

Abstract
Prostatic acid phosphatase (PAP) is expressed in nociceptive dorsal root ganglion (DRG) neurons, functions as an ectonucleotidase, and generates adenosine extracellularly. Here, we found that PAP inhibits noxious thermal sensitivity and sensitization that is associated with chronic pain through sustained activation of the adenosine A1 receptor (A1R) and phospholipase C-mediated depletion of phosphatidylinositol 4,5-bisphosphate (PIP2). In mice, intrathecal injection of PAP reduced PIP2 levels in DRGs, inhibited thermosensation through TRPV1, and enduringly reduced thermal hyperalgesia and mechanical allodynia caused by inflammation, nerve injury, and pronociceptive receptor activation. This included inhibitory effects on lysophosphatidic acid, purinergic (ATP), bradykinin, and protease-activated (thrombin) receptors. Conversely, PIP2 levels were significantly elevated in DRGs from Pap−/− mice, and this correlated with enhanced thermal hyperalgesia and mechanical allodynia in Pap−/− mice. To directly test the importance of PIP2 in nociception, we intrathecally injected PIP2 into mice. This transiently (2 h) elevated PIP2 levels in lumbar DRGs and transiently (2 h) enhanced thermosensation. Additionally, thermal hyperalgesia and mechanical allodynia were enduringly enhanced when PIP2 levels were elevated coincident with injury/pronociceptive receptor stimulation. Nociceptive sensitization was not affected if PIP2 levels were elevated in the absence of ongoing pronociceptive receptor stimulation. Together, our data suggest that PIP2 levels in DRGs directly influence thermosensation and the magnitude of nociceptive sensitization. Moreover, our data suggest there is an underlying “phosphoinositide tone” that can be manipulated by an adenosine-generating ectonucleotidase. This tone regulates how effectively acute nociceptive insults promote the transition to chronic pain.
Introduction
Nociceptive neurons in the dorsal root ganglia (DRGs) sense noxious thermal and mechanical stimuli and can be sensitized by diverse pronociceptive chemicals (Hucho and Levine, 2007; Basbaum et al., 2009). Once sensitized, animals often display long-lasting thermal hyperalgesia and mechanical allodynia—two common symptoms of chronic pain.
Recently, we found that nociceptive neurons express two molecularly distinct ectonucleotidases that generate adenosine extracellularly by dephosphorylating AMP. These ectonucleotidases include the transmembrane (TM) isoform of prostatic acid phosphatase (PAP) (also known as ACPP) and ecto-5′-nucleotidase (NT5E) (also known as CD73) (Zylka et al., 2008; Sowa et al., 2010). Interestingly, PAP knock-out (Pap−/−) mice, Nt5e−/− mice, and adenosine A1 receptor knock-out (A1R−/−) mice all display enhanced nociceptive responses after inflammation or nerve injury (Wu et al., 2005; Zylka et al., 2008; Sowa et al., 2010). These observations suggest that deficiencies in adenosine production or A1R signaling enhance nociceptive sensitization; however, the mechanism underlying these enhanced nociceptive responses is currently unknown.
The secretory isoform of PAP (S-PAP) also generates adenosine by dephosphorylating AMP (Vihko, 1978; Zylka et al., 2008; Sowa et al., 2009). When injected intrathecally, S-PAP has long-lasting (3 d) thermal antinociceptive effects in naive mice as well as long-lasting antihyperalgesic and antiallodynic effects in sensitized animals (Zylka et al., 2008; Sowa et al., 2009). These antinociceptive effects were transiently blocked by an A1R antagonist and were eliminated in A1R−/− mice, indicating that S-PAP activates A1R over a sustained time period.
Adenosine and A1R agonists have antinociceptive effects when administered acutely to rodents and humans (Eisenach et al., 2003; Sawynok, 2007). Additionally, acute A1R activation inhibits neurotransmitter release from nociceptive neurons, voltage-gated calcium channels, and postsynaptic neurons in spinal cord (Dolphin et al., 1986; Li and Perl, 1994; Lao et al., 2001). Although these inhibitory mechanisms may account for some aspects of A1R-mediated antinociception, inhibition of neurotransmission cannot readily explain why sustained A1R activation by PAP selectively inhibits noxious thermal sensitivity in naive mice without affecting mechanical sensitivity (Zylka et al., 2008; Sowa et al., 2009). This selectivity suggests that PAP might regulate thermal nociception by acting through a specific thermosensory channel.
