was normalized to Hsp90 intensity to control for equal loading. Error bars represent SEM.
Additional evidence to support binding of CaBP1/G2A to CaV2.1 channels in intact cells derives from comparison of line scans of the subcellular localization of CaBP1/G2A when expressed alone with the localization of CaBP1/G2A when coexpressed with CaV2.1 channels. Line scans across the diameter of the cell through the center of the nucleus show a plateau of fluorescence across the cell at both resting and elevated intracellular Ca2+ levels for cells transfected with CaBP1/G2A alone (Fig. 5Q,S). In contrast, cells coexpressing CaBP1/G2A and CaV2.1 show substantial peaks of fluorescence at or near the plasma membrane (Fig. 5R,T). This quantitative change in the subcellular localization of CaBP1/G2A caused by overexpression of a target for binding indicates that CaBP1/G2A binds to CaV2.1 channels in intact cells.
Differential modulation of CaV2.1 expression by CaBP1 and VILIP-2
In addition to the previously observed effects of CaBP1 on the function of CaV2.1 channels (Lee et al., 2002), we characterized a new modulatory effect of CaBP1 on expression of CaV2.1 channels. In cells cotransfected with CaBP1 and CaV2.1 channels, the amplitude of the Ca2+ tail current (Fig. 6A, open circles) was smaller than that for cells transfected with CaV2.1 alone (Fig. 6A, filled circles) over the entire range of voltages tested. This effect is Ca2+ independent, because amplitudes of IBa in cells transfected with CaBP1 and CaV2.1 are also smaller than in cells transfected with CaV2.1 channels only (data not shown). These data indicate that CaBP1 substantially decreases peak currents conducted by CaV2.1 channels. In cells cotransfected with CaBP1/G2A and CaV2.1 channels, the peak Ca2+ tail current (Fig. 6A, open squares) is not different from cells transfected with CaV2.1 channels only (Fig. 6A, filled circles). A similar effect of CaBP1/G2A is observed for IBa (data not shown). Therefore, consistent with its other modulatory effects, myristoylation of CaBP1 is required to decrease expression of functional CaV2.1 channels.

Myristoylation of VILIP-2 is required for distinct modulation of CaV2.1 channels. A, B, ICa elicited by a 1 s depolarization from -80 to +10 mV in cells transfected with CaV2.1, β2a, α2δ (A, black trace, n = 17), and VILIP-2 (A, B, light gray trace, n = 12), or VILIP-2/G2A (B, dark gray trace, n = 7). C, Amplitudes of the residual Ca2+ currents at the ends of the depolarizations (Ires) were normalized to the peak (Ipk) and plotted for the data in A and B. The data are shown as smoothed averages ± SEM with every 100th error bar plotted for clarity (*p < 0.05; **p < 0.01).
To examine the mechanism by which CaBP1 decreases peak currents, we measured the level of CaV2.1 protein in lysates of transfected cells compared with the constitutively expressed housekeeping protein Hsp90 (heat shock protein 90). Immunoblots of lysates from tsA-201 cells transfected with CaBP1 and CaV2.1 show a decrease in the intensity of staining for α12.1 compared with cells transfected with CaV2.1 alone (Fig. 6B). To determine whether this decrease in CaV2.1 expression was an artifact of cotransfection, we cotransfected a different nCaBP that modulates CaV2.1 channels, VILIP-2 (Lautermilch et al., 2005). Lysates from cells expressing CaV2.1 and VILIP-2 did not show a decrease in staining for α12.1 (Fig. 6B), indicating that the reduction of expression is a specific effect of CaBP1.
