

Abstract
Oxidative stress has been implicated as a key trigger of neuronal apoptosis in stroke and neurodegenerative conditions such as Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis. The Bcl-2 homology 3 (BH3)-only subfamily of Bcl-2 genes consists of multiple members that can be activated in a cell-type- and stimulus-specific manner to promote cell death. In the present study, we demonstrate that, in cortical neurons, oxidative stress induces the expression of the BH3-only members Bim, Noxa, and Puma. Importantly, we have determined that Puma−/− neurons, but not Bim−/− or Noxa−/− neurons, are remarkably resistant to the induction of apoptosis by multiple oxidative stressors. Furthermore, we have determined that Bcl-2-associated X protein (Bax) is also required for oxidative stress induced cell death and that Puma plays a dominant role in regulating Bax activation. Specifically, we have established that the induction of Puma, but not Bim or Noxa, is necessary and sufficient to induce a conformational change in Bax to its active state, its translocation to the mitochondria and mitochondrial membrane permeabilization. Finally, we demonstrate that whereas both Puma and BimEL can bind to the antiapoptotic family member Bcl-XL, only Puma was found to associate with Bax. This suggests that in addition to neutralizing antiapoptotic members, Puma may play a dominant role by complexing with Bax and directly promoting its activation. Overall, we have identified Puma as a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis, and suggest that Puma may be an effective therapeutic target for the treatment of a number of neurodegenerative conditions.
Introduction
There is substantial evidence indicating that a significant portion of affected neurons in acute and chronic neurodegenerative conditions die through a process known as apoptosis (Vila and Przedborski, 2003; Chan, 2004). Apoptosis is a genetically programmed cell death pathway that typically involves caspase activation and distinct morphological changes in the cell. Neurons exhibiting apoptotic features have been observed in postmortem tissue of individuals affected by stroke and from patients diagnosed with Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS) (Tatton et al., 1998; Martin, 1999; Gastard et al., 2003; Chan, 2004). Furthermore, a number of studies have demonstrated that molecular and pharmacological inhibitors of caspases can reduce or delay neuronal cell death in animal models of cerebral ischemia and neurodegenerative disease (Li, 2000; Le et al., 2002). However, caspase activation occurs at a relatively late step in the cell death process and specific inhibition of these proteases is typically not sufficient to prevent neuronal cell death (Miller et al., 1997; Johnson et al., 1999). This is likely because of compromised mitochondrial function and the activation of caspase-independent death effectors (Cregan et al., 2004b). Therefore, to effectively prevent neuronal cell death and retain neurological function it will be critical to identify the molecular pathways that regulate apoptotic events upstream of mitochondrial dysfunction.
The Bcl-2 protein family consists of proapoptotic and anti-apoptotic members that interact at both the physical and functional level to regulate mitochondrial integrity and apoptotic cell death (Tsujimoto, 2003). After activation, proapoptotic family members such as Bcl-2-associated X protein (Bax) and Bcl-2 homologous antagonist killer (Bak) target the mitochondria and cause membrane permeabilization and the release of proapoptotic effectors, including cytochrome-c and apoptosis-inducing factor (Kluck et al., 1997; Cregan et al., 2002). When released into the cytoplasm cytochrome-c facilitates activation of the apoptotic activating factor-1 (Apaf-1)/Caspase-9 apoptosomal complex that triggers the caspase cascade and subsequent cell degradation (Li, 1997). Bax/Bak activation in response to apoptotic stimuli can be regulated by the actions of a third class of Bcl-2 family members known as the Bcl-2 homology 3 (BH3)-domain-only proteins. BH3-only proteins are thought to promote apoptosis by binding to prosurvival Bcl-2 family members, thereby unleashing Bax/Bak. The BH3-only protein family consists of eight known members that can be activated through a variety of transcriptional and post-translational mechanisms depending on the cell type and death stimulus involved (Puthalakath and Strasser, 2002). Therefore, it is believed that the different BH3-only proteins provide a link between the diverse upstream signaling pathways and the convergent downstream effectors in apoptotic cell death.
Oxidative stress has been implicated as a key trigger of neuronal cell death in stroke and several chronic neurodegenerative disorders including Alzheimer's disease, Parkinson's disease, and ALS (Jenner, 1998; Simpson et al., 2003; Sugawara and Chan, 2003; Tieu et al., 2003; Aslan and Ozben, 2004; Andersen, 2004). Therefore, identifying the molecular regulators of oxidative stress induced neuronal apoptosis will be an important step in the development of therapeutic strategies to treat these neurodegenerative conditions. In this study we identify the critical Bcl-2 family members involved in the regulation of oxidative stress induced neuronal apoptosis.
Materials and Methods
Animals.
Apaf-1, Bax, and p53 mice (The Jackson Laboratory, Bar Harbor, ME) were maintained on a C57BL/6 background and genotyped as described previously (Cregan et al., 1999; Fortin et al., 2001). Mice carrying targeted null mutations for Bim, Noxa, and Puma were generated on a C57BL/6 background in the laboratory of Dr. Andreas Strasser (WEHI, Victoria, Australia) and the genotyping of these mice was performed as described previously (Bouillet et al., 1999; Villunger et al., 2003).
Neuronal cell cultures.
Cortical neurons were dissociated from E14.5–E15.5 mice and cultured in Neurobasal medium containing B27 and N2 supplements (Invitrogen, Eugene, OR) as described previously (Fortin et al. 2001). Drug treatments were initiated after 4 d in culture. For treatments with hydrogen peroxide (Sigma, St. Louis, MO) and tert-butyl hydroperoxide (Sigma), neurons were exposed to drugs in HBSS for 60 or 90 min respectively, and then returned to their conditioned medium. In the indicated studies RNA or protein synthesis inhibitors actinomycin-D (ActD) and cycloheximide (CHX) (Sigma) were added to cultures after return to conditioned media. For NOC-12 (Calbiochem, La Jolla, CA) or 1-methyl-4-phenylpyridinium iodide (MPP+; Sigma) treatments, drugs were added directly to the culture medium and were not washed out. Preparation, titration, and transduction of neurons with recombinant adenoviral vectors expressing HA-PUMA or green fluorescent protein (GFP) were performed as previously described (Cregan et al., 2004a).
Cell death and survival assays.
Neuronal apoptosis was assessed by examining nuclear morphology in Hoechst 33258 stained cells. Neurons were fixed in 4% paraformaldehyde and stained with Hoechst 33258 (1 μg/ml) and the fraction of cells exhibiting an apoptotic nuclear morphology (chromatin condensation and/or apoptotic bodies) was determined. Neuronal survival was also determined by Live/Dead viability assay (Invitrogen) as described previously (Fortin et al., 2001). Briefly, cells were stained with Calcein-AM (2 μm) and ethidium homodimer (2 μm) for 15 min and the fraction of live (Calcein-AM positive) and dead (ethidium positive) cells was determined. In both assays, neurons were visualized by fluorescence microscopy (IX70; Olympus, Tokyo, Japan) and images were captured with a CCD camera (Q-imaging, Burnaby, British Columbia, Canada) and Northern Eclipse software (Empix Imaging, Mississauga, Ontario, Canada). Images were captured and scored by a blinded observer and a minimum of 400 cells were analyzed per well.
