

Abstract
Previous work demonstrated that a brief, sublethal excitotoxic insult strikingly increased the sensitivity of cortical neurons to the cytotoxic effects of the terminal pathway of complement, a process termed “excitotoxic sensitization.” Here, we sought to elucidate the cellular mechanism of excitotoxic sensitization in embryonic rat cortical neurons in vitro. Excitotoxic sensitization did not increase membrane attack complex deposition on cortical neurons and produced only a small reduction of membrane attack complex removal, because of a selective decrease of endocytic elimination. Membrane attack complexes and other osmotic stressors, namely hypotonic stress and glutamate, induced transient swelling of cortical neurons, followed by return to normal volume despite persistence of the stressor, a homeostatic response termed regulatory volume decrease (RVD). A minimal excitotoxic insult impaired this homeostatic response and sensitized neurons to cytotoxic effects of diverse osmotic stressors. Structurally distinct membrane-impermeable osmolytes, dextran and polyethylene glycol, prevented excitotoxic sensitization to diverse osmotic stressors including membrane attack complexes. Paraquat, a reactive oxygen species generator, alone was sufficient to impair RVD, and MnTBAP [Mn(III)tetrakis(4-benzoic acid)porphyrin chloride], a reactive oxygen species scavenger, prevented excitotoxin-mediated impairment of RVD. Together, these findings demonstrate that impairment of RVD is the mechanism of excitotoxic sensitization, that reactive oxygen species alone are sufficient to impair RVD, and that reactive oxygen species are necessary for excitotoxic sensitization-mediated impairment of RVD.
- complement
- membrane attack complexes
- regulatory volume decrease
- hypotonic
- excitotoxic sensitization
- homeostatic
Introduction
Complement is an important member of the innate immune system and also one of the major effectors of the humoral component of the adaptive immune system (Walport, 2001). Although diverse mechanisms can activate complement, each activation pathway culminates in the formation of C5b, the first component of the terminal pathway. Subsequent steps within the membrane attack pathway involve the sequential binding of C6, C7, C8, and multiple C9s with C5b to form a multimolecular complex, the membrane attack complex (MAC). The transmembrane channels formed by MAC allow ion influx, resulting in colloid osmotic imbalance and ultimately cytolysis (Morgan, 1995). Although its normal function is critical to the health of the organism, inappropriate activation of complement can itself damage tissue and contribute to disease of many organs. In the CNS, immunohistochemical evidence of MAC has been detected in brains of humans with multiple neurodegenerative diseases, including Alzheimer's (Itagaki et al., 1994; Webster et al., 1997) and Huntington's disease (Singhrao et al., 1999), Rasmussen's encephalitis (Whitney et al., 1999), and multiple sclerosis (Compston et al., 1989; Scolding et al., 1989).
One mechanism of cellular injury unique to the nervous system is excitotoxicity whereby excessive activation of ionotropic glutamate receptors can destroy neurons (Huang and McNamara, 2004). Evidence from in vitro models and multiple animal models have implicated excitotoxicity as a mechanism of neuronal injury after diverse insults, including ischemia, hypoxia, seizures, trauma, hypoglycemia, and others (Choi, 1988; Doble, 1999). Clinical studies indicate that fleeting insults thought to involve excitotoxic mechanisms such as ischemia and flurries of seizures are associated with an accelerated rate of neurological deterioration in Alzheimer's disease (Volicer et al., 1995) and Rasmussen's encephalitis (Oguni et al., 1991), diseases in which inappropriate activation of complement has been implicated. These studies led to our discovery that a brief, sublethal activation of ionotropic glutamate receptors resulted in a dramatic increase in the cytotoxic effects of complement, a process termed “excitotoxic sensitization” (Xiong and McNamara, 2002). The objective of the present study was to elucidate the cellular mechanism of excitotoxic sensitization.
Materials and Methods
Neuronal cultures.
Cortices from embryonic day 18 (E18) gestation rats were trypsinized (10 min, 37°C), washed, and gently triturated by passing the tissue through a Pasteur pipette with fire-polished tip. The cells were plated at a density of 100,000 cells/well in poly-d-lysine-coated 24-well polystyrene plates or 60,000 cells/poly-d-lysine-coated glass coverslips and maintained at 37°C in a humidified incubator with 5% CO2/95% O2 in growth medium. The cells were plated in Neurobasal medium containing 10% fetal bovine serum, B27, and glutamax I. Twenty-four hours after plating, the culture medium was completely replaced with Neurobasal medium without serum. These serum-free conditions supported the growth of neurons but not glial cells (Ganguly et al., 2001). Cultures were fed once a week by adding Neurobasal medium. Neurons were maintained for 12–16 d before experiments in a humidified incubator at 37°C in 95% O2/5% CO2.
Terminal complement proteins (C5b6 complex, C7, C8, C9) were obtained from Advanced Research Technologies (San Diego, CA). Equal concentration of each component in μg/ml was sequentially added, which led to the formation of membrane attack complexes on cell surfaces, without the need for enzymatic activation. The sublethal dose of complement was 1 μg/ml, as determined by lactate dehydrogenase (LDH) cell assay and confirmed with trypan blue exclusion assay (data not shown). Hypotonic solution was prepared by diluting culture media with the corresponding percentage of water (30% hypotonic, 223 mOsm; 60% hypotonic, 127 mOsm; 70% hypotonic, 94 mOsm).
Cell death assay.
Cell death was assessed by measuring LDH activity in culture supernatants by a spectrophotometric method (Vassault, 1983). These results were validated by an independent measure of cell death using the trypan blue dye (0.4%) exclusion assay (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Data was presented as the means and SEM of determinations made in six wells per condition of a representative experiment. Each experiment was performed at least three times.
Protein iodination.
Proteins (anti-neoC9 and C9; Advanced Research Technologies) were iodinated by the IodoBead (Pierce, Rockford, IL) method, as suggested by the manufacturer. Briefly, between 0.1 and 0.5 mg of protein was incubated with 0.5 mCi of NaI125 (Amersham Biosciences, Uppsala, Sweden) and two IodoBeads (Chloramine T-derivatized polystyrene) at 4°C for 30 min. The iodinated protein was separated from free iodine by chromatography on Sephadex G-25. Specific radioactivity approximated 0.5 Ci/g.
MAC endocytosis and shedding assay.
The specificity of 125I-anti-neoC9 was tested by determining antibody binding after sequential addition of each of the proteins of the terminal pathway (C5b6 complex, C7, C8, C9; Advanced Research Technologies) in comparison to the sequential addition of each of all of these components except C7. Once the specificity had been established, subsequent experimental procedure consisted of washing cultured cortical neurons with MEM and purified complement proteins were added sequentially and allowed to bind at 37°C for 10 min. Unbound complement was removed by washing with MEM. Next, the surface MACs were labeled by addition of 125I-anti-neoC9 on ice for 5 min and the unbound antibody was removed by washing. Thereafter, the neurons were incubated at 37°C for varying periods of time to allow MAC endocytosis and shedding. Shedded MACs were quantified by removing the extracellular media and measuring the radioactivity with a gamma counter. The antibody bound to the surface MACs was then removed by acid wash (Carroll et al., 1999) using two washes over 8 min. This procedure selectively strips bound antibody from the cell surface and leaves intracellular antibody-receptor complexes intact. The counts in the acid wash fraction reflected the amount of MACs on the cell surface. The remaining counts on the cells after the acid wash reflected the endocytosed MACs.