Effect of myristoylation on modulation of CaV2.1 channels by VILIP-2
Coexpression of VILIP-2 has the opposite effect on the rate of inactivation of CaV2.1 channels as CaBP1 (Lautermilch et al., 2005). To determine whether myristoylation, which is a conserved feature of the nCaBP family, is also required for the distinct modulation of CaV2.1 channels by VILIP-2, we used a myristoyl-deficient mutant of VILIP-2, VILIP-2/G2A. ICa in cells coexpressing CaV2.1 and VILIP-2 inactivates more slowly [Ires/Ipk = 0.65 ± 0.04 (Fig. 7A, light gray trace)] than ICa in cells expressing only CaV2.1 channels modulated by endogenous CaM (Ires/Ipk = 0.40 ± 0.04) (Fig. 7A, black trace), consistent with previous findings (Lautermilch et al., 2005). However, ICa in cells coexpressing VILIP-2/G2A and CaV2.1 channels inactivates more rapidly [Ires/Ipk = 0.44 ± 0.07 (Fig. 7B, dark gray trace)] than ICa in cells coexpressing VILIP-2 and CaV2.1 (Fig. 7B, light gray trace) and is not significantly different from modulation of CaV2.1 inactivation by endogenous CaM (Fig. 7C). These data indicate that myristoylation of VILIP-2 is required to slow the rate of CaV2.1 inactivation and suggest that myristoylation, a conserved feature of nCaBPs, may be essential to allow these proteins to differentially regulate CaV2.1 channels.
Discussion
Myristoylation of CaBP1 and VILIP-2 is required for their distinct modulation of inactivation
Our results with the myristoylation-deficient mutants CaBP1/G2A and VILIP-2/G2A show that myristoylation of CaBP1 and VILIP-2 is required to produce their characteristic regulation of inactivation of CaV2.1 channels, which is distinct from regulation by CaM. CaBP1 increases inactivation of CaV2.1 channels whether Ca2+ or Ba2+ is the permeant ion, whereas VILIP-2 decreases inactivation whether Ca2+ or Ba2+ is the permeantion. In contrast, coexpression of the myristoylation-deficient mutants with CaV2.1 channels has no effect on Ca2+-dependent inactivation. Thus, myristoylation is required for the characteristic effects of these nCaBPs on CaV2.1 channels, although they have opposite effects on inactivation.
Myristoylation of CaBP1 is required for block of paired-pulse facilitation and for modulation of expression and activation
CaBP1 blocks Ca2+/CaM-dependent facilitation of CaV2.1 channels, but VILIP-2 does not (Lee et al., 2002; Lautermilch et al., 2005). As for inactivation, block of paired-pulse facilitation of CaV2.1 channels by CaBP1 requires myristoylation, although partial block of facilitation by CaBP1/G2A is observed late in trains of stimuli. Similarly, CaBP1 positively shifts voltage-dependent activation and reduces functional expression of CaV2.1 channels, whereas VILIP-2 does not. These effects of CaBP1 also require myristoylation. Thus, myristoylation is required for the characteristic effects of CaBP1 on expression, activation, inactivation, and paired-pulse facilitation of CaV2.1 channels and for the normal level of block of facilitation in trains of stimuli.
Although the effects of CaBP1 on CaV2.1 channel function were described previously, the decrease in the amplitude of peak CaV2.1 currents because of reduced expression of the α12.1 subunit is a new effect. Our results do not distinguish whether this reduction in protein expression is caused by decreased transcription and/or translation, decreased posttranslational processing and targeting, or increased degradation. However, effects on transcription and translation are less likely, because these processes are driven by the promoter and translation start signals in the cDNA vector rather than those specified by the CaV2.1 gene. Although VILIP-2 does not change the expression of CaV2.1, the related nCaBP, NCS-1, reduces expression of the α1 subunits of CaV1.2, CaV2.1, and CaV2.2 channels when expressed with β1, β2, and β4 subunits but not with β3 subunits (Rousset et al., 2003). This suggests that regulation of CaV2.1 peak current amplitude by CaBP1 may also require specific β subunit(s). However, we found that CaBP1 reduces peak CaV2.1 currents when coexpressed with β1b (data not shown) as well as with β2a.