Caspase activity assay.
Neurons were harvested in caspase lysis buffer (1 mm KCl, 10 mm HEPES, pH 7.4, 1.5 mm MgCl2, 1 mm DTT, 1 mm PMSF, 5 μg/ml leupeptin, 2 μg/ml aprotinin, and 10% glycerol) and 10 μg of protein was used in caspase activity assay as described previously (Cregan et al., 1999). Briefly, protein samples were added to caspase reaction buffer [25 mm HEPES, pH 7.4, 10 mm DTT, 10% sucrose, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), and 10 μm N-acetyl-Asp-Glu-Val-Asp-(7-amino-4-trifluoromethyl coumarin) (Ac-DEVD-AFC)] and fluorescence produced by DEVD-AFC cleavage was measured on a SpectraMax M5 fluorimeter (excitation 400 nm, emission 505 nm) over a 1 h interval. Caspase-3-like activity is reported as the ratio of the fluorescence output in treated samples relative to corresponding untreated controls.
Quantitative RT-PCR.
RNA was isolated using Trizol reagent as per manufacture's instructions (Invitrogen) and 20 ng of RNA was used in one-step Sybr green reverse transcription (RT)-PCR (QuantiTect; Qiagen, Hilden, Germany). RT-PCR was carried out on a Chromo4 system (MJ Research, Watertown, MA; Bio-Rad, Hercules, CA) and changes in gene expression were determined by the Δ(ΔCt) method using S12 transcript for normalization. Data is reported as fold increase in mRNA levels in treated samples relative to corresponding untreated control cells for each transcript. All PCRs exhibited high amplification efficiency (>90%) and the specificity of PCR products was confirmed by sequencing. Primer sequences used for gene specific amplification are available on request.
Cytochrome-c and MAP-2 immunostaining.
Neurons were fixed in 4% paraformaldehyde, washed in three changes of PBS and then incubated for 2 h with monoclonal antibodies directed against cytochrome-c (BD PharMingen, San Diego, CA) or microtubule-associated protein-2 (MAP-2; Roche, Welwyn Garden City, UK) as described previously (Cregan et al., 2002). Cells were then washed and incubated for 45 min with Alexa-488 conjugated goat anti-mouse IgG secondary antibody (Invitrogen) and counterstained with Hoechst 33258 (1 μg/ml). To evaluate mitochondrial membrane permeabilization cells were visualized by fluorescence microscopy and cells exhibiting punctate, cytoplasmic cytochrome-c staining were considered to have maintained membrane integrity. Images were captured and scored by a blinded observer and a minimum of 300 cells were analyzed per well.
Western blot analysis.
To prepare whole-cell lysates, neurons were incubated in lysis buffer (10 mm HEPES, 150 mm NaCl, 0.5 mm EDTA, 0.5% CHAPS, 0.5 mm PMSF, 5 μg/ml aprotinin, 5 μg/ml leupeptin) for 30 min on ice and soluble extract was recovered by centrifugation. For subcellular fractionation neurons were harvested in isotonic buffer (10 mm HEPES, 210 mm mannitol, 70 mm sucrose, 0.5 mm EDTA, and protease inhibitor mixture) and lysed by 20 passes through a 25-gauge syringe. Unbroken cells and nuclei were cleared by 5 min centrifugation at 750 g and the supernatant was then centrifuged for 20 min at 10,000 × g to separate the cytosol and heavy membrane (HM) fractions. The HM fractions were incubated for 20 min in 0.1 m Na2CO3 and then extracted with standard lysis buffer. Protein concentration was determined by BCA assay (Pierce, Rockford, IL) and 40 μg of protein was separated on 12.5% SDS-PAGE gels and then transferred to nitrocellulose membrane. Membranes were blocked for 2 h in TBST (10 mm Tris, 150 mm NaCl, 0.1% Tween 20) containing 5% skim milk and then incubated overnight with primary antibodies to Bax, Actin (Santa Cruz), BIM (Stressgen), PUMA or cytochrome c oxidase subunit IV (COX-IV) (both from Cell Signaling Technology, Beverly, MA) in TBST containing 3% skim milk. Membranes were then washed in TBST and then incubated for 1 h with appropriate HRP-conjugated goat anti-rabbit IgG secondary antibodies. Membranes were again washed and then developed by an enhanced chemiluminescence system according to manufacturer's instructions (Amersham Biosciences, Arlington Heights, IL).
Immunoprecipitation assay.
Neuronal lysates were prepared in lysis buffer (10 mm HEPES pH 7.4, 150 mm NaCl) containing 1% CHAPS and protease inhibitors. For immunoprecipitation, 250 μg of cell lysate was incubated with the conformation specific anti-Bax mouse monoclonal antibody 6A7 (BD PharMingen) and protein G-Sepharose beads for 4 h at 4°C. Precipitated complexes were released by boiling in SDS sample buffer and subjected to Western blot analysis as described above using anti-Bax rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
GST pulldown assays.
PUMA and BIMEL cDNAs were inserted into pGEX4T1 plasmid (Amersham) to generate a glutathione S-transferase (GST)-PUMA and GST-BIMEL fusion proteins. Expression of GST-fusion proteins was induced in DH5α bacterial cells with 1 mm isopropyl β-d-1-thiogalactopyranoisde and GST-PUMA/BIM proteins were purified from cell lysates on glutathione-agarose columns. Neurons were harvested and extracted for 1 h in ice-cold lysis buffer and lysates (500 μg of protein) were incubated with GST-PUMA, GST-BIM, or unfused GST (50 μg) for 1 h at 37°C. Equilibrated GST-Sepharose beads were added and after 2 h incubation complexes were recovered by centrifugation. The beads were washed twice in lysis buffer and then interacting proteins were eluted in 50 mm Tris-HCl, pH 8.0 containing 10 mm reduced glutathione. Aliquots were then separated by SDS-PAGE and immunoblotted for Bcl-xL or Bax as described above.
Electrophysiology.
Electrophysiology measurements were performed on neurons after 10 d in culture. Culture dishes were placed in an Olympus IX-70 inverted microscope and neurons were continuously perfused in Ringer's solution consisting of (in mm) 145 NaCl, 5 KCl, 10 HEPES, 2 CaCl2, 2 MgCl2, and 10 glucose (300–310 mOsm, pH adjusted to 7.3). Patch-clamp recordings from individual cells were made using electrodes made from Sutter Instruments borosilicate glass (outer diameter, 1.5 mm; inner diameter, 0.86 mm) in electrode solution composed of (in mm) 145 KCl, 10 NaCl, 10 HEPES, 1 Na2EDTA, and 20 glucose (adjusted to 300–320 mOsm with sucrose, pH 7.3, free Ca2+ was 80 nm). A Molecular Devices Axopatch 200B amplifier was used to record voltage–current relationships and the spontaneous synaptic currents (5 kHz low pass filtered) were digitized (10 kHz) using a Molecular Devices Digidata 1200A interface. All recordings were performed at room temperature (20–22°C) on visually identified pyramidal neurons. Neurons were clamped at a membrane potential of −60 mV and electrode resistance ranged from 3 to 6 MΩ. Series resistance was typically in the range of 7–15 MΩs with compensation up to 80%. Sodium current density was estimated from measuring the peak of the sodium current in V–I mode and then normalizing this value based on an estimate of the cell size from the total cellular capacitance. For an estimate of the synaptic activity present the frequency of spontaneous synaptic currents was estimated from 20 min long recordings where the events were counted by the operator off line and is expressed in events per second (hertz).