To ensure that the acid stripping procedure removed surface-bound antibody, immunofluorescence was performed using a fluorescence secondary antibody against anti-neoC9. In unstripped cells, surface MACs were visualized in numerous puncta. After acid stripping, labeling of surface MACs was abolished (data not shown), thus confirming the efficiency of acid stripping.
MAC deposition assay.
The cultured neurons were washed with MEM and the terminal complement proteins, C5b6, C7, C8, 125I-C9 were added sequentially. The cells were then incubated at 37°C for varying periods of time. Controls were performed by the sequential addition of all terminal complement proteins except C7. After which, unbound complement proteins were washed off, cells were solubilized with lysis buffer [0.01 m Tris-HCl, pH 7.4, 1 mm EDTA, 8 m urea, 0.05 m dithiothreitol, 10% (v/v) glycerol, 5% (v/v) Nonidet P-40], and the amount MAC deposited was quantified by measuring radioactivity with a gamma counter.
Cell volume measurements.
To monitor changes in cell volume, cultured neurons on glass coverslips were loaded for 40 min at room temperature with calcein-acetoxymethyl ester (5 μm) (Crowe et al., 1995). Coverslips were then mounted onto a RC-25 recording chamber (Warner Instruments, Hamden, CT) and the intracellular calcein fluorescence changes were measured at room temperature with the 488 nm excitation line using a Leica (Nussloch, Germany) TCS SL confocal system set up on a Leica DMIRE2 inverted microscope equipped with epifluorescent illumination and a 63× oil-immersion objective (numerical aperture, 1.4). Images were taken at 1 min intervals and two line averages were performed to improve image quality. Only live cells show calcein fluorescence, which was insensitive to changes in pH and intracellular ions (Haugland, 1996). Photobleaching was minimized by reducing the intensity of the excitation light (laser power, 2%). At this laser intensity, control experiments revealed no detectable photobleaching during the course of the experiments. The change in fluorescence intensity was measured by tracing cell borders on the computer monitor with the use of a mouse and a computer-generated cursor. Each image was traced twice and the average fluorescence intensity of the traced regions was quantified using Leica confocal image analysis software. The relative change in cell volume was obtained by taking an inverse of It/I0, where It and I0 were the fluorescence intensity at time t and time 0 respectively.
To verify that changes in calcein fluorescence intensity accurately reflected changes in cell volume, independent measures of cell size were obtained using differential interference contrast (DIC) microscopy. That is, cells were visualized on a Leica DMIRE2 inverted microscope equipped with a 63× oil-immersion objective (numerical aperture, 1.4). The cross-sectional area of cells was calculated by adjusting the plane of focus to the level at which the neuron soma was widest and the change in somal cross-sectional area with time was measured by tracing cell borders on the monitor (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Each image was traced twice and the values were averaged. The cell somal cross-sectional area of the traced regions was quantified using Leica confocal image analysis software and corrected for volume using the following equation: relative cell volume = Vt/V0 = (cross-sectional areat/cross-sectional area0)3/2. As shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material), there was an excellent correlation in estimates of cell volume by these two independent methods after osmotic perturbations consisting of sublethal complement, hypotonic solution, or glutamate. In contrast, for lethal complement attack, the estimates of cell volume increase obtained from calcein fluorescence greatly exceeded the estimates by DIC microscopy (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material); we suspect that this discrepancy is attributable, at least in part, to leakage of calcein out of the cell through the MAC channels.
Results
Our previous study demonstrated that a minimal excitotoxic stress sensitizes cortical neurons to death by complement and that the complement molecule effecting cytotoxicity is the MAC (Xiong and McNamara, 2002). We extended the previous results in the following experiments. Briefly (10 min) treating cortical cultures with a low concentration of glutamate (30 μm) resulted in minimal cell death (Fig. 1A, compare first open bar and first filled bar). In contrast, pretreatment with a minimally toxic concentration of glutamate (30 μm, 10 min) potentiated the cytotoxicity of MACs in a concentration-dependent manner (Fig. 1A). Similarly, excitotoxic sensitization was also dependent on the concentration of glutamate as evident in experiments using an otherwise minimally toxic concentration of complement (1 μg/ml) (Fig. 1B). Thus, our results demonstrate that pretreatment with glutamate acted synergistically with complement to destroy cortical cells, as evident in the massive increase in LDH release after excitotoxic sensitization. Selective activation of NMDA receptors (NMDA, 20 μm for 5 min in the presence of CNQX, 50 μm) or kainate/AMPA receptors (KA, 100 μm, 30 min in the presence of d-APV, 50 μm) produced sensitization similar to that of glutamate (data not shown).
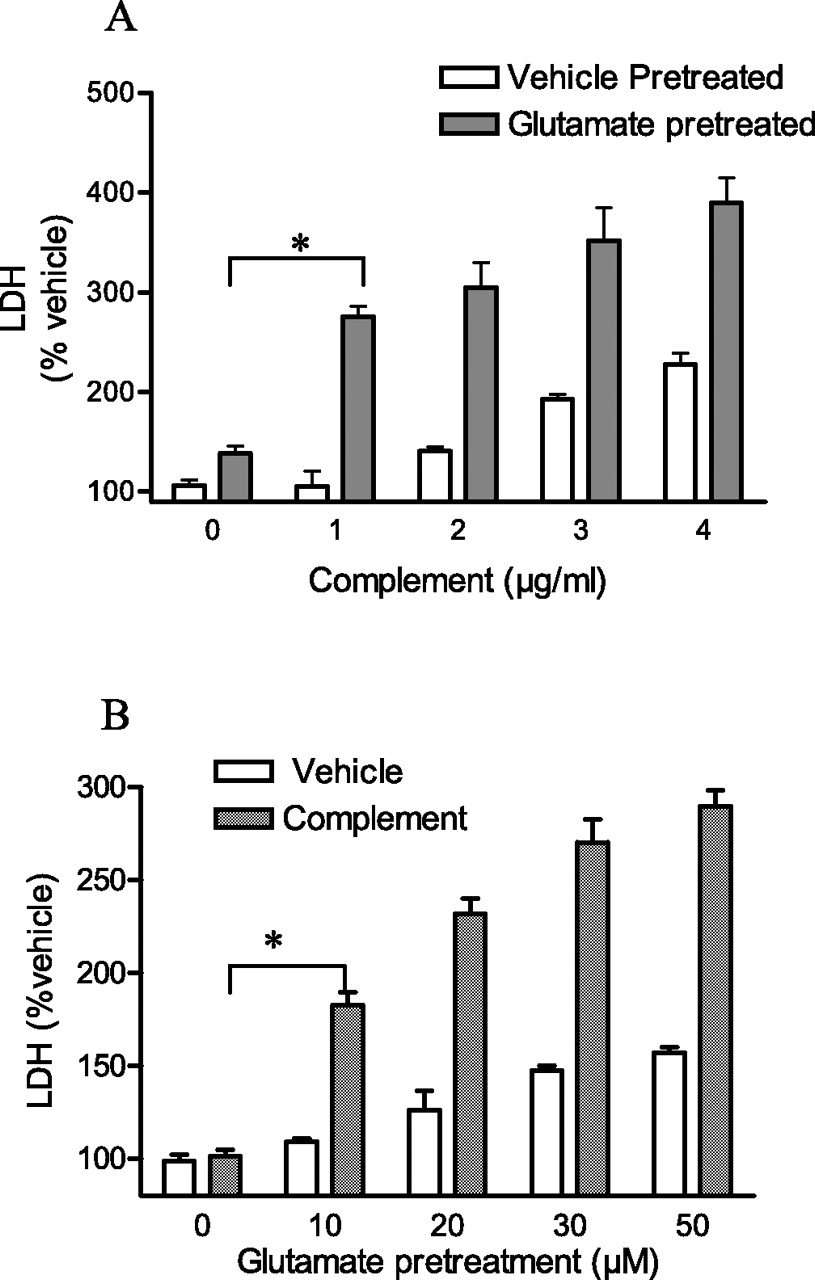
Excitotoxic insults sensitize cortical neurons to complement attack. A, Cortical neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, after which individual proteins of the terminal pathway were added sequentially and incubated for 2 h. Filled and open bars denote glutamate or vehicle pretreatment, respectively. *p < 0.001 comparing glutamate pretreatment followed by vehicle and glutamate pretreatment followed by terminal pathway proteins (ANOVA with Student's t tests). B, Cultures were pretreated with increasing concentrations of glutamate for 10 min, followed by C5b6 plus C7/C8/C9 (1 μg/ml each). LDH released into the bathing medium was assessed 2 h after addition of terminal pathway proteins. Hatched and open bars denote presence or absence of terminal pathway proteins, respectively. *p < 0.001 comparing vehicle pretreatment followed by terminal pathway proteins and glutamate pretreatment followed by terminal pathway proteins (ANOVA with Student's t tests). Values in this figure are the means and SEM of six wells per condition of a single experiment. Each experiment was performed at least three times, and similar results were obtained.