Indeed, in our present study, we found that PAP acts via A1R to reduce the levels of phosphatidylinositol 4,5-bisphosphate (PIP2) in cultured cells and in vivo. In turn, this reduction in PIP2 inhibits thermosensation, in part through the capsaicin and noxious heat receptor TRPV1 (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) (Caterina et al., 1997, 2000; Davis et al., 2000). This mechanism is consistent with several studies showing that PIP2 is required for TRPV1 to function optimally in vitro (for review, see Rohacs et al., 2008). Pronociceptive G-protein-coupled receptors (GPCRs) also require PIP2 to signal, suggesting alterations in PIP2 might regulate sensitization through these receptors. Indeed, we found that PAP inhibited signaling and sensitization through diverse pronociceptive GPCRs by depleting PIP2 (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), with PIP2 levels at the time of stimulation/injury enduringly influencing the level of nociceptive sensitization. Collectively, our experiments suggest PIP2 plays a central role in nociceptive mechanisms.
Materials and Methods
All procedures and behavioral experiments involving vertebrate animals were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
Molecular biology.
Full-length expression constructs for mouse TM-PAP (nucleotides 64-1314 from GenBank accession number NM_207668) and human TM-PAP (nucleotides 51-1304 from GenBank accession number BC007460) were generated by reverse transcription (RT)-PCR amplification using C57BL/6 mouse trigeminal cDNA or human placental cDNA (Clontech) as template and Phusion polymerase. The red fluorescent protein mCherry was then fused in-frame to the C terminus of all TM-PAP constructs. Mouse TM-PAP(H12A) was generated by PCR-based mutagenesis using mouse TM-PAP as template (His12 corresponds to His43 of the mPAP preprotein). This active site mutant was previously described and lacks catalytic activity (Schneider et al., 1993; Ostanin et al., 1994). All constructs have a Kosak consensus sequence, were cloned into pcDNA3.1, and were sequence verified. We obtained additional constructs from others (see acknowledgments). We confirmed that adenosine receptors were expressed in Rat1 fibroblasts by RT-PCR (A1R primers, 5′-CATTGGGCCACAGACCTACT and 5′-GGCAGAAGAGGGTGATACA).
Calcium imaging.
Cell lines were grown on glass bottom culture dishes (MatTek; P35G-0-10-C) in DMEM containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin, and transfected with Lipofectamine Plus (Invitrogen) according to manufacturer's protocol. The total amount of DNA per transfection was adjusted to 1 μg by adding pcDNA 3.1. After transfection (18–24 h), cells were loaded for 1 h at room temperature with 2 μm fura-2 AM (Invitrogen; F-14185) in Hanks buffered salt solution (HBSS plus calcium and magnesium) assay buffer (HBSS plus 9 mm HEPES plus 11 mm d-glucose plus 0.1% fatty-acid free BSA, pH 7.4). Cells were then washed three times with HBSS assay buffer and sat for at least 30 min before imaging. A Nikon TE2000U microscope and Sutter DG4 light source were used to image calcium responses (excitation, 340 nm/380 nm; emission, 510 nm). We manually pipetted and aspirated solutions for all calcium imaging experiments. Cells were stimulated with 1 μm capsaicin, 100 nm lysophosphatidic acid (LPA), 1 U/ml thrombin (Thr), 10 μm ATP, or 1 μm bradykinin (BK) for 1–5 min, washed in HBSS assay buffer for 1 min, and then stimulated with 0.006% SDS to evoke maximal calcium responses for normalization. We did not use ionomycin to normalize responses because this calcium ionophore activates calcium-dependent phospholipase C (PLC) enzymes. As a result, the magnitude of the ionomycin-induced calcium response is also proportional to PIP2 levels in cells.
Calcium responses were normalized by calculating the area under the curve during ligand stimulation for each cell, and then dividing by the maximum SDS-evoked calcium response in each cell. These values were averaged over all cells for a given condition and then normalized relative to untransfected cells in the same field of view or relative to control cells (with the untransfected or control cell response set to 100%).