Data analysis.
Data is reported as mean and SD. The value n represents the number of independent neuron cultures or number of embryos of indicated genotype from which independent neuron cultures were prepared. Differences between groups were determined by one-way ANOVA followed by post hoc Tukey test and were considered statistically significant when p < 0.05.
Results
Oxidative damage triggers neuronal apoptosis through a mitochondrial pathway
To characterize oxidative stress induced neuronal apoptosis we treated cortical neurons with agents known to generate hydroxyl- (H2O2), peroxyl- (t-butyl hydroperoxide), or nitric oxide- (NOC-12) free radicals, all of which have been implicated in neurodegenerative conditions (Andersen, 2004; Margaill et al., 2005). We also treated neurons with MPP+ the active metabolite of the drug 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) that is known to cause Parkinson's syndrome in rodents and humans (Przedborski and Ischiropoulos, 2005). MPP+ is an inhibitor of the respiratory chain complex-I and its toxicity has been shown to be caused by the consequent production of oxygen derived free radicals (Fallon et al., 1997; Przedborski and Ischiropoulos, 2005; Richardson et al., 2007). All of these oxidative stressors triggered a significant increase in the fraction of cells exhibiting an apoptotic nuclear morphology characterized by chromatin condensation and/or nuclear fragmentation (Fig. 1A). After treatment with H2O2, tert-butyl hydroperoxide (TBH), and NOC-12 the fraction of apoptotic neurons began to increase after ∼12 h and continued to rise for at least 24 h (Fig. 1A). Cell death in response to MPP+ treatment was notably slower as the fraction of apoptotic cells only began to increase after ∼24 h and continued to rise over the next 48 h. To determine whether oxidative stress induced apoptosis is dependent on gene induction, we examined the effect of the transcription and translation inhibitors actinomycin-D and cycloheximide. As shown in Figure 1B, both of these inhibitors essentially blocked oxidative stress induced neuronal cell death suggesting that de novo gene expression is required for activation of the apoptotic program.

Oxidative damage triggers neuronal apoptosis through a mitochondrial pathway. A, Cortical neurons were treated with NOC-12 (200 μm), TBH (200 μm), H2O2 (20 μm), or MPP+ (100 μm) and the fraction of apoptotic cells was determined by Hoechst 33258 staining at the indicated times (n ≥ 4). Representative images of Hoechst 33258 stained neurons captured 48 h after indicated treatments are shown. B, Cortical neurons were treated with the oxidative stressors as above in the presence of CHX (5 μg/ml), ActD (5 μg/ml), or DMSO (solvent control) and the fraction of apoptotic cells was determined by Hoechst 33258 staining at 40 h (n = 4; *p < 0.001). C, Cortical neurons derived from wt and Apaf-1−/− littermates were treated with the indicated oxidative stressors as above and assayed for caspase-3-like activity at 24 h (or 40 h in the case of MPP+ treatment) (n ≥ 3; *p < 0.001).
Apaf-1 is known to be a key regulator of caspase activation in mitochondrial driven apoptotic pathways (Yoshida et al., 1998). To determine whether oxidative stress induces caspase activation through the mitochondrial pathway we treated neurons derived from Apaf-1−/− mice and wild-type (wt) control littermates with NOC-12, H2O2, TBH, and MPP+. All of these oxidative stressors triggered a significant induction of caspase-3-like activity in wt control neurons (>10-fold), but not in Apaf-1−/− neurons, consistent with the involvement of the intrinsic (mitochondrial) apoptotic pathway (Fig. 1C).
Oxidative stress induces the expression of the BH3-only members Bim, Noxa, and Puma
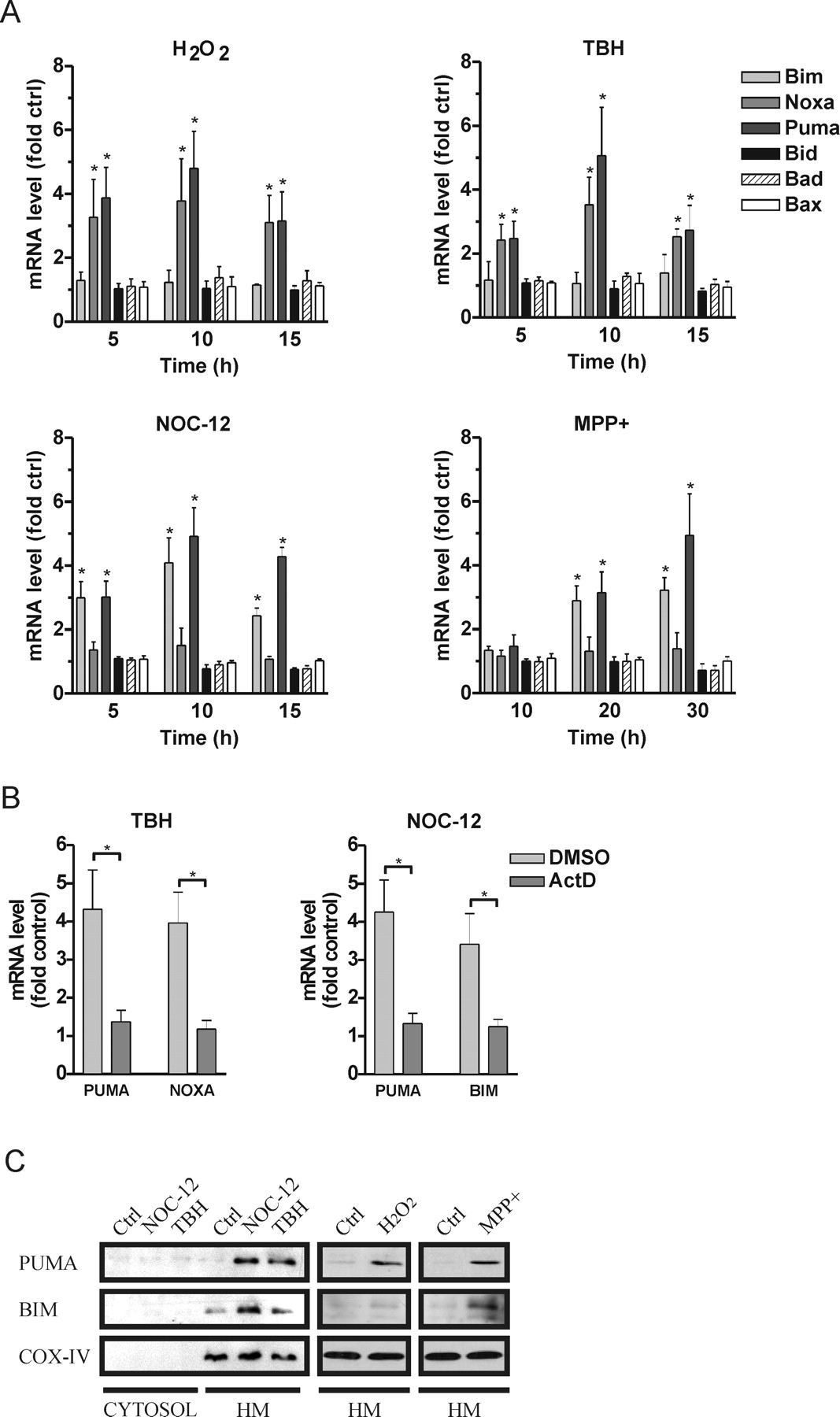
Bcl-2 family proteins can be activated in a cell type and stimulus specific manner and are known to be key regulators of mitochondrial apoptotic pathways. Because de novo gene expression appeared to be required for activation of cell death (Fig. 1B), we examined the expression profiles of various proapoptotic Bcl-2 family members by quantitative RT-PCR. As shown in Figure 2A, treatment with H2O2 or TBH led to a significant increase in the expression of the BH3-only genes Noxa and Puma beginning ∼5 h and peaking at ∼10 h after drug exposure. However, these treatments did not affect the expression of other BH3-only family members including Bim, Bid, and Bad or the multi-BH domain proapoptotic Bax. Interestingly, NOC-12 and MPP+ treatment led to a different pattern of BH3-only gene induction. Specifically, Puma and Bim gene expression was upregulated, whereas the expression of Noxa and other BH3-only members was not. It was noted that the induction of Puma and Bim after MPP+ treatment was slower and only became evident after ∼20 h, consistent with the delayed onset of apoptosis observed in this death paradigm (Fig. 1A). We also noted that the increase in Puma, Bim and Noxa mRNA levels was essentially blocked in the presence of actinomycin-D consistent with enhanced transcription of these BH3-only genes (Fig. 2B).