Subsequent experiments examined several hypotheses in an effort to elucidate the mechanism by which the brief, minimally excitotoxic stimulus sensitized cortical neurons to death by complement.
MAC expression on cortical neurons after an excitotoxic stimulus
The mechanism by which MACs destroy cells requires insertion of MACs into the surface membrane of the cell. Previous studies of non-neuronal cells revealed that surface membrane-inserted MACs could be eliminated by both endocytosis and membrane shedding, permitting recovery from complement attack (Morgan et al., 1987; Iida et al., 1991). We therefore asked whether neurons eliminated MACs from the cell surface by endocytosis and/or shedding, and if so, whether these defense mechanisms were compromised by a previous excitotoxic stimulus.
To examine the fate of MACs after membrane deposition, neurons were treated with a nonlethal concentration of complement (1 μg/ml) for 10 min at room temperature, and then MACs were labeled with 125I-anti-neoC9 antibody on ice for 5 min. The cells were then incubated at 37°C to allow MAC elimination. MACs shed from cell surface were quantified by measuring 125I-anti-neoC9 antibody in culture media. MACs remaining on the cell surface were removed by acid stripping and quantified. MACs internalized were quantified by measuring the 125I-anti-neoC9 antibody remaining in the cells after acid stripping. The effectiveness of acid stripping in removal of cell surface anti-neoC9 antibody was verified by immunocytochemistry (data not shown).
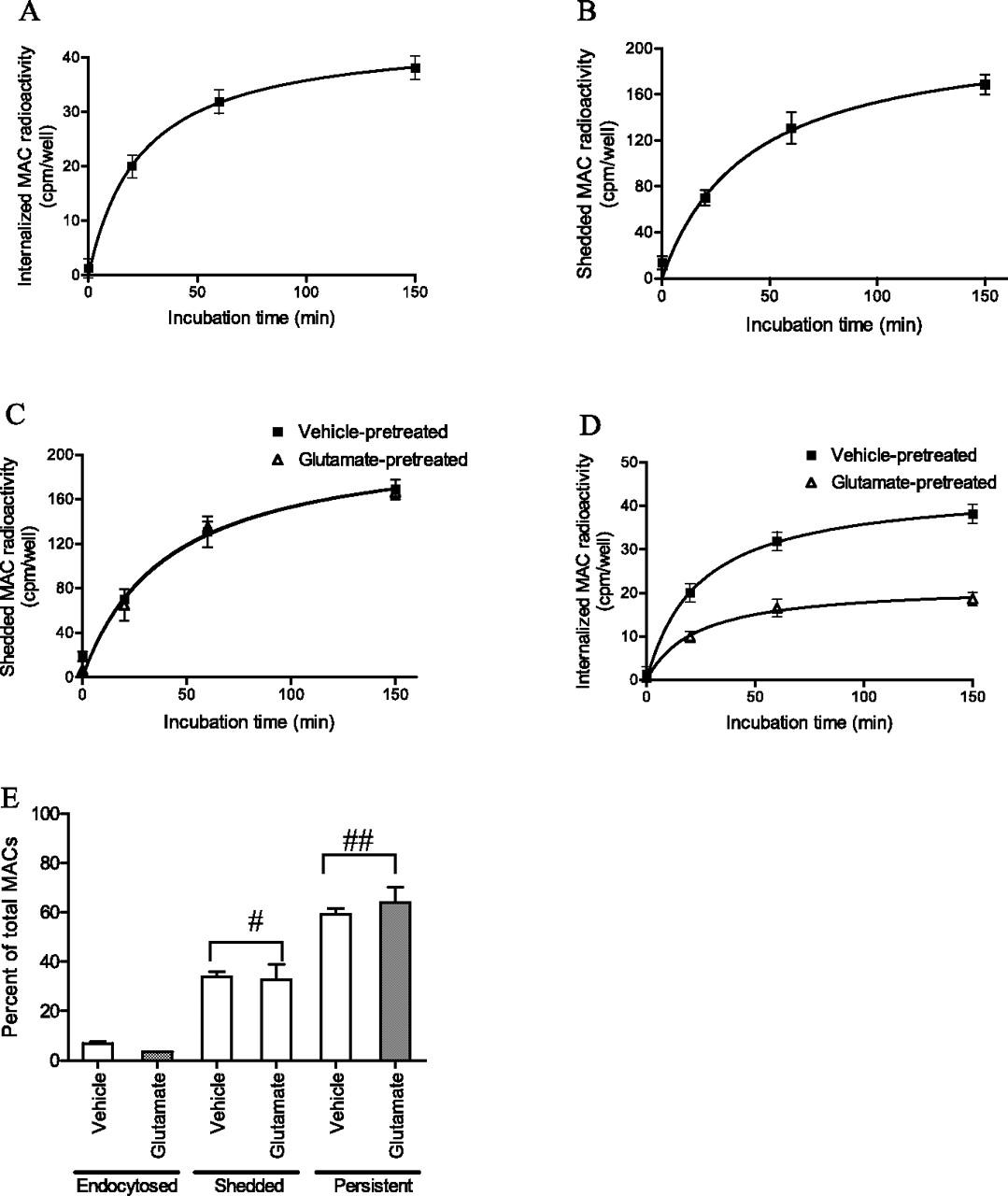
These experiments revealed that neurons did remove MACs from the cell surface by endocytosis in a time-dependent manner (Fig. 2A) but that only a small portion of the MACs (∼7%) inserted into the cell membrane were removed by this mechanism. Neurons also eliminated MACs from the cell surface by shedding into the media (Fig. 2B), the amount exceeding that internalized by almost fivefold (∼35%). Remarkably, the neurons survived despite persistence of more than half of the MACs on the cell surface.