For experiments with capsaicin, Rat1 fibroblasts were transfected with TRPV1-green fluorescent protein (GFP) alone or TRPV1-GFP plus various constructs. The same amount of TRPV1-GFP was used for each transfection and the total amount of DNA per transfection was adjusted to 1 μg by adding pcDNA 3.1. Cells were stimulated with 1 μm capsaicin (from 100 mm stock in 100% DMSO, dissolved to final concentration in HBSS assay buffer) for 1 min, followed by a 5 min wash in HBSS assay buffer, and then stimulation with 0.006% SDS.
For experiments with the PIP2 shuttle, Rat1 fibroblasts were stimulated with 100 nm LPA for 1 min, followed by a 15 min wash with HBSS assay buffer plus 3 nm PIP2 plus 3 nm Carrier 2 (PIP2 Shuttle kit; Echelon; P-9045) or HBSS assay buffer plus 3 nm Carrier 2 alone. Cells were then stimulated with 100 nm LPA again, washed for 1 min with HBSS assay buffer, and stimulated with 0.006% SDS.
For thapsigargin experiments, HBSS assay buffer lacking calcium and containing 1 mm EGTA was used to eliminate extracellular calcium. The 10 μm thapsigargin was added for 5 min, and the cells were then washed for 2 min with HBSS assay buffer and stimulated with 0.006% SDS. For pertussis toxin (PTX) experiments, Rat1 fibroblasts were incubated for 18 h with 500 ng/ml PTX before loading with fura-2 AM and stimulation with 100 nm LPA. For experiments with adenosine receptor antagonists, PLC inhibitor [1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122)], protein kinase C (PKC) inhibitor (staurosporine), or protein kinase A (PKA) inhibitor [(9S,10S,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester (KT5720)], cells were incubated with antagonists for 3–4 h, loaded in the presence of antagonists/inhibitors with fura-2 AM for 1 h, and then stimulated with pronociceptive ligands.
Electrophysiology.
A HEK293-TRPV1 stable cell line (Kim et al., 2008) was transfected with TM-PAP-mCherry or TM-PAP(H12A)-mCherry. Patch-clamp recordings were made from mCherry-expressing cells using a Multiclamp 700B amplifier and pClamp 9.2 software as described previously (Campagnola et al., 2008). Heat ramps were generated by exchanging bath solution with a preheated solution via a two-to-one port. Solution was preheated with an in-line heater controlled by a TC-324B temperature controller modified for high temperature (Warner Instruments). Only one current recording was made per coverslip. The bath solution consisted of the following (in mm): 140 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 Na-HEPES, 5 glucose, pH 7.4, mOsm 295–310, and was perfused at a rate of 2–3 ml/min by gravity flow. Electrodes were pulled from borosilicate glass on a Sutter Instrument P-2000 and filled with intracellular solution that contained the following (in mm): 135 KCl, 3 MgATP, 10 HEPES, 0.5 Na2ATP, 1.1 CaCl2, 2 EGTA, 5 glucose, with pH adjusted to 7.5 with HCl and osmolarity adjusted to 300 mOsm with sucrose. Tip resistances ranged from 2.5 to 5 MΩ. Series resistance was not compensated; however, recordings with series resistances >15 MΩ were discarded.
PIP2 quantification.
For quantification of PIP2 in vitro, HEK293 cells or Rat1 fibroblasts were plated onto glass coverslips and transfected with the construct PLCδ-PH-GFP along with indicated constructs using Lipofectamine Plus (Invitrogen), according to the manufacturer's protocol. Eighteen to 24 h later, the cells were fixed with 4% paraformaldehyde–PBS. Cells were imaged on a confocal microscope. GFP fluorescence on the plasma membrane (PM) of cells compared with the cytoplasm was quantified using ImageJ by taking cross-sectional averages of pixel intensity at the plasma membrane and dividing by the average of pixel intensity in the cytoplasm.
For quantification of PIP2 in DRGs, age-matched, adult male mice were injected intrathecally with 5 μl of 15% lidocaine plus 50 U/ml hPAP (250 mU total), 15% lidocaine plus 3 nm Carrier 2 (Echelon; P-9C2), 15% lidocaine plus 3 nm PIP2 (Echelon; P-9045) plus 3 nm Carrier 2 or 15% lidocaine alone. Before injection, an equimolar mixture of PIP2 plus Carrier 2 was incubated for 15 min at room temperature. Lidocaine causes transient (5–20 min.) paralysis of both hindlimbs, permitting us to visually determine whether each mouse received a successful intrathecal injection (we only quantified PIP2 levels in mice that showed transient bilateral paralysis). One day later, mice were killed and lumbar 3 (L3) to L6 DRGs were dissected bilaterally (n = 8 ganglia/sample) and placed in PBS on ice. For each sample, DRG wet weight was determined, and then lipids were extracted and quantified using the PI(4,5)P2 Mass ELISA kit from Echelon (K-4500) following the manufacturer's protocol. PIP2 levels were normalized by dividing by the wet weight of DRG tissue.