Myristoylation enhances, but is not required for, binding of CaBP1
CaBP1 and CaM bind to the CaM-binding domain (CBD) in the C terminus of CaV2.1 channels (Lee et al., 2002). Coimmunoprecipitation experiments with in situ cross-linking indicate that myristoyl-deficient CaBP1 binds to CaV2.1 channels but with reduced affinity and/or stability in the absence of Ca2+. Colocalization studies indicate that CaV2.1 channels bind myristoyl-deficient CaBP1 in intact cells and redirect its distribution to or near the plasma membrane. The failure of CaBP1/G2A to alter activation, inactivation, and paired-pulse facilitation of CaV2.1 channels, although it binds to them in intact cells, indicates that myristoyl-deficient CaBP1 acts functionally like CaM when bound to their common binding site.
In previous work (Lee et al., 2002) and in these experiments, we found that CaBP1 binding and action is independent of Ca2+ from the resting level in tsA-201 cells to the high level achieved in the presence of Ca2+ ionophore. In contrast, Haynes et al. (2004) have shown that CaV2.1 channels in brain extracts interact with immobilized glutathione S-transferase/CaBP1 in a Ca2+-dependent manner. One explanation for this apparent discrepancy may be that binding of CaBP1 occurs in intact cells at 37°C and resting intracellular Ca2+ (∼100 nm) in our experiments, whereas binding occurs in vitro at 4°C and nominally zero Ca2+ in the experiments by Haynes et al. (2004).
Control of CaBP1 localization by N-terminal myristoylation
Myristoylation has been shown to control membrane localization of several nCaBPs, including NCS-1, KChip1, hippocalcin, VILIP-1, and recoverin (Dizhoor et al., 1993; Kobayashi et al., 1993; Spilker et al., 1997; O'Callaghan and Burgoyne, 2003; O'Callaghan et al., 2003). Although myristoylation of CaBP1 is necessary for localization at or near the plasma membrane when it is expressed alone [Haynes et al. (2004) and the present study], we conclude that myristoylation of CaBP1 is not essential for association with a membrane-bound target protein like CaV2.1 channels. Overexpression of other target proteins changes the localization of nCaBPs (O'Callaghan et al., 2003). For example, overexpression of Kv4.2 localizes KChip1 to the plasma membrane, whereas KChip1 is located in punctate intracellular structures when expressed alone (O'Callaghan et al., 2003).
Myristoyl groups can impart regulated membrane association to nCaBPs via a Ca2+-myristoyl switch. However, the constitutive membrane association of CaBP1 in the absence of Ca2+ indicates that either CaBP1 does not have a functional Ca2+-myristoyl switch or its Ca2+-myristoyl switch is activated at the resting intracellular Ca2+. NCS-1/frequenin and KChip1 are also membrane-associated in their Ca2+-free and Ca2+-bound states (O'Callaghan and Burgoyne, 2003). The NMR structure of frequenin, a Drosophila homolog of NCS-1, shows that the myristoyl group is exposed to an aqueous environment in Ca2+-free and Ca2+-bound forms (Ames et al., 2000), which provides a structural basis for lack of a Ca2+-myristoyl switch. A similar structural mechanism may explain the lack of CaBP1 translocation in response to increases in intracellular Ca2+. The presence or absence of a Ca2+-myristoyl switch may determine the time scale on which nCaBPs respond to Ca2+. Proteins that undergo a Ca2+-myristoyl switch may require longer, more widespread elevations in Ca2+ to translocate to membranes and regulate their Ca2+-dependent targets, whereas nCaBPs that are constitutively bound to their targets may regulate them constitutively or may quickly respond to local Ca2+ elevations (O'Callaghan et al., 2002; O'Callaghan and Burgoyne, 2003).
Control of CaBP1 and VILIP-2 function by N-terminal myristoylation
Although myristoylation is not essential for binding of CaBP1 to CaV2.1 channels, it is required for normal functional activity. How might myristoylation control the function of CaBP1 and VILIP-2? In addition to their role in targeting to specific membrane compartments, myristoyl groups often regulate the functional activity of nCaBPs. For example, myristoylation of recoverin is not necessary to inhibit rhodopsin kinase (Chen et al., 1995). However, by inducing cooperativity in Ca2+ binding to the two functional EF-hands in recoverin, myristoylation increases its inhibition of rhodopsin kinase (Kawamura et al., 1994; Calvert et al., 1995; Senin et al., 1995). Myristoylation also enhances cooperativity of Ca2+ binding to neurocalcin δ (Ames et al., 1995; Ladant, 1995). Thus, conformational changes in nCaBPs can be controlled by N-terminal myristoylation.