Excitotoxic sensitization has no effect on MAC shedding and minimal effects on endocytic elimination. A, Time course of MAC internalization, assayed with 125I-antineo-C9 antibody. Neurons were treated with terminal pathway proteins (1 μg/ml each) for 10 min at room temperature and labeled with 125I-anti-neoC9 antibody on ice for 5 min. The antibody-labeled cells were then incubated at 37°C to allow MAC elimination. The amount of MACs internalized was determined by measuring the radioactivity remaining in the cells after mild acetic acid wash, which removed 125I-anti-neoC9 antibody bound to surface MACs. B, Time course of MAC shedding, assayed with 125I-anti-neoC9 antibody. Neurons were treated with terminal pathway proteins (1 μg/ml each) for 10 min at room temperature and labeled with 125I-anti-neoC9 antibody on ice for 5 min. The antibody-labeled cells were then incubated at 37°C to allow MAC elimination. The amount of MACs shed was assayed by measuring the amount of radioactivity shed into the bathing medium. C, Effect of glutamate on MAC shedding, assayed with 125I-anti-neoC9 antibody. Cells were incubated in glutamate (30 μm) or vehicle for 10 min and then incubated with terminal pathway proteins (1 μg/ml each) for 10 min before labeling with 125I-anti-neoC9 antibody. Preincubation with glutamate (30 μm) for 10 min did not affect MAC shedding. D, Effect of glutamate on MAC internalization, assayed with 125I-anti-neoC9 antibody. Cells were incubated in glutamate (30 μm) or vehicle for 10 min and then incubated with terminal pathway proteins (1 μg/ml each) for 10 min before labeling with 125I-anti-neoC9 antibody. Preincubation with glutamate (30 μm) for 10 min decreased MAC internalization by 51.06 ± 2.75%. E, Graph of MACs endocytosed, shedded versus persistent on cell surface as a percentage of total MACs in vehicle-pretreated versus glutamate-pretreated groups. #p = 0.4279 comparing glutamate pretreatment versus vehicle pretreatment on MAC shedding. ##p = 0.2747 comparing glutamate versus vehicle pretreatment on amount of persistent MACs (ANOVA with Student's t tests).
Next, we asked whether a minimal excitotoxic stimulus inhibited removal of MACs by either endocytosis or shedding. To address these questions, cultures were briefly (10 min) treated with either glutamate (30 μm) or vehicle and then complement proteins and 125I-anti-neoC9 antibody were added and the elimination of MACs quantified as described in the preceding paragraph. These experiments revealed that preincubation with glutamate did not affect shedding of MACs (Fig. 2C). In contrast, pretreatment with glutamate reduced MAC internalization by 51 ± 3% (Fig. 2D). Because internalization eliminates only 7% of the MACs deposited on the neuronal surface, the net reduction of surface MAC content caused by excitotoxic stimulus approximates 3.5% of the total (Fig. 2E).
The absence of detectable difference in shedding and the small contribution of endocytosis led us to ask whether an excitotoxic stimulus might increase the total amount of MACs that had been deposited on the surface membrane of neurons initially. Addressing this question is complicated because deposition on the cell surface and elimination are almost certainly occurring simultaneously. That said, we quantified the MACs deposited on the cell membrane using a method distinct from that described in the two preceding paragraphs. Instead of labeling MACs with an 125I-anti-neoC9 antibody, here MACs were labeled by adding 125I-C9 after sequential addition of purified C5b, C6, C7, and C8. This circumvents the time required for the 125I-anti-neoC9 antibody to bind to the MAC. The sequential addition of purified C5b, C6, C7, C8 and 125I-C9 to cultured cortical neurons, followed by 37°C incubation, resulted in MAC deposition in a time-dependent manner. Moreover, preincubation with glutamate (30 μm) for 10 min did not modify the MAC content of cells when compared with vehicle alone (data not shown). The lack of effect of an excitotoxic stimulus on MAC deposition or shedding together with the small effect on endocytosis led us to search for alternative mechanisms that might mediate excitotoxic sensitization.
Nonlethal complement attack induces RVD in cortical neurons
Previous studies revealed that human neutrophils and erythrocytes undergoing nonlethal complement attack swelled and subsequently returned to apparently normal volume, despite the continued presence of complement (Morgan, 1988, Halperin et al., 1993). This led us to ask whether a nonlethal complement attack of a neuron resulted in swelling and subsequent recovery of volume despite the persistence of complement. To address this question, relative neuronal cell volume was measured by loading the cells with calcein AM. After addition of sublethal concentrations of terminal pathway proteins (1 μg/ml), neurons swelled by ≈5% and subsequently returned to their initial volume despite continued presence of the proteins (Fig. 3A). The latency to peak swelling after addition of sublethal concentrations of terminal pathway proteins was 11 ± 0.4 min. To independently verify that changes in calcein fluorescence intensity accurately reflected changes in cell volume, the cross-sectional area of the cell soma was measured in parallel experiments using DIC microscopy, a useful technique for assessing changes in cell volume (Kirk et al. 1984; Strange and Spring, 1986). Estimates of cell volume with these two independent methods revealed an excellent correlation (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material), supporting the validity of each measure. The mechanism by which a sublethal attack by terminal pathway proteins induces neuronal swelling presumably results from insertion of MACs in the surface membrane followed by ions moving down their electrochemical gradient, in turn followed by water to maintain osmotic equilibrium. In contrast to a sublethal attack, the addition of lethal concentrations of terminal pathway proteins (5 μg/ml) induced massive swelling and lysis of neurons (Fig. 3B). The induced swelling of neurons and subsequent recovery of volume despite the persistence of terminal pathway proteins at sublethal concentrations is similar to the effect observed in neutrophils (Morgan, 1988). This type of homeostatic response has been described previously in diverse cell types and termed “regulatory volume decrease” (RVD) (Strange et al., 1996).

Cortical neurons undergo RVD after various volume perturbations. A, Time course of cell swelling and regulatory volume decrease in response to sublethal terminal pathway proteins (1 μg/ml each). Eliminating C7 abolished cell swelling and regulatory volume decrease. B, Time course of cell swelling in response to lethal complement attack, C5b6 plus C7/C8/C9 (5 μg/ml each). C, Time course of cell swelling and regulatory volume decrease in response to 30 and 60% hypotonic challenge. D, Time course of cell swelling in response to 70% hypotonic challenge. E, Time course of cell swelling and regulatory volume decrease in response to sublethal glutamate insult (30 μm). Values in this figure are mean ± SEM for five cells per condition of a single experiment.
Next, we asked whether neurons are capable of RVD after an osmotic perturbation other than terminal pathway proteins. We simply challenged neurons with hypotonic solutions. Stimulation of cortical neurons with a 30% hypotonic solution resulted in a 12.8 ± 1.1% increase of cell volume followed by RVD, despite continued presence of the hypotonic solution (Fig. 3C). Maximum swelling was evident at the earliest time point sampled after addition of hypotonic solution, namely 1 min. Return of iso-osmotic medium from hyposmotic solution resulted in cell shrinkage followed by recovery to its initial volume (data not shown). Incubation with a 60% hypotonic solution induced a 29.2 ± 2.5% increase of cell volume followed by return to initial volume (Fig. 3C). Similar results were obtained when cell volume was estimated by measuring cell soma cross-sectional area (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Incubation with a 70% hypotonic solution induced massive cell swelling and osmotic lysis (Fig. 3D).