Behavior.
Pap−/−, A1R−/−, and Trpv1−/− (B6.129X1-Trpv1tm1Jul/J) mice were backcrossed to C57BL/6 mice for at least 10 generations. For all other experiments, male, C57BL/6 mice were purchased from The Jackson Laboratory. Male 2- to 4-month-old mice were used for all behavioral studies. All mice were acclimated to testing room, equipment, and experimenter for 1–3 d before behavioral testing. The experimenter was blind to genotype and drug treatment during behavioral testing. Thermal and mechanical sensitivity were measured as described previously (Zylka et al., 2008). For intrathecal drug delivery, 5 μl was injected into unanesthetized mice using the direct lumbar puncture method (Fairbanks, 2003). The complete Freund's adjuvant (CFA) model of inflammatory pain and the spared nerve injury (SNI) model of neuropathic pain were performed as described previously (Shields et al., 2003; Zylka et al., 2008).
Drugs.
Secreted human PAP (S-hPAP) (Sigma-Aldrich; P1774) and heat-inactivated S-hPAP were prepared as described previously (Zylka et al., 2008). The 18:1 lysophosphatidic acid (Avanti Polar Lipids; 857130) was dissolved in 0.9% ethanol and then diluted to final concentrations in either HBSS assay buffer (calcium imaging) or 0.9% saline (injections). ATP (Sigma-Aldrich; A26209) was dissolved in either HBSS assay buffer (calcium imaging) or 0.9% saline (injections). U73122 (Tocris; 1268) was first dissolved into DMSO, and then further diluted in 0.9% saline for intrathecal injection. The PI(4,5)P2 Shuttle PIP kit (Echelon; P-9045) was used to increase PIP2 levels in vivo. PtdIns(4,5)P2 di-C16 was first dissolved into 10% DMSO in 0.9% saline. Carrier 2 (Histone H1) was dissolved into 0.9% saline. Before injection, PIP2 and Carrier 2 were mixed in a 1:1 molar ratio and incubated at room temperature for 15 min. Thrombin (Sigma-Aldrich; T4648) was first dissolved to 100 U/ml in 0.1% BSA and further diluted in HBSS assay buffer to final concentrations. BK was dissolved to 1 mm in DMSO and further diluted in HBSS assay buffer to final concentrations. PTX (Sigma-Aldrich; P7208) and caffeine (Sigma-Aldrich; C0750) were dissolved in water. 8-Cyclopentyl-1,3-dimethylxanthine (CPT) (Sigma-Aldrich; C102), 8-cyclopentyl-1,3-dipropylxanthine (CPX) (Sigma-Aldrich; C101), 7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1,2,4-triazolo [1,5-c]pyrimidine (SCH 58261) (Sigma-Aldrich; S4568), 8-[4-[((4-cyanophenyl)carbamoylmethyl)oxy]phenyl]-1,3-di(n-propyl)xanthine hydrate (MRS 1754) (Sigma-Aldrich; M6316), 3-propyl-6-ethyl-5-[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine carboxylate (MRS 1523) (Sigma-Aldrich; M1809), staurosporine (Sigma-Aldrich; S4400), KT5720 (Tocris; 1288), and U73122 (Tocris; 1268) were dissolved in DMSO and further diluted in HBSS assay buffer to final concentrations.