Our results provide a preliminary indication of how altered conformational changes in myristoylated CaBP1 and VILIP-2 may cause their distinct regulation of CaV2.1 channels. EF-hands 1 and 2 in the N-terminal lobe of CaM interact with the CBD in the C terminus of Cav1.2 channels to initiate Ca2+-dependent inactivation (Lee et al., 2003). EF-hand 2 of CaBP1 is inactive in Ca2+ binding, whereas EF-hand 1 of VILIP-2 is inactive. Our results suggest that interaction of these N-terminal EF-hands with CaV2.1 channels is altered by N-terminal myristoylation. In the absence of myristoylation, these EF-hands bind to CaV2.1 channels and act like CaM, causing Ca2+-dependent inactivation, as we observed for CaBP1/G2A and VILIP-2/G2A. We propose that myristoylation of CaBP1 allows enhancement of inactivation at resting intracellular Ca2+ mediated by its inactive EF-hand 2, whereas myristoylation of VILIP-2 allows inhibition of inactivation at resting intracellular Ca2+ mediated by its inactive EF-hand 1. The active EF-hands 3 and 4 of CaBP1 and VILIP-2 likely cause CaM-like Ca2+-dependent facilitation in both wild-type and myristoyl-deficient forms of these proteins. However, this facilitation is immediately reversed by the enhanced inactivation caused by myristoylated CaBP1 leading to apparent block of facilitation (Fig. 3F), whereas it is enhanced by the slowed inactivation caused by myristoylated VILIP-2 (Lautermilch et al., 2005).
Differential regulation of CaV2.1 channels by nCaBPs and synaptic plasticity
nCaBPs that undergo large conformational changes after binding Ca2+ (Cox et al., 1994; Burgoyne et al., 2004) and bind Ca2+ with submicromolar affinities (Burgoyne and Weiss, 2001) are poised to act as potent regulators of synaptic transmission. nCaBPs may modulate synaptic responses by acting as local buffers that regulate residual Ca2+ that accumulates with multiple action potentials (Zucker, 2003). In addition, the Ca2+-dependent conformational changes that nCaBPs experience may allow these proteins to act as regulators to produce facilitation and depression of neurotransmission (Zucker, 2003). By regulating presynaptic CaV2.1 channels, CaBP1, VILIP-2, and NCS-1 can directly shape presynaptic Ca2+ transients, which will have profound effects on the time course and amount of neurotransmitter release. Because CaBP1 and VILIP-2 are differentially expressed throughout the nervous system (Braunewell and Gundelfinger, 1999; Haeseleer et al., 2000), they will regulate the output of neurons in a cell-type-specific manner. Myristoylation, by changing the conformation, cooperativity of Ca2+-binding, and targeting of nCaBPs, can tune the functional activity of these proteins.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS22625 (W.A.C.), F32 NS11099 (N.J.L.), and T32 GM07270 (A.P.F.). We thank Drs. F. Haeseleer and K. Palczewski, Departments of Opthalmology and Pharmacology, University of Washington, for cDNAs encoding CaBP1 and CaBP1/G2A.
Correspondence should be addressed to William Catterall, Department of Pharmacology, University of Washington School of Medicine, 1959 Northeast Pacific Street, Room F427, Seattle, WA 98195. E-mail: wcatt{at}u.washington.edu.
N. J. Lautermilch's present address: MDS Pharma Services, 22011 30th Drive Southeast, Bothell, WA 98021-4444.
Copyright © 2005 Society for Neuroscience 0270-6474/05/257071-10$15.00/0





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}