Because glutamate itself is known to cause cell swelling (Choi, 1987; Inglefield and Schwartz-Bloom, 1998), we further queried whether a sublethal concentration of glutamate itself produced RVD. Addition of a sublethal concentration of glutamate (30 μm) resulted in a 9.0 ± 1.3% increase of cell volume followed by return to initial volume (Fig. 3E). Like osmotic challenges with sublethal complement and hypotonic solution, assessment of cell volume by measurement of cell soma cross-sectional area revealed an excellent correlation with measures of calcein fluorescence (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). The latency to peak swelling was 6.3 ± 0.3 min.
In sum, these findings demonstrate that cortical neurons are capable of RVD, a homeostatic response to an osmotic stress. Moreover, neurons exhibit RVD in response to diverse stimuli that induce neuronal swelling, some of which are triggered by influx of ions (complement and glutamate) and others triggered simply by lowering extracellular osmolarity (hypotonic solution).
Excitotoxic stimulus compromises RVD
The fact that neurons exhibit RVD when challenged by any one of diverse stimuli that induces neuronal swelling raised the question as to whether a minimal excitotoxic stimulus might impair this homeostatic response. That is, if the minimal excitotoxic stimulus impaired RVD, then glutamate should sensitize neurons to an osmotic perturbation potentially resulting in water influx, massive cell swelling, and death. To test this idea, neurons were first treated with either vehicle or a minimal excitotoxic stimulus and subsequently exposed to hypotonicity. These experiments confirmed our reasoning because a minimally toxic concentration of glutamate potentiated the cytotoxicity of a hypotonic stimulus in a concentration-dependent manner (Fig. 4A). In comparison to vehicle, hypotonic solution (60%) alone produced minimal cell death, as evident by the ≈20% increase in LDH release. In contrast, pretreatment with glutamate acted synergistically with hypotonic solution to destroy cortical cells in a concentration-dependent manner (Fig. 4A). These findings demonstrate that a minimal excitotoxic stimulus can sensitize cortical neurons to death by an osmotic stimulus that induces otherwise reversible cell swelling. We further asked whether excitotoxic sensitization to hypotonicity was associated with impaired RVD. Pretreatment with glutamate (30 μm, 10 min) followed by hypotonic solution (60%) led to massive and irreversible cell swelling (Fig. 4B). These results demonstrated that an excitotoxic sensitization eliminated RVD and raised the possibility that impairment of RVD is the mechanism of excitotoxic sensitization.

Impaired volume regulation sensitizes cortical neurons to death by various osmotic perturbations. A, Cultures were exposed to increasing concentrations of glutamate (0–50 μm) for 10 min, followed by 60% hypotonic solution. LDH released into the bathing medium was assessed 2 h later. B, Excitotoxic sensitization leads to irreversible cell swelling after an innocuous hyposmotic insult. Neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, washed, and treated with 60% hypotonic solution while recording neuronal cell volume. C, Excitotoxic sensitization leads to irreversible cell swelling after an innocuous complement insult. Neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, washed, and treated with sublethal terminal pathway proteins (1 μg/ml each) while recording neuronal cell volume. Inset, Regulatory volume decrease after sublethal complement attack in vehicle-pretreated group. D, Cortical neurons were pretreated with varying concentrations of glutamate for 10 min, washed with MEM, and incubated with glutamate (30 μm) for 2 h. Filled bars were with second glutamate treatment, whereas open bars were with subsequent vehicle treatment. E, Excitotoxic sensitization leads to irreversible cell swelling after an innocuous glutamate insult. Neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, washed, and treated with glutamate (30 μm) while recording neuronal cell volume. Values in A and D are the means and SEM of six wells per condition of a single experiment. Each experiment was performed at least three times, and similar results were obtained. Values in B, C, and E are mean ± SEM for five cells per condition of a single experiment.
In addition, these findings imply that a minimal excitotoxic stimulus may impair RVD induced by other osmotic stressors that induce cell swelling such as terminal pathway proteins. To test this idea, neurons were pretreated with glutamate (30 μm) or vehicle and subsequently exposed to sublethal concentrations of terminal pathway proteins, while measuring cell volume. Pretreatment with glutamate, but not vehicle, eliminated RVD after sublethal concentrations of terminal pathway proteins (Fig. 4C), demonstrating that an excitotoxic stimulus impaired RVD that was activated by diverse stimuli that led to cell swelling. Selective activation of NMDA receptors (NMDA, 20 μm for 5 min in the presence of CNQX, 50 μm) or kainate/AMPA receptors (KA, 100 μm, 30 min in the presence of d-APV, 50 μm) impaired RVD similar to that of glutamate (data not shown).
Because neurons undergo cell swelling and RVD after sublethal glutamate attack (Fig. 3E), we asked whether a previous sublethal glutamate insult might sensitize neurons to death by subsequent sublethal glutamate attack and whether it does by paralyzing RVD. Pretreatment with vehicle followed by treatment with a sublethal concentration of glutamate (30 μm) resulted in RVD and no detectable cell death. In contrast, pretreatment with a minimal excitotoxic stimulus (glutamate, 30 μm, 10 min) followed by a second sublethal glutamate insult led to massive cell swelling and death (Fig. 4D,E).
The fact that glutamate itself induced cell swelling followed by RVD raised the possibility that a variety of stimuli that can induce RVD could themselves sensitize neurons to death by a subsequent stimulus that induced RVD. To test this possibility, neurons were exposed briefly to a hypotonic solution and subsequently exposed to either complement or hypotonic solution and cell death was monitored by measuring LDH release. No evidence of sensitization was detected (supplemental Fig. 3A,C, available at www.jneurosci.org as supplemental material), implying that sensitization was specific to an excitotoxic stimulus. Likewise, pretreatment with a sublethal concentration of terminal pathway proteins alone did not sensitize neurons to a subsequent stimulus by either complement or hypotonic solution (supplemental Fig. 3B,D, available at www.jneurosci.org as supplemental material). Together, sensitization is specific to an excitotoxic stimulus; cell swelling, be it caused by hypotonic stress or by the action of complement, is insufficient to sensitize cortical neurons to impaired volume regulation in response to osmotic stress.
Membrane-impermeable osmolytes rescue cortical neurons from excitotoxin-mediated impaired RVD
Our results demonstrate that an excitotoxic stimulus is sufficient to impair RVD induced by each of three distinct osmotic stressors. These results raise the question as to whether impairment of RVD is the mechanism by which an excitotoxic stimulus sensitizes cortical neurons to death by an osmotic stress. If impairment of RVD is the mechanism mediating excitotoxic sensitization, then membrane-impermeable osmolytes, which oppose the increase in hydrostatic pressure, should prevent cell swelling and rescue the neurons from osmotically induced cell death after a minimal excitotoxic stimulus.
To determine whether membrane-impermeable osmolytes could prevent the cell swelling induced by an osmotic perturbation after excitotoxic sensitization, a brief excitotoxic stimulus was applied followed by hypotonic solution and cell volume was quantified by calcein imaging. To counteract the increase in hydrostatic pressure, the effects of either dextran or polyethylene glycol (PEG) were compared with vehicle together with the hypotonic solution. Addition of either dextran or PEG but not vehicle together with the hypotonic solution resulted in no change of cell volume despite previous excitotoxic sensitization (Fig. 5A). Likewise, both dextran and PEG prevented the increase of cell swelling induced by terminal pathway proteins or glutamate after an excitotoxic prestimulus (Fig. 5C,E). Importantly, both dextran and PEG also prevented the cell death induced by excitotoxic sensitization to hypotonic solution, terminal pathway proteins, or glutamate measured either 2 h (Fig. 5B,D,F) or 24 h (supplemental Fig. 2B,D,F, available at www.jneurosci.org as supplemental material) after osmotic insults. Together, the fact that two distinct membrane-impermeable osmolytes prevented both the cell swelling and cell death support the conclusion that impairment of RVD is the mechanism by which an excitotoxic stimulus sensitizes cortical neurons to death by diverse osmotic stressors.