Results
PAP inhibits activation of TRPV1
We previously found that S-PAP reduced noxious thermal sensitivity by activating A1R for a sustained 3 d time period (Zylka et al., 2008; Sowa et al., 2009). Since TRPV1 functions as a noxious heat and capsaicin receptor (Caterina et al., 1997, 2000), we hypothesized that PAP might reduce thermal sensitivity by inhibiting TRPV1. To test this hypothesis, we transfected Rat1 fibroblasts with TRPV1 and mouse TM-PAP, and then measured capsaicin-evoked responses with the calcium indicator fura-2 AM. We observed that both the amplitude and duration of the capsaicin-evoked calcium response was reduced by 25% in TM-PAP-transfected cells relative to cells expressing TRPV1 alone (Fig. 1A). Inhibition was dependent on TM-PAP catalytic activity because capsaicin-evoked calcium responses were not inhibited in cells transfected with TM-PAP(H12A), a phosphatase-dead mutant of TM-PAP (Fig. 1B). Moreover, the A1R-selective antagonist CPX blocked the effect of TM-PAP on capsaicin-evoked signaling (Fig. 1B) (Rat1 cells express A1R) (data not shown). Last, heat-evoked current density through TRPV1 was reduced in cells expressing TM-PAP relative to cells expressing catalytically inactive TM-PAP(H12A) (Fig. 1C). All TM-PAP constructs were fused to the red fluorescent protein mCherry and were expressed at similar levels, but only the H12A mutant lacked catalytic activity (as assessed using enzyme histochemistry) (data not shown). Collectively, our data suggest that TM-PAP acts via A1R to reduce capsaicin- and heat-evoked activation of TRPV1.

PAP reduces activity of the capsaicin and noxious heat receptor TRPV1. A, Capsaicin (1 μm)-evoked calcium responses in Rat1 fibroblasts expressing TRPV1 alone or with TM-PAP. The 340/380 ratio is directly proportional to calcium concentration; n = 80 cells per condition. B, Normalized capsaicin-evoked calcium responses in Rat1 fibroblasts transfected with the indicated constructs. Where indicated, cells were incubated with CPX (5 μm) for 3 h before stimulation; n = 40–60 cells per condition; t tests relative to TRPV1 only condition. C, Noxious heat-evoked currents and current density in HEK293-TRPV1 stable cells transfected with TM-PAP or the catalytically inactive H12A mutant; n = 20 cells per condition. D, The hindpaws of WT (black) and Trpv1−/− mice (red) were tested for noxious thermal sensitivity before [baseline (BL)] and after intrathecal injection of S-hPAP (250 mU). Paired t tests were used to compare responses within each genotype to BL (black asterisks) and between genotypes (red asterisks); n = 10 mice per genotype. D, Inset, Detailed time course to determine onset of antinociception. E, F, Mechanical (E) and thermal (F) sensitivity of WT and Trpv1−/− mice before (BL) and after injection of CFA into one hindpaw. One day later, the indicated mice were injected intrathecally with S-hPAP (250 mU) or heat-inactivated S-hPAP (0 mU). CFA-injected and noninjected (control) hindpaws were tested. Paired t tests were used to compare responses at each time point between WT mice and Trpv1−/− mice injected with S-hPAP (red asterisks); n = 8 mice per group; paired t test, *p < 0.05, **p < 0.005, ***p < 0.0005. All data are presented as means ± SEM.
Next, to determine whether PAP reduced noxious thermal sensitivity by acting through TRPV1 in vivo, we measured thermal withdrawal latencies in wild-type (WT) and Trpv1−/− mice before and after intrathecal injection of S-hPAP. As previously observed (Caterina et al., 2000), there were no significant differences at baseline between WT and Trpv1−/− mice when stimulating the hindpaw with radiant heat (Fig. 1D). After S-hPAP injection, paw withdrawal latency significantly increased (relative to baseline) at the 30 min time point and remained elevated for 3 d in WT mice (Fig. 1D), reproducing previous results (Zylka et al., 2008). In contrast, the thermal antinociceptive effect of S-hPAP was blunted in magnitude (p < 0.001 by two-way ANOVA; relative to WT) and duration (2 d; relative to baseline) in Trpv1−/− mice (Fig. 1D) (this difference between genotypes was independently reproduced in Fig. 1F, control paw). S-hPAP was equally effective at reducing mechanical allodynia in WT and Trpv1−/− mice after CFA-induced inflammation (Fig. 1E, black dashed line vs red dashed line), ruling out the trivial possibility that Trpv1−/− mice were less sensitive to all antinociceptive effects of S-hPAP. Although S-hPAP had long-lasting thermal antinociceptive effects in WT mice (Fig. 1F), we were unable to study the thermal antinociceptive effects of S-hPAP in Trpv1−/− mice because Trpv1−/− mice did not develop thermal hyperalgesia after inflammation (Fig. 1F), as previously found by others (Caterina et al., 2000; Davis et al., 2000). Together, our cell-based and in vivo data suggest PAP reduces thermal sensitivity, in part, by inhibiting TRPV1.