Oxidative stress induces the expression of the BH3-only members Bim, Noxa, and Puma. A, RNA was isolated from cortical neurons at the indicated times after treatment with TBH (200 μm), H2O2 (20 μm), NOC-12 (200 μm) or MPP+ (100 μm) and mRNA levels of Bcl-2 family members was determined by quantitative RT-PCR. Expression was normalized to S12 mRNA levels and is reported as fold increase over corresponding untreated control cells (n ≥ 4). Asterisks indicate significant increase in mRNA level in treated versus untreated neurons for each transcript (*p < 0.05). B, Neurons were treated with TBH (200 μm) or NOC-12 (200 μm) in the presence of ActD (5 μg/ml) or DMSO (solvent control) and after 10 h the fold increase in Puma, Noxa, and Bim mRNA was determined by qRT-PCR (n = 3; *p < 0.05). C, Bax-deficient cortical neurons were treated with NOC-12 (200 μm), TBH (200 μm), H2O2 (20 μm), or MPP+ (100 μm) and after 24 h (or 48 h in the case of MPP+) Puma and Bim protein levels were assessed in the cytosolic and HM fractions by Western blot analysis.
As illustrated in Figure 2C, oxidative stress also triggered an increase in Puma and Bim protein levels specifically within the mitochondrial-enriched HM fraction. Similar to the mRNA profiles, Puma protein levels were increased in response to all oxidative stressors whereas Bim protein was upregulated predominantly in response to NOC-12 and MPP+ treatments. The Bim antibody used in these studies can detect all three Bim isoforms (BimEL, BimL, and BimS), however, similar to other cell death paradigms (Bouillet et al., 1999; Putcha et al., 2001; Whitfield et al., 2001), oxidative damage appeared to preferentially induce the expression of BimEL. Although Noxa mRNA levels were increased in response to H2O2 and TBH treatments, we were unable to detect Noxa protein using several different commercially available antibodies. However, we believe that this is an antibody issue because ectopically expressed Flag-Noxa could be detected with antibodies directed against the Flag-epitope, but not with any of the Noxa antibodies tested (data not shown).
Puma is essential for oxidative stress induced neuronal apoptosis
Because the BH3-only proteins Bim, Noxa, and Puma were selectively induced by oxidative stress, we sought to determine whether these proteins contributed to neuronal cell death. We first examined the potential role of Puma as it was upregulated in response to several different types of reactive oxygen species. We treated Puma+/+ (wt), Puma+/−, and Puma−/− neurons with NOC-12, H2O2, TBH, or MPP+ and measured the extent of apoptosis as a function of time (Fig. 3A). In contrast to wt and Puma+/− neurons, treatment of Puma−/− neurons with these different oxidative stressors did not result in a significant induction of apoptosis relative to untreated controls even after 48–72 h. Consistent with the marked reduction in apoptotic cells, caspase-3 like activity was also found to be dramatically reduced in Puma−/− neurons (Fig. 3B). It was also noted that the apoptotic frequency was generally reduced in Puma+/− neurons as compared with wt neurons suggesting a possible gene dosage effect (Fig. 3A).

Puma is essential for oxidative stress induced neuronal apoptosis. A, Cortical neurons cultured from wt, Puma+/−, and Puma−/− littermates were treated with TBH (200 μm), H2O2 (20 μm), NOC-12 (200 μm), or MPP+ (100 μm), and the fraction of apoptotic cells was determined by Hoechst 33258 staining at the indicated times (n ≥ 6). Asterisks indicate significant increase over untreated neurons of the same genotype (*p < 0.01). B, Wt and Puma−/− neurons were treated with oxidative stressors as above and assayed for caspase-3 activity at 24 h (or 40 h in the case of MPP+ treatment) (n = 4; *p < 0.01).
To rule out the possibility that Puma−/− neurons were dying via an alternative (nonapoptotic) mechanism, we also measured neuronal survival in Puma+/+ and Puma−/− neurons by Calcein-AM/ethidium homodimer staining (live/dead assay). As shown in Figure 4A, treatment with NOC-12, TBH, or MPP+ reduced survival of wt neurons at 48 h to 38.7, 45.3, and 46.4%, respectively. In contrast, after these same treatments survival of Puma−/− neurons remained similar to untreated controls at >90% indicating that Puma-deficient neurons were not dying via an alternate mechanism. Furthermore, MAP-2 immunostaining revealed that unlike wt neurons the vast majority of Puma−/− neurons maintained their neuritic extensions after exposure to oxidative stress (Fig. 4B).

Puma-deficient neurons remain functional after oxidative injury. A, Wt and Puma−/− neurons were treated with NOC-12 (200 μm), TBH (200 μm), or MPP+ (100 μm) and neuronal survival was determined by live/dead staining at 48 h (n ≥ 3; *p < 0.01). Representative images of NOC-12 treated wt and Puma−/− neurons stained using the live (green)/dead (red) assay. B, Wt and Puma−/− neurons were treated with NOC-12 (200 μm) and then immunostained for MAP-2 (green) and counterstained with Hoechst 33258 (pseudocolored orange). C, Voltage-clamp recordings were performed on untreated neurons (wt or Puma−/−) or NOC-12-treated (200 μm) Puma−/− neurons and corresponding voltage–current relationships are shown. The circles represent the average inward current at various holding potentials and squares show the current required at steady state (n ≥ 9). D, Representative recordings of spontaneous activity in untreated (wt and Puma−/−) neurons and NOC-12-treated Puma−/− neurons.