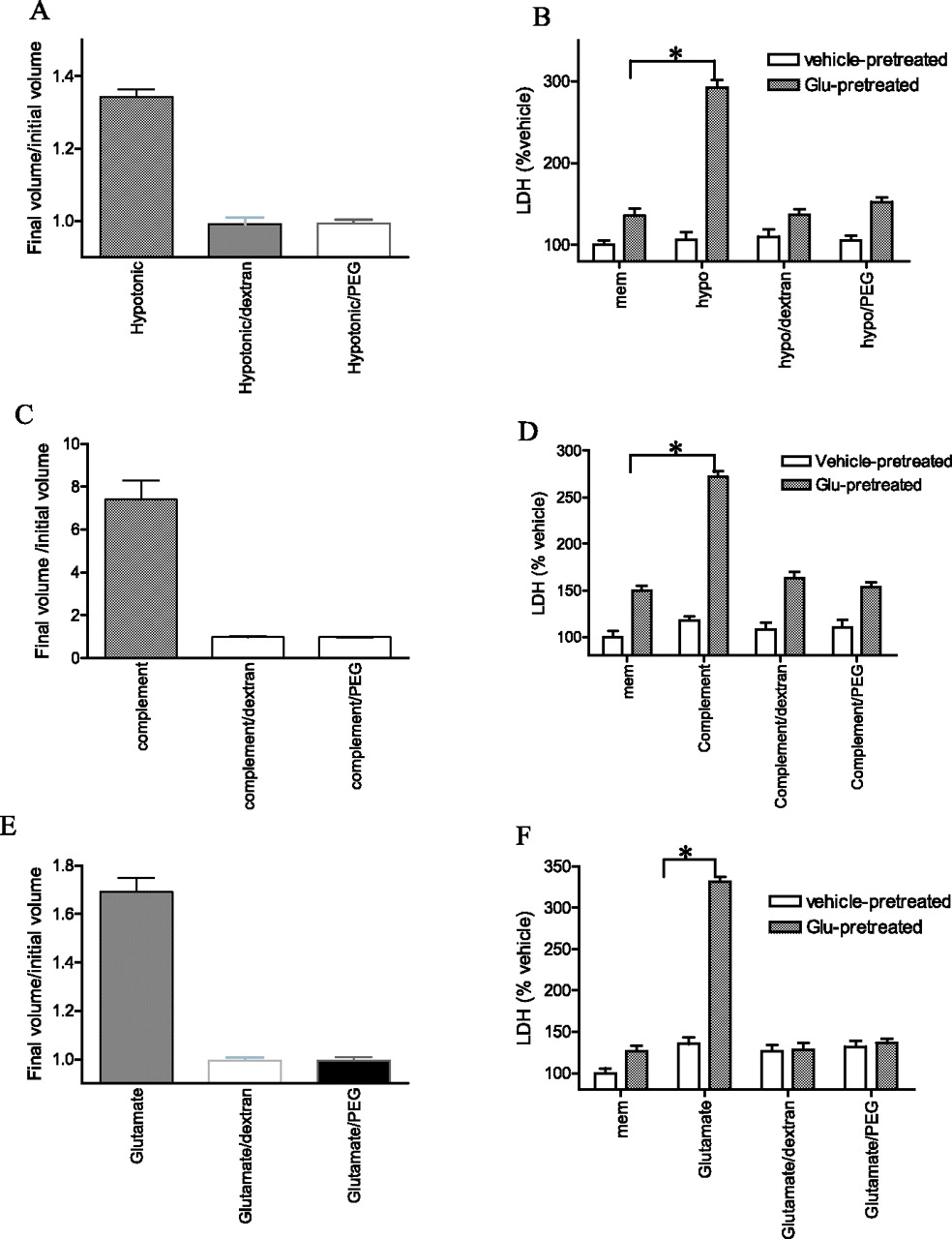
Membrane-impermeable osmolytes rescued cortical neurons from death by excitotoxic sensitization. A, Membrane-impermeable osmolytes rescued neurons from irreversible cell swelling after excitotoxic sensitization. Neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, washed, and treated with 60% hypotonic solution in the presence or absence of dextran (1 mm)/PEG (50 mm) while recording neuronal cell volume. B, Cortical neurons were pretreated with glutamate (30 μm) or vehicle for 10 min and incubated with 60% hypotonic solution in the presence or absence of dextran (1 mm)/PEG (50 mm) for 2 h. Filled bars were with glutamate pretreatment, whereas open bars were with vehicle pretreatment. *p < 0.001 compared with glutamate pretreatment followed by vehicle. C, Membrane-impermeable osmolytes rescued neurons from irreversible cell swelling after excitotoxic sensitization. Neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, washed, and treated with terminal pathway proteins (1 μg/ml each) in the presence or absence of dextran (1 mm)/PEG (50 mm) while recording neuronal cell volume. D, Cortical neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, followed by C5b6 plus C7/C8/C9 (1 μg/ml each) in the presence or absence of dextran (1 mm)/PEG (50 mm) for 2 h. Filled bars were with glutamate pretreatment, whereas open bars were with vehicle pretreatment. *p < 0.001 compared with glutamate pretreatment followed by vehicle. E, Membrane-impermeable osmolytes rescued neurons from volume deregulation after excitotoxic sensitization. Neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, washed, and treated with glutamate (30 μm) in the presence or absence of dextran (1 mm)/PEG (50 mm) while recording neuronal cell volume. F, Cortical neurons were pretreated with glutamate (30 μm) or vehicle for 10 min, washed with MEM, and incubated with a second glutamate (30 μm) treatment in the presence or absence of dextran (1 mm)/PEG (50 mm) for 2 h. Filled bars were with glutamate pretreatment, whereas open bars were with vehicle pretreatment. *p < 0.001 compared with vehicle pretreatment followed by glutamate. Values in B, D, and F are the means and SEM of the determinations made in six wells of a single experiment; each experiment was performed at least three times, and similar results were obtained. Values in A, C, and E are mean ± SEM for five cells per condition of a single experiment.
Reactive oxygen species are required for impairment of RVD
To begin to investigate the signaling events by which an excitotoxic insult impairs RVD, we first analyzed its time course. Varying the interval between glutamate exposure and the addition of terminal pathway proteins revealed that cytotoxicity was maximal when complement was added immediately after glutamate exposure, was greatly diminished after 6 h, and was absent after 12 h (Fig. 6A). Likewise, the impaired volume regulation was maximal immediately after glutamate exposure, had largely resolved by 6 h, and was absent after 12 h (Fig. 6B). This time course supports a mechanism involving posttranslational modification of existing proteins and suggests that alterations in gene expression or protein synthesis are not required.