Activation of A1R by TM-PAP depletes PIP2
Our findings raised the question of how sustained, PAP-dependent activation of A1R inhibited TRPV1 at a mechanistic level. A1R stimulation inhibits PKA via PTX-sensitive Gαi/o-proteins. In addition, A1R stimulation activates PLC (including PLCβ3) via PTX-sensitive Gβγ subunits (Murthy and Makhlouf, 1995; Dickenson and Hill, 1998). PLC enzymes then hydrolyze PIP2 in the membrane to diacylglycerol (DAG) and inositol triphosphate (IP3). These facts suggested that sustained activation of A1R might inhibit TRPV1 activity by inhibiting PKA, activating PKC (via DAG), depleting intracellular calcium stores (via IP3) or depleting PIP2 (via PLC activation). Although TRPV1 can be modulated by PKC and PKA (Bhave et al., 2002, 2003; Huang et al., 2006), TM-PAP did not inhibit TRPV1 through PKC or PKA pathways (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). Nor did TM-PAP deplete intracellular calcium stores (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material).
TRPV1 is also modulated by PIP2 (Prescott and Julius, 2003; Liu et al., 2005; Stein et al., 2006; Lishko et al., 2007; Lukacs et al., 2007; Klein et al., 2008; Rohacs et al., 2008; Yao and Qin, 2009). At high capsaicin concentrations (≥1 μm) and in the presence of extracellular calcium, PIP2 is required for TRPV1 channel activity, whereas depletion of PIP2 desensitizes the channel. This requirement for PIP2 suggested TM-PAP might inhibit TRPV1 by reducing cellular levels of PIP2 through sustained A1R activation.
To test this possibility, we quantified the levels of PIP2 in cells using the biosensor PLCδ-PH-GFP (Várnai and Balla, 1998). When PIP2 levels are high, PLCδ-PH-GFP is primarily localized to the PM. When PIP2 levels are reduced, PLCδ-PH-GFP translocates from the membrane to the cytosol. This translocation can be quantified by measuring the GFP signal intensity on the PM relative to the cytosol (expressed as the ratio PM/cytosol). We used HEK293 cells for these experiments because this biosensor was difficult to visualize in Rat1 fibroblasts (although we reproduced our key finding in Rat1 cells) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
In HEK293 cells expressing only PLCδ-PH-GFP, the majority of the GFP signal was in the PM, giving a PM/cytosol ratio of 3.43 ± 0.35 (Fig. 2A,E). In contrast, PLCδ-PH-GFP was redistributed to the cytosol in cells cotransfected with TM-PAP or PLCβ3 (PM/cytosol ratio of 1.60 ± 0.06 and 1.70 ± 0.9, respectively) (Fig. 2B,E). This finding suggested TM-PAP and PLCβ3 depleted PIP2 to a similar extent when expressed for an extended (∼24 h) time period. Importantly, the A1R antagonist CPX and the PLC inhibitor U73122 blocked the TM-PAP-mediated redistribution of PLCδ-PH-GFP to the cytosol (Fig. 2C,E), suggesting that TM-PAP depleted PIP2 by acting through A1R and PLC. Additionally, the TM-PAP- and PLCβ3-mediated redistribution of PLCδ-PH-GFP was blocked by overexpressing phosphatidylinositol-4-phosphate-5-kinase (PIPK) (Fig. 2D,E). PIPK increases PIP2 levels in transfected cells (Lin et al., 2005; Milosevic et al., 2005), suggesting TM-PAP and PLCβ3 altered PLCδ-PH-GFP membrane localization by depleting PIP2.

TM-PAP reduces capsaicin-evoked calcium responses by decreasing PIP2. A–D, Subcellular localization of the PIP2 biosensor PLCδ-PH-EGFP (PLCδ-PH) in HEK293 cells, imaged by confocal microscopy. PLCδ-PH alone (A), cotransfected with TM-PAP (B), cotransfected with TM-PAP and incubated with 5 μm CPX for 3 h before fixation (C), and cotransfected with TM-PAP and PIPK (D). Scale bar, 10 μm. E, Quantification of PLCδ-PH subcellular localization after cotransfection with the indicated constructs or after incubation with CPX. Fluorescence values in the PM and cytosol were quantified from cell cross-sections using ImageJ and expressed as a ratio; n = 30–70 cells per condition. F, Normalized capsaicin (1 μm)-evoked calcium responses in Rat1 fibroblasts transfected with the indicated constructs; n = 40–100 cells per condition; t tests were used to compare PLCδ-PH-transfected cells to cotransfected cells (E) and TRPV1-transfected cells to cotransfected cells (F). ***p < 0.0005. All data are presented as means ± SEM.