We next examined whether Puma-deficient neurons remain functional after oxidative damage. Individual pyramidal neurons are known to respond to hyperpolarizing and depolarizing voltage in a characteristic manner in culture (Hutcheon et al., 2000). Therefore, we performed patch-clamp recordings to determine whether Puma-deficient neurons retain their electrophysiological properties after oxidative injury. In untreated wt and Puma−/− neurons we found the peak sodium current to have a density of 23.2 ± 9.8 pA/um2 and 22.3 ± 7.8 pA/μm2, respectively (Fig. 4C). The magnitude of the sodium current in Puma−/− neurons treated with NOC-12 was found to be 17.9 ± 4.2 pA/μm2 and this was not significantly different from untreated control neurons (p = 0.29). However, the vast majority of wt neurons treated with NOC-12 had undergone cell death at this point and the few surviving neurons were too fragile to obtain stable recordings. Thus, Puma-deficient neurons appear to retain normal voltage dependent current activity after oxidative injury. Cortical neurons in culture are also known to make functional synaptic connections, and so we assayed the spontaneous synaptic activity in these cultures (Fig. 4D). We found that the activity between the untreated and NOC-12 treated Puma−/− neurons was also indistinguishable. The frequency of spontaneous events in untreated wt, untreated Puma−/−, and NOC-12-treated Puma−/− cultures was 1.7 ± 0.6, 1.1 ± 0.2 and 1.2 ± 0.3 Hz, respectively (p = 0.65). Overall our data indicate that Puma-deficient neurons survive and retain normal neuronal function after oxidative injury.
Because the BH3-only family members Bim and Noxa were also induced by oxidative stress, we then examined whether they also contribute to neuronal cell death. As shown in Figure 5A and 5B, NOC-12, and MPP+ treatments induced similar levels of apoptosis and caspase activity in wt and Bim−/− neurons. Likewise, we observed no significant change in caspase activity or apoptotic frequency in wt and Noxa−/− neurons after treatment with H2O2 or TBH (Fig. 5C,D). Thus, although Puma, Bim, and Noxa expression is induced in response to oxidative stress, only Puma induction appears to be essential for cell death.

Bim and Noxa are not required for oxidative stress induced neuronal apoptosis. A, Wt and Bim−/− neurons were treated with NOC-12 (200 μm) or MPP+ (100 μm) and the fraction of apoptotic cells was determined by Hoechst staining at 24 and 48 h, respectively (n ≥ 4). B, Wt and Bim−/− neurons were treated as above and caspase-3 activity was assayed at 24 h (NOC-12) or 48 h (MPP+) (n = 3). C, Wt and Noxa−/− neurons were treated with TBH (200 μm) or H2O2 (20 μm) and the fraction of apoptotic cells was determined by Hoechst staining at 24 h (n ≥ 3). D, Wt and Noxa−/− neurons were treated with TBH (200 μm) or H2O2 (20 μm) and caspase-3 activity was assayed aft 24 h (n = 3).
Puma regulates Bax activation after oxidative injury
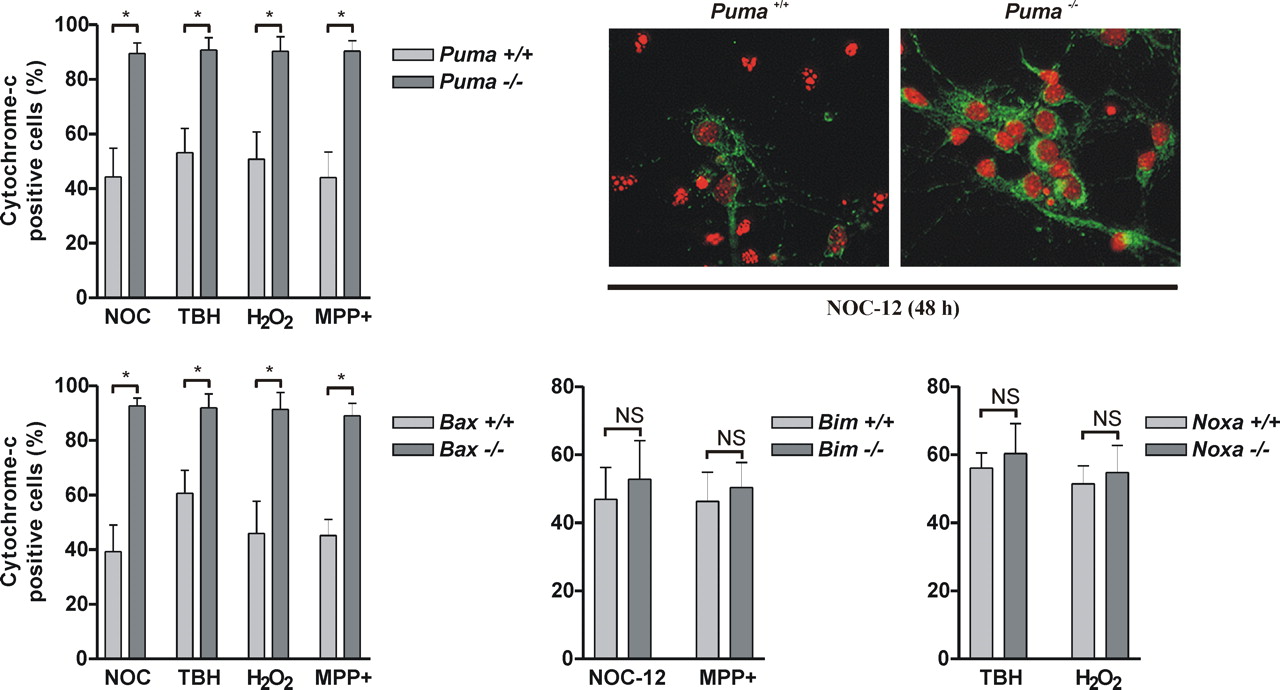
BH3-only proteins are thought to co-operate with multidomain proapoptotic Bcl-2-family members to induce apoptosis (Harris and Johnson, 2001; Zong et al., 2001). Consistent with this we have determined that similar to Puma-deficient neurons, Bax-deficient neurons are remarkably resistant to oxidative stress induced apoptosis (Fig. 6A). Therefore, to address the mechanism responsible for the different effects of Puma, Bim and Noxa on neuronal cell death we examined whether these BH3-only proteins differentially affect Bax activation. In response to apoptotic stimuli, Bax is known to translocate from the cytoplasm to the mitochondrial membrane (Wolter et al., 1997; Goping et al., 1998). As shown in Figure 6B, in wt neurons treatment with NOC-12 or TBH decreased Bax levels in the cytosolic fraction and simultaneously increased the amount of Bax associated with the heavy membrane fraction consistent with its translocation to the mitochondria. However, these oxidative treatments did not appreciably alter the distribution of Bax in Puma−/− neurons, indicating that Puma is required for Bax translocation. In contrast, neither Bim-deletion nor Noxa-deletion prevented oxidative stress induced Bax translocation to the mitochondria (Fig. 6C). During activation Bax is known to undergo a conformational change that exposes an epitope within its N terminus that is recognized by the 6A7 monoclonal antibody (Hsu and Youle, 1998). Consistent with a requirement for Puma in mediating oxidative stress induced Bax activation we found that immunoprecipitation of Bax with the 6A7 antibody was markedly reduced in Puma−/− neuronal extracts as compared with wt extracts (Fig. 6D). Finally, as a measure of Bax function we also examined the effect of Puma, Bax, Bim, and Noxa deletion on oxidative stress induced mitochondrial membrane permeabilization. We and others have shown previously that cytochrome-c immunoreactivity is rapidly lost in neurons after its release from the mitochondria and therefore can be used as an effective measure of outer membrane permeabilization (Deshmukh and Johnson, 1998; Neame et al., 1998; Cregan et al., 2002). In wt neurons, treatment with NOC-12, TBH, H2O2, or MPP+ led to a marked reduction in the fraction of cells retaining mitochondrial cytochrome-c as compared with untreated control cells (Fig. 7). However, after these same treatments the vast majority (>90%) of Puma−/− and Bax −/− neurons retained mitochondrial cytochrome-c indicating that Puma and Bax cooperate to induce mitochondrial membrane permeabilization. In contrast, neither Bim deletion nor Noxa deletion prevented oxidative stress induced cytochrome-c release (Fig. 7).