Time course of excitotoxin-mediated cytotoxicity and impaired regulatory volume decrease after complement attack. A, Cultures were pretreated with glutamate (30 μm) or vehicle for 10 min, and terminal pathway proteins (1 μg/ml each) were added at varying time points, after 10 min of glutamate exposure. LDH released into the media was then assayed. Values are the means and SEM of six wells per condition of a single experiment. B, Cultures were pretreated with glutamate (30 μm) for 10 min, and terminal pathway proteins (1 μg/ml) were added at varying time points after 10 min of glutamate exposure. Neuronal volume was recorded during complement exposure. Data were plotted with final volume/initial volume (Vf/V0) on the y-axis. Values are mean ± SEM for five cells per condition of a single experiment.
Given its pivotal role in signaling via glutamate receptors, we next asked whether calcium was required for excitotoxic sensitization. To test this idea, Ca2+o was removed during glutamate exposure and both cell volume and cell viability were measured. Exposure of cortical neurons to low concentrations of glutamate in calcium-free media prevented the increased cytotoxicity induced by various osmotic stimuli (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Also blocked was the massive cell swelling and lack of volume regulation after various osmotic stimuli (supplemental Fig. 4B–D, available at www.jneurosci.org as supplemental material). Together, these results demonstrate that that the presence of extracellular Ca2+ is necessary for excitotoxic sensitization.
Because previous studies have demonstrated that an excitotoxic stimulus of cortical neurons generated reactive oxygen species (ROS) in a calcium-dependent manner (Xiong and McNamara, 2002), we asked whether reactive oxygen species generated by an excitotoxic insult might be required for the impaired volume regulation and cytotoxicity after an osmotic stressor. To test this idea, cells were treated with Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP) (250 μm), a superoxide dismutase mimetic (Patel et al., 1996), 30 min before and during exposure to glutamate, and subsequently incubated with hypotonic solution. Inclusion of MnTBAP prevented the impaired volume regulation and cytotoxicity induced by hypotonic solution after an excitotoxic stimulus (Fig. 7A,B). In contrast, TBAP, a structural analog of MnTBAP that lacks superoxide dismutase mimetic actions, was ineffective. Together, these findings indicate that reactive oxygen species are necessary for excitotoxin-mediated impairment of RVD.

Reactive oxygen species are necessary for excitotoxic sensitization. A, Blockade of glutamate-induced hypotonic sensitization by reactive oxygen species scavenger MnTBAP. Cells were pretreated with glutamate (30 μm) for 10 min in the presence of 250 μm MnTBAP or TBAP (present 30 min before and throughout the duration of glutamate treatment), followed by wash and addition of hypotonic solution (60%) while measuring cell volume. B, Blockade of glutamate-induced hypotonic sensitization by ROS scavenger, MnTBAP. Cells were pretreated with glutamate (30 μm) for 10 min in the presence of 250 μm MnTBAP or TBAP (present 30 min before and throughout the duration of glutamate treatment), followed by wash and addition of hypotonic solution (60%) for 2 h. C, Paraquat sensitized neurons to hypotonic solution by an impairment of RVD. Neurons were pretreated with paraquat (150 μm) in the presence or absence of 250 μm MnTBAP for 2 h, washed, and then exposed to 60% hypotonic solution while measuring cell volume. D, Paraquat sensitized cortical neurons to death by hypotonic insult. Neurons were pretreated with vehicle or paraquat (150 μm) in the presence or absence of MnTBAP for 2 h, washed, and then exposed to hypotonic solution (60%) for additional 2 h. *p < 0.001 compared with vehicle pretreatment followed by hypotonic solution. E, Membrane-impermeable osmolytes abolished paraquat-mediated sensitization. Neurons were pretreated with vehicle or paraquat (150 μm) for 2 h, washed, and then exposed to hypotonic solution (60%) for additional 2 h in the presence or absence of dextran (1 mm)/PEG (50 mm). *p < 0.001 compared with vehicle pretreatment followed by hypotonic solution. Values in A and C are mean ± SEM for five cells per condition of a single experiment. Values in B, D, and E are the means and SEM of the determinations made in six wells of a single experiment.
The necessity of reactive oxygen species for excitotoxin-mediated impairment of RVD raised the question of whether reactive oxygen species are sufficient to impair RVD. To test this idea, cells were treated with either vehicle or paraquat (150 μm, 2 h), a redox cycling agent capable of generating reactive oxygen species intracellularly (Patel et al., 1996), and subsequently challenged with otherwise sublethal concentrations of hypotonic solution. Both cell volume and cell viability were measured. Pretreatment with paraquat abolished RVD after 60% hypotonic solution (Fig. 7C). Inclusion of MnTBAP during pretreatment with paraquat blocked the effect of paraquat as indicated by the recovery of cell volume (Fig. 7C). Paraquat (150 μm, 2 h) alone did not trigger cell swelling (data not shown). Together these observations indicate that reactive oxygen species are necessary and sufficient to impair RVD.
Additional experiments examined the effects of paraquat on cell death as measured by LDH release. Pretreatment with paraquat (150 μm, 2 h) followed by incubation with vehicle (MEM) did not produce any detectable cell death (Fig. 7D, compare first open bar with second open bar). However, preincubation with paraquat followed by hypotonic stimulation produced marked sensitization to the cytotoxic effects of hypotonicity. Blocking of ROS by MnTBAP during treatment with paraquat eliminated paraquat-induced sensitization to hypotonic-mediated cytotoxicity (Fig. 7D). In addition, the sensitization produced by paraquat was eliminated by two distinct membrane- impermeable osmolytes, dextran and PEG, thereby demonstrating that the mechanism by which paraquat sensitized neurons to cytotoxicity involved impairment of RVD (Fig. 7E).
Discussion
The goal of this study was to elucidate the mechanism by which an excitotoxin sensitizes cortical neurons to the cytotoxic effects of the terminal pathway of complement. Four principal findings emerged: (1) a minimal excitotoxic insult impaired RVD and sensitized neurons to cytotoxic effects of diverse osmotic stressors, one of which was complement; (2) excitotoxic sensitization was prevented by structurally distinct membrane-impermeable osmolytes, each of which prevented the cell swelling and the enhanced cytotoxicity; (3) a reactive oxygen species generator (paraquat) alone was sufficient to impair RVD and a reactive oxygen species scavenger (MnTBAP) prevented excitotoxin-mediated impairment of RVD; (4) excitotoxic sensitization did not modify MAC deposition and produced only a small reduction of MAC removal, because of a selective reduction of endocytic elimination. Together, these findings demonstrate that impairment of RVD is the mechanism of excitotoxic sensitization. Moreover, reactive oxygen species are necessary for excitotoxic sensitization and alone are sufficient to impair RVD and sensitize cortical neurons to harmful consequences of osmotic stress.