TM-PAP reduces TRPV1 activity by depleting PIP2
Next, we genetically manipulated PIP2 levels to determine whether increasing or decreasing PIP2 affected capsaicin-evoked calcium responses. Both TM-PAP and PLCβ3 deplete PIP2 to a similar extent (Fig. 2E), but only PLCβ3 hydrolyzes PIP2 directly. Likewise, both TM-PAP and PLCβ3 reduced capsaicin-evoked calcium responses to a similar extent (Fig. 2F), suggesting indirect or direct depletion of PIP2 was sufficient to reduce TRPV1 activity. Conversely, increasing PIP2 levels by overexpressing PIPK (which regenerates PIP2) blocked TM-PAP and PLCβ3 from inhibiting capsaicin-evoked calcium responses (Fig. 2F). These findings suggested signaling through TRPV1 was reduced as a direct result of PIP2 depletion, consistent with the findings of others using cultured cells (Liu et al., 2005; Stein et al., 2006; Lishko et al., 2007; Lukacs et al., 2007; Klein et al., 2008; Rohacs et al., 2008; Yao and Qin, 2009). In addition, TM-PAP did not affect the capsaicin-evoked calcium response in cells expressing TRPV1Δ42(777-820) (data not shown), a TRPV1 mutant that is missing a putative PIP2 binding domain (Prescott and Julius, 2003; Kwon et al., 2007; Kim et al., 2008). Together, these data show that TM-PAP reduces TRPV1 activity in vitro through sustained activation of A1R and subsequent depletion of PIP2.
PAP regulates PIP2 levels in vivo
Since PAP regulated PIP2 levels in cultured cells, we hypothesized PAP might also regulate PIP2 levels in vivo. To test this hypothesis, we measured PIP2 levels in L3–L6 DRGs from WT mice that were injected (intrathecally) with vehicle or S-hPAP. We collected DRGs 1 d after injections because S-hPAP has maximal A1R-dependent antinociceptive effects at this time (Zylka et al., 2008). Injection of S-hPAP significantly reduced PIP2 levels by 40 ± 9% (Fig. 3A), and this reduction was dependent on A1R activation [as evidenced by the observation that PIP2 levels in L3–L6 DRGs were not significantly different in A1R−/− mice injected (intrathecally) 1 d earlier with 250 mU of S-hPAP (250 mU) relative to heat-inactivated S-hPAP (0 mU); 159.6 ± 23.5 pmol/mg DRG vs 174.4 ± 28.0 pmol/mg DRG, respectively; n = 4 mice per condition]. Conversely, PIP2 levels were elevated by 89 ± 23% in DRGs from Pap−/− mice (Fig. 3A). Together, these data suggest PIP2 levels are inversely related to the amount of PAP ectonucleotidase activity and A1R stimulation.

Manipulation of PIP2 levels in vivo. A, PIP2 levels in L3–L6 DRGs were quantified in WT mice, Pap−/− mice, or WT mice injected (intrathecally) 1 d earlier with 250 mU of S-hPAP. Values were compared with WT by paired t test; n = 3 mice per condition. B, C, The hindpaws of WT mice were tested for noxious thermal sensitivity before [baseline (BL)] and after intrathecal injection of S-hPAP (250 mU) or saline. Two days later, the indicated mice were injected intrathecally with either U73122 (5.4 nmol) or vehicle (V) and thermal sensitivity was measured every hour for the first 7 h as well as for the next 2 d. Data in C are from the boxed area in B. Mice injected with U73122 were compared with vehicle-injected mice at each time point by paired t test; n = 8 mice per group. D, PIP2 in L3–L6 DRGs was quantified in WT mice at the given time points after intrathecal injection with PIP2 (3 nmol) plus carrier. Mice from later time points were compared with time 0 values by paired t test; n = 4 mice per condition. E, F, The hindpaws of WT mice were tested for noxious thermal sensitivity before (BL) and after intrathecal injection of S-hPAP (250 mU) or saline. Two days later, the indicated mice were injected intrathecally with carrier (Car) or PIP2 (3 nmol) plus carrier, and then thermal sensitivity was measured every hour for the first 6 h after injection as well as for the next 2 d. Data in F are from the boxed area in E. Mice injected with PIP2 after S-hPAP were compared with vehicle-injected mice at each time point by paired t test. G, Model showing how PAP interferes with TRPV1 channel activity (see text for details). *p < 0.05, **p < 0.005, ***p < 0.0005. All data are presented as means ± SEM.