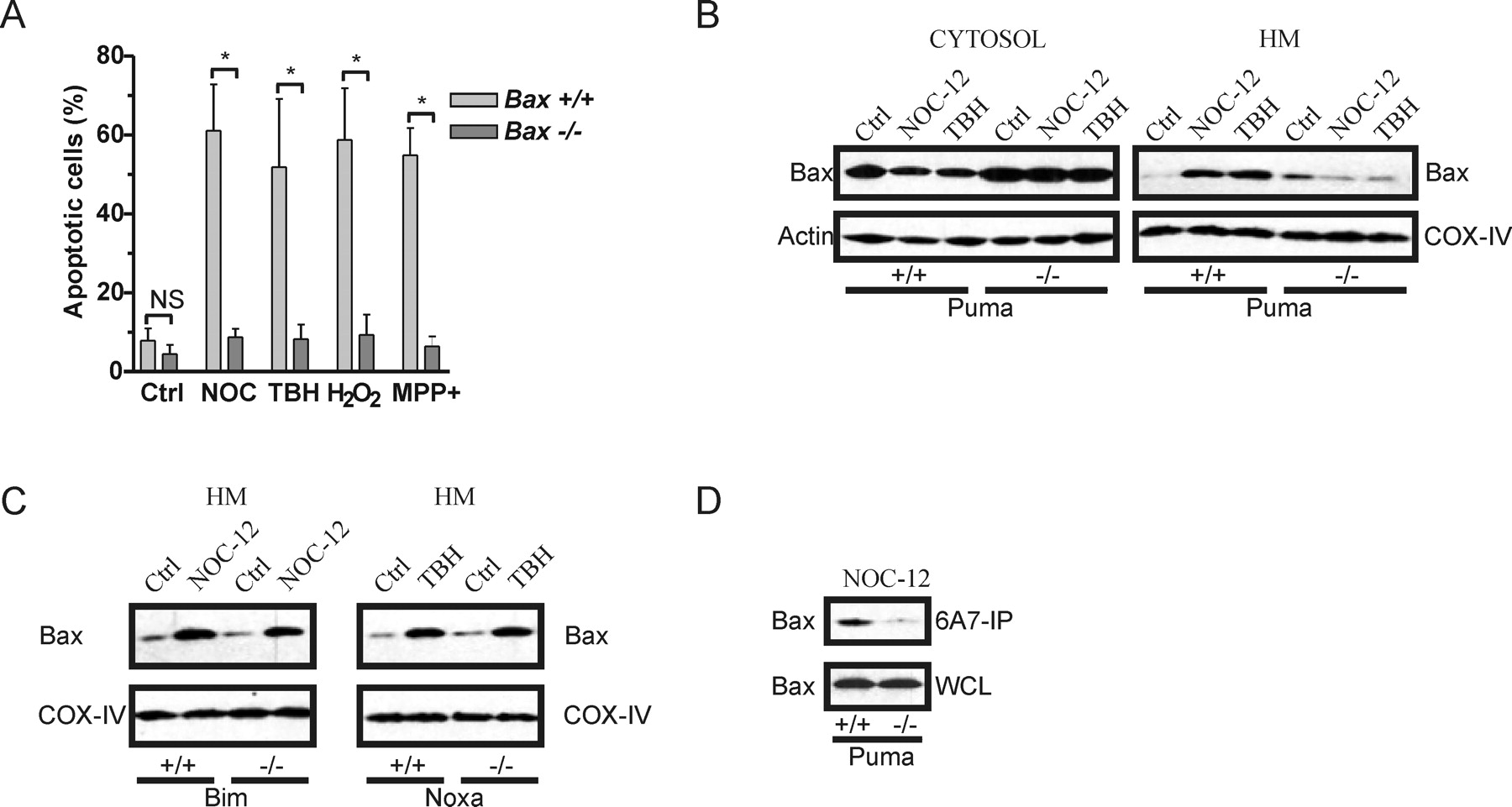
Puma is required for oxidative stress induced Bax activation. A, Wt and Bax−/− neurons were treated with NOC-12 (200 μm), TBH (200 μm), H2O2 (20 μm), or MPP+ (100 μm) and the fraction of apoptotic cells was determined by Hoechst staining at 48 h (n ≥ 5; *p < 0.001). B, Wt and Puma−/− neurons were treated with NOC-12 (200 μm) or TBH (200 μm) and after 24 h Bax protein levels were assessed in cytosolic and HM fractions by Western blot analysis. Immunoblots for β-Actin and COX-IV are shown for normalization of cytosolic and heavy membrane fractions respectively. C, Wt, Bim−/−, and Noxa−/− neurons were treated with NOC-12 (200 μm) or TBH (200 μm) and after 24 h Bax protein levels were assessed in the HM fraction by Western blot. D, Wt and Puma−/− neurons were treated with NOC-12 (200 μm) and after 24 h Bax was immunoprecipitated from neuronal extracts with an antibody that specifically recognizes activated Bax (6A7). Immunoprecipitates (IP) and whole-cell lysates (WCL) were resolved by SDS-PAGE and immunoblotted for Bax. WCL represents 20% of protein used in IP reaction.

Puma and Bax cooperate to regulate oxidative stress induced mitochondrial membrane permeabilization. Cortical neurons derived from Puma+/+ and Puma−/− littermates (n = 4; *p < 0.001), Bax+/+, and Bax−/− littermates (n = 4; *p < 0.001), Bim+/+ and Bim−/− littermates (n ≥ 3) and Noxa+/+ and Noxa−/− littermates (n = 3) were treated with NOC-12 (200 μm), H2O2 (20 μm), TBH (200 μm), or MPP+ (100 μm) and the fraction of cells retaining mitochondrial cytochrome-c was determined at 24 h (or 48 h in the case of MPP+). Representative images of wt and Puma−/− neurons treated for 24 h with NOC-12 (200 μm) and immunostained for cytochrome-c (green) and counterstained with Hoechst (pseudocolored orange).