One striking result from the experiments examining deposition and removal of membrane attack complexes is that the neurons survived despite persistence of ∼50% of the complexes deposited (Fig. 2E). This is remarkable because insertion of MACs leads to influx of cations and osmotically obliged water follows, leading to cell swelling. Survival of this insult implies the presence of a powerful homeostatic mechanism that functions critically in the defense against this osmotic stressor. Indeed, previous studies revealed that some non-neuronal cells undergoing nonlethal complement attack swelled and subsequently returned to apparently normal volume despite persistence of the complement (Morgan, 1988, Halperin et al., 1993). We found that a sublethal complement attack induced a slight, yet significant neuronal swelling (5%) followed by RVD despite continued presence of the complement (Fig. 3A). Likewise, a minimal excitotoxic stimulus also induced neuronal swelling (10%) followed by RVD despite persistence of the glutamate in the media (Fig. 3E). In contrast to complement and glutamate in which neuronal swelling is presumably triggered by influx of ions followed by osmotically obliged water, simply reducing extracellular osmolality also triggered rapid and massive increases of cell swelling, followed by RVD despite continued presence of hypotonicity (Fig. 3C). Interestingly, the kinetics of cell swelling are much faster for hypotonic stressor compared with complement and glutamate as evident in the different latencies from addition of osmotic stressor to peak of cell swelling (Fig. 3, compare A, C, E); this raises the possibility that the molecular route of water entry into the cell may vary with different osmotic stressors. In contrast, the kinetics of the return to original volume are quite similar for each of these osmotic stressors (Fig. 3A,C,E). These results confirm and extend previous findings that established the presence of RVD in mouse cortical neurons challenged with hypotonic solution (Inoue et al., 2005) or the sodium channel activator veratridine (Churchwell et al., 1996). Notably, the homeostatic mechanisms are powerful because the neurons reestablished their original cell volume despite persistence of the osmotic stressor.
Our findings demonstrate that a minimal excitotoxic prestimulus impaired RVD induced by each osmotic stressor studied, namely complement, a second excitotoxic stimulus, or hypotonic solution (Fig. 4). Moreover, only after an excitotoxic prestimulus did each of these osmotic stimuli destroy the neurons (Fig. 4). Importantly, the time course of impaired RVD and sensitization to the cytotoxic effects exhibited striking similarity. Finally, the sensitization to an osmotic insult induced by the excitotoxin was prevented by structurally distinct membrane-impermeable osmolytes, which facilitated restoration of the original cell volume and prevented cytotoxicity, thereby demonstrating that impairment of RVD is the mechanism of excitotoxic sensitization.
Our findings confirm a previous report by Churchwell et al. (1996) who demonstrated that MK-801 (dizocilpine maleate) promoted recovery of cell volume of cultured cortical neurons treated with veratridine, leading to the idea that veratridine-induced release of glutamate activated NMDA receptors that in turn interfered with volume recovery mechanisms activated by sodium influx through voltage-gated sodium channels. The present study extends these findings by demonstrating that fleeting activation of glutamate receptors is sufficient to impair volume recovery induced by diverse osmotic stimuli and, importantly, this impairment of RVD contributes to the demise of the neurons when challenged with an osmotic stress. Our investigation of the signaling events revealed that excitotoxic insults are not the only insults capable of impairing RVD. The protective effects of the superoxide dismutase mimic MnTBAP, but not its inactive structural analog TBAP, demonstrate that generation of reactive oxygen species is required for glutamate-mediated impairment of RVD. This in turn led to the discovery that generation of reactive oxygen species alone is sufficient to impair RVD. That is, exposure to paraquat, a compound that increases intracellular reactive oxygen species was sufficient to impair RVD and induce sensitization to an osmotic stressor. The paraquat-mediated sensitization and impairment of RVD were abolished by MnTBAP, but not TBAP. Finally, membrane-impermeable osmolytes facilitated restoration of the cell volume and prevented cytotoxicity induced by osmotic insults after paraquat treatment, demonstrating that reactive oxygen species alone are sufficient to impair RVD and thereby sensitize neurons to death by an osmotic stressor. Thus, excitotoxins represent just one of a potential multitude of stimuli that generate reactive oxygen species and could thereby impair RVD and sensitize neurons to death by an osmotic insult.
The role of intracellular Ca2+ in both RVD and impairment of RVD are deserving of comment. Removal of extracellular Ca2+ and/or chelation of intracellular Ca2+ have been shown to impair RVD, in a variety of cell types, including astrocytes and neurons (McCarty and O'Neil, 1992; O'Connor and Kimelberg,1993; Churchwell et al., 1996), implying that intracellular Ca2+ is required for this homeostatic response. Paradoxically, reduction of extracellular Ca2+ during the brief exposure to glutamate in the present experiments prevented impairment of RVD, implying that intracellular Ca2+ is required for impairment of this homeostatic response. One possible explanation for this paradox is that the source and/or locale of the intracellular Ca2+ responsible for promoting and impairing RVD may differ. Alternatively, a threshold level of Ca2+ may be required for induction of RVD and an excitotoxin-mediated increase above this threshold level may impair RVD.
The present findings predict that a diversity of otherwise subclinical pathological events of the human brain may compromise volume regulatory mechanisms and thereby render CNS neurons susceptible to an osmotic insult. One category of such pathological events would consist of excitotoxic insults, such as transient ischemic attacks or isolated epileptic seizures. Here, the initial excitotoxic insult would impair volume regulatory mechanisms, but a second excitotoxic insult would not only impair these homeostatic mechanisms but itself also present an osmotic challenge. For example, the occurrence of multiple transient ischemic attacks at short intervals could impair homeostatic volume regulatory mechanisms and thereby promote destruction of cortical neurons. The interval between the transient ischemic attacks is likely to be a critical variable in that the sensitization observed here is maximal at intervals of a few hours or less and has dissipated by 12 h; this contrasts sharply with the protective effects referred to as ischemic tolerance that occur as a consequence of excitotoxic insults occurring at intervals approximating 24–72 h (Kirino, 2002). Another example might be that “flurries” or multiple isolated epileptic seizures occurring at intervals of tens of minutes to hours may impair volume recovery mechanisms and also provide an osmotic insult, thereby leading to osmotic lysis of cortical neurons.
In addition to clinical settings in which multiple acute insults may occur at short intervals, other instances might be imagined in which an acute insult that impairs RVD occurs in the setting of a chronic insult that provides a persistent osmotic challenge. One such chronic insult providing a persistent osmotic challenge is that of chronic, low-level attack by complement that occurs in multiple neurodegenerative diseases (Morgan, 1995; Shin et al., 1998; Gasque et al., 2000). Evidence of membrane attack complex immunoreactivity decorating cortical neurons has been identified in diseases as diverse as Alzheimer's disease (McGeer et al., 1989; Itagaki et al., 1994; Webster et al., 1997) and Rasmussen's encephalitis (Whitney et al., 1999). The present findings predict that the occurrence of excitotoxic insults in the context of an assault by complement would impair RVD and lead to swelling of cortical neurons initiated by ion influx through MAC channels. The occurrence of repeated focal seizures in Rasmussen's encephalitis may provide the excitotoxic insults that impair volume recovery mechanisms and transform an otherwise sublethal osmotic insult by complement into osmotic lysis and death, thereby contributing to the progressive destruction of a single cerebral hemisphere (McNamara et al., 1999) in this disease. If so, pharmacological intervention with membrane-impermeable osmolytes or reactive oxygen species scavengers may prevent impairment of volume recovery mechanisms and thereby limit the progressive destruction of cortical neurons in this disease.
Footnotes
-
This work was supported by National Institutes of Health Grant NS-036808-08 and the Agency for Science, Technology and Research, Singapore. We thank Ms. Weihua Qian for her excellent technical help and Drs. Wolfgang Liedtke, Yangzhong Huang, Soren Leonard, and Robert Kotloski for their comments on this manuscript.
- Correspondence should be addressed to James O. McNamara, Department of Neurobiology, Campus box 3676, Duke University Medical Center, Durham, NC 27710. jmc{at}neuro.duke.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}