Since our cell-based data suggested that PAP depleted PIP2 by activating PLC, we next evaluated whether the thermal antinociceptive effect of S-hPAP could be blocked using the PLC inhibitor U73122. This inhibitor was previously injected intrathecally (at 5.4 nmol) to block PLC activation by a δ-opioid receptor ligand (Narita et al., 2000). Indeed, intrathecal injection of U73122 at the same dose transiently and completely blocked the thermal antinociceptive effect of S-hPAP, providing evidence that S-hPAP acted through PLC to reduce thermal sensitivity in vivo (Fig. 3B,C). Importantly, the 5.4 nmol dose had no effect on thermal sensitivity when injected alone (Fig. 3B,C) (Narita et al., 2000). However, a higher U73122 dose (12.5 nmol) transiently enhanced thermal sensitivity (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Since strong PLC inhibition would be predicted to elevate PIP2 levels, this latter result suggests that the PLC/PIP2 pathway tonically modulates thermal thresholds.
To directly assess whether S-hPAP reduced thermal sensitivity by depleting PIP2, we transiently replenished PIP2 in lumbar DRGs using a PIP2 shuttle (Ozaki et al., 2000). We found that injection (intrathecally) of PIP2 (complexed with carrier) transiently (2 h) elevated PIP2 levels well above normal levels in lumbar DRGs (Fig. 3D), whereas injection of carrier alone (the control) had no effect (based on the observation that PIP2 levels in L3–L6 DRGs were not significantly different in WT mice relative to WT mice injected intrathecally with Car; 112.0 ± 14.6 pmol/mg DRG vs 102.5 ± 10.5 pmol/mg DRG, respectively; n = 4 mice per condition). Strikingly, intrathecal injection of PIP2 (complexed with carrier) also transiently (2 h) reversed S-hPAP-mediated thermal antinociception, whereas carrier alone had no effect (Fig. 3E,F). The fact that behavior was significantly altered only during the time at which PIP2 was significantly elevated makes it unlikely these effects on thermal sensitivity were a coincidence. Instead, these results strongly suggest that PAP inhibits thermal sensitivity as a direct result of PIP2 depletion. Moreover, control animals injected with PIP2 displayed transient thermal hyperalgesia, suggesting thermal sensitivity can be enhanced when PIP2 levels are elevated above normal levels. The magnitude of this effect on thermal sensitivity in control animals was smaller (2.6 s) than in animals that were injected with S-hPAP and PIP2 (4.0 s). This argues that PIP2 replenishment was sufficient to block the thermal antinociceptive effect of S-hPAP independent of how PIP2 affects thermal sensitivity in control animals.
Together, our data strongly support a mechanism (Fig. 3G) in which (1) TM- and S-PAP function as ectonucleotidases that generate adenosine. Adenosine then stimulates (2) A1R in a sustained fashion, followed by (3) PLC activation and (4) PIP2 hydrolysis. (5) Sustained reductions in PIP2 levels decreased TRPV1 activity and decreased noxious thermal sensitivity. In addition, our data suggest PIP2 levels are regulated by tonic ectonucleotidase-dependent adenosine production and A1R activation.
PAP inhibits pronociceptive LPA receptor signaling by reducing PIP2 levels
Diverse chemicals are released after injury and inflammation and sensitize nociceptive neurons, in many cases, by activating pronociceptive GPCRs (Hucho and Levine, 2007; Basbaum et al., 2009). Since many pronociceptive receptors signal through PLC (and hence require PIP2 for signaling), we hypothesized that PAP might reduce signaling through pronociceptive receptors via sustained activation of A1R and PIP2 depletion. Such a mechanism could have important physiological implications because reduced signaling through pronociceptive receptors would be predicted to reduce nociceptive sensitization