To determine whether Puma is sufficient to induce Bax activation we transduced wt and Bax−/− neurons with recombinant adenoviruses (Ad) expressing Puma (Ad-PUMA) or GFP (Ad-GFP) as a control. As illustrated in Figure 8A, enforced expression of Puma significantly enhanced the amount of Bax immunoprecipitated with the activated conformation specific antibody (6A7) consistent with the induction of Bax activation. Furthermore, we found that enforced expression of Puma could induce cytochrome-c release and caspase activation in wt neurons, but not Bax−/− neurons (Fig. 8B,C). Together these results suggest that Puma is both necessary and sufficient to induce Bax activation in neurons.

Puma expression is sufficient to induce Bax activation. A, Neurons were transduced with Ad-Puma or Ad-GFP at 20 multiplicity of infection and after 24 h Bax was immunoprecipitated from neuronal extracts with an antibody (6A7) that specifically recognizes activated Bax. Immunoprecipitates (IP) and whole-cell lysates (WCL) were resolved by SDS-PAGE and immunoblotted for Bax. WCL represents 20% of protein used in IP reaction. B, Wt and Bax−/− neurons were transduced with Ad-Puma or Ad-GFP and the fraction of cells retaining mitochondrial cytochrome-c was measured after 40 h (n = 3; *p < 0.001). C, Caspase-3 activity was measured 40 h after transduction with Ad-Puma or Ad-GFP (n = 3; *p < 0.01).
BH3-only proteins are generally thought to promote apoptosis by binding to and neutralizing anti-apoptotic Bcl-2 family members (Moreau et al., 2003). However, it has been suggested that certain BH3-only proteins may also directly interact with multidomain proapoptotic members to promote their apoptotic function (Eskes et al., 2000; Letai, 2002; Kuwana et al., 2005). Because Puma, but not Noxa or Bim, appears to be essential for oxidative stress induced Bax activation we explored the possibility that in addition to interacting with anti-apoptotic Bcl-2 proteins, Puma may interact with Bax. To address this question, we examined the ability of GST-Puma and GST-BimEL to pulldown Bax or the anti-apoptotic member Bcl-XL from cell lysates obtained from NOC-12 treated Puma−/− cortical neurons. Extracts from Puma−/− neurons were used in these experiments to avoid potential complications associated with cell death. As shown in Figure 9A, GST-Puma precipitated both endogenous Bcl-XL and Bax with high efficiency. These interactions were specific for Puma as binding was not observed in pulldowns performed with GST alone. In contrast, GST-BimEL efficiently precipitated Bcl-XL, but not Bax (Fig. 9B). These results suggest that Puma may play a dominant role in regulating Bax activation and neuronal apoptosis by associating with Bax and targeting it to the mitochondria.

Puma physically associates with Bcl-XL and Bax. A, B, Lysates from Puma−/− neurons were incubated with GST-Puma (A) or GST-BimEL (B) and complexes were precipitated with glutathione-sepharose beads. Binding of GST-Puma or GST-Bim to Bcl-XL and Bax was determined by immunoblot. Lanes labeled lysate correspond to 10% of protein used in pulldown assay.
p53 is a key regulator of oxidative stress induced puma expression and neuronal cell death
We have shown previously that Puma is a key transcriptional target in p53 mediated neuronal cell death (Cregan et al., 2004a). Therefore, we examined whether p53 regulates Puma induction and neuronal apoptosis in response to oxidative stress. As shown in Figure 10A, the induction of Puma expression in response to various oxidative stressors was decreased in p53−/− neurons as compared with wt neurons. Furthermore, we found that TBH, H2O2, and NOC-12 induced apoptosis was significantly diminished in p53−/− cortical neurons (Fig. 10B). Together these results indicate that p53 is a key regulator of oxidative damage induced Puma expression and neuronal cell death.

p53 is a key regulator of oxidative stress induced Puma expression and neuronal cell death. A, Cortical neurons from wt and p53−/− littermates were treated with TBH (200 μm), H2O2 (20 μm) or NOC-12 (200 μm) and Puma mRNA induction was determined by quantitative RT-PCR after 10 h (n = 6; *p < 0.01). B, Wt and p53−/− neurons were treated with the indicated oxidative stressors and the fraction of apoptotic cells was determined by Hoechst staining after 24 h (n = 5; *p < 0.01).
Discussion
Puma is required for oxidative stress induced neuronal apoptosis
It is widely recognized that apoptosis contributes to the neurological dysfunction that occurs in acute neuronal injuries and in certain neurodegenerative diseases (Vila and Przedborski, 2003; Chan, 2004). A multitude of studies implicate oxidative stress as a major trigger for neuronal cell death in these neurodegenerative conditions (Sugawara and Chan, 2003; Andersen, 2004). Indeed, elevated levels of several reactive oxidant species have been reported in animal models of cerebral ischemia and neurodegenerative disease, and oxidative damage has been observed in postmortem tissue of affected humans (Lyras et al., 1998; Eliasson et al., 1999; Hsu et al., 2000). Furthermore, it has been demonstrated that free radical reducing agents are neuroprotective in animal models of cerebral ischemia, and that transgenic mice that overexpress the antioxidant enzymes superoxide-dismutase (SOD1) or glutathione peroxidase (GSHPx) exhibit reduced infarct volumes (Margaill et al., 2005). Likewise, it has been shown that neuronal cell death is enhanced in SOD1- and GSHPx-deficient mice in the MPTP model of Parkinson's disease as well as in models of ischemic injury (Zhang et al., 2000; Crack et al., 2001). Importantly, we have determined that transcriptional induction of the Bcl-2 family member Puma is essential for oxidative stress induced neuronal apoptosis suggesting that Puma may be an important therapeutic target for the treatment of several neurodegenerative conditions. Interestingly, it has been reported that after cerebral ischemia in rat, Puma expression is upregulated in regions of the hippocampus known to exhibit significant levels of apoptosis, suggesting that Puma may be an important target in stroke (Reimertz et al., 2003). Previous data also suggests that Puma may contribute to neuronal cell death in Parkinson's disease. For example, Puma has been implicated in apoptosis in PC12 cells induced by the Parkinson's related toxin 6-hydroxydopamine (Biswas et al., 2005). Furthermore, in the present study we demonstrated that Puma-deficient neurons are remarkably resistant to cell death induced by MPP+, the active metabolite of the Parkinson inducing agent MPTP. However, it will be important in future studies to assess the role of Puma in dopaminergic neuron cell death in animal models of Parkinson's disease in vivo.
The BH3-only subgroup of the Bcl-2 protein family consists of multiple family members that could potentially contribute to neuronal cell death. In addition to Puma, oxidative stress triggered induction of the BH3-only member BimEL. However, Bim- deletion did not affect oxidative stress induced mitochondrial permeabilization, caspase activation, or neuronal cell death. This was curious as BimEL has been reported to contribute to the induction of neuronal apoptosis after NGF deprivation, potassium/serum deprivation, and p75NTR signaling (Putcha et al., 2001; Whitfield et al., 2001; Becker et al., 2004). In these developmental neuronal death paradigms Jun N-terminal kinase-mediated phosphorylation of BimEL has been shown to promote its apoptotic activity (Putcha et al., 2003; Becker et al., 2004). However, it has been shown that phosphorylation of Bim by ERK1/2 in non-neuronal cells can promote its ubiquitination and degredation and thereby reduce its apoptotic activity (Ley et al., 2003; Harada et al., 2004). Although we have not determined whether oxidative stress affects the phosphorylation status of BimEL, we found that BimEL localized to the mitochondria and was able to bind to Bcl-X, suggesting that this is not sufficient to promote neuronal apoptosis in the absence of Puma.
In addition to Bim and Puma, we also observed a robust induction of Noxa mRNA in response to certain types of oxidative stress. However, similar to Bim-deficient neurons, Noxa-deficient neurons were not protected against oxidative stress-induced cell death. Consistent with this, we have shown previously that Noxa is upregulated in cortical and cerebellar granule neurons after DNA damage or enforced expression of p53, but that ectopic expression of Noxa was not sufficient to induce neuronal apoptosis (Cregan et al., 2004a). Similarly, it has been shown previously that Noxa mRNA levels are induced after DNA damage in sympathetic neurons, but that cell death was not diminished in the absence of Noxa (Wyttenbach and Tolkovsky, 2006). However, it has been reported that axotomy induced motor neuron cell death is reduced in Noxa-deficient mice (Kiryu-Seo et al., 2005). Therefore, it is possible that Noxa may function in certain neuronal populations, but not others. We also recognize that BH3-only proteins activated through transcription-independent mechanisms might also contribute to oxidative stress induced neuronal apoptosis. For example, Bid is known to be activated in response to death receptor (e.g., Fas) activation when cleaved by caspase-8 into its truncated form tBid (Li et al., 1998). Interestingly, tBid has been implicated in neuronal cell death induced by oxygen-glucose deprivation and in a rodent model of focal ischemia (Plesnila et al., 2001). Although we cannot rule out the possible involvement of Bid or other post-translationally activated BH3-only family members our results indicate that regardless of the their potential contribution, Puma induction is essential for cell death.
Puma regulates Bax activation in response to oxidative damage
BH3-only proteins are thought to function by promoting the activity of multidomain proapoptotic Bcl-2 family members (Harris and Johnson, 2001; Zong et al., 2001). Indeed we have demonstrated that not only Puma but also Bax is required for mitochondrial outer membrane permeabilization and neuronal apoptosis in response to oxidative injury. Furthermore, we have determined that Puma is required for oxidative stress induced Bax translocation and conversion of Bax to its activated conformation. Moreover, we found that ectopic expression of Puma is sufficient to induce Bax activation and to trigger Bax-mediated mitochondrial membrane permeabilization. Together, these results suggest that Puma is a key regulator of Bax activation in response to oxidative damage.
It is generally thought that BH3-only proteins function by binding and neutralizing anti-apoptotic family members (Moreau et al., 2003). Interestingly, it has been reported that certain BH3-only proteins can bind to multiple prosurvival Bcl-2 family members whereas others are more restricted (Chen et al., 2005). Moreover, the authors of this study indicated that the cytotoxic potential of different BH3-only proteins when ectopically expressed correlates with their degree of promiscuity. For example, it was shown that Noxa interacts with Mcl-1, but not other prosurvival members including Bcl-xL or Bcl-2 and as a result has limited cell killing potential. This is consistent with our previous findings demonstrating that ectopic expression of Noxa is not sufficient to induce neuronal apoptosis (Cregan et al., 2004a), and our current findings that although Noxa is induced by oxidative stress, its sole loss does not inhibit cell death. However, it was shown that both Puma and Bim exhibit high affinity for all prosurvival Bcl-2 family proteins (Chen et al., 2005). However, we found that Puma-deletion but not Bim-deletion prevents oxidative stress induced Bax activation and neuronal apoptosis indicating that Puma and Bim do not perform redundant functions in this context. Although controversial, it has been suggested that in addition to neutralizing prosurvival members certain BH3-only proteins may also directly interact with multidomain proapoptotic members to promote their activity (Eskes et al., 2000; Letai, 2002; Kuwana et al., 2005; Kim et al., 2006; Willis et al., 2007). Indeed, we have demonstrated using GST-pulldown assays that Puma but not Bim can associate with Bax in cell extracts isolated from neurons exposed to oxidative stress. However, we cannot rule out the possibility that Puma associates with Bax as part of a complex and not through direct physical interaction. Unfortunately, we were unable to examine the interaction between Puma and Bax after oxidative stress in situ as Puma protein levels were only appreciably elevated in Bax-deficient neurons presumably because of its potent cell killing capacity. These results suggest that Puma could play a dominant role in regulating neuronal cell death by virtue of its ability to both neutralize anti-apoptotic Bcl-2 proteins and directly promote Bax activation.
Oxidative stress triggers Puma induction and neuronal cell death via p53-dependent and p53-independent mechanisms
In this study, we demonstrate that the transcriptional activator p53 plays a key role in regulating oxidative stress induced Puma expression and neuronal apoptosis. Consistent with this we have previously demonstrated that Puma is an essential transcriptional target in p53 mediated neuronal cell death (Cregan et al., 2004a). The contribution of p53 in this setting likely reflects the ability of oxidative stress associated free radicals to induce DNA damage. Indeed, previous studies have demonstrated that p53 is a critical regulator of DNA damage induced neuronal apoptosis (Wood and Youle, 1995; Xiang et al., 1996; Enokido et al., 1996). Interestingly, however, we found that the induction of Puma and neuronal apoptosis after oxidative injury was only partially blocked in p53−/− neurons suggesting that other factors also contribute to these processes. At this stage the nature of the p53-independent activation pathway remains unclear, although several interesting candidates exist. For example, it has been reported that in colorectal cancer cells Puma expression can be induced by the p53-family member p73 (Melino et al., 2004). Another group has shown that the transcription factor E2F1 can bind to the Puma promoter and drive its expression in fibroblasts (Hershko and Ginsberg, 2004). Most recently, it was reported that Foxo3a can activate Puma expression in hematopoietic cells after cytokine deprivation (You et al., 2006). Therefore, in future studies it will be important to identify additional transcriptional activators that contribute to oxidative stress induced Puma induction and neuronal cell death. In summary, we have established that Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis suggesting that Puma may be a key therapeutic target for neuroprotection.
Footnotes
-
This work was supported by The Heart and Stroke Foundation Grant NA 5758, the Canadian Institutes of Health Research Grant MOP68995, and the Krembil Foundation. The authors are grateful to Gillian Bayley and Meggan Brine for their technical assistance in certain aspects of these studies, and to Profs. J. Adams and S. Cory for generously providing the Bim, Noxa, and Puma heterozygous mouse lines.
- Correspondence should be addressed to Dr. Sean P. Cregan, Robarts Research Institute, 100 Perth Drive, London, Ontario, Canada, N6A 5K8. scregan{at}robarts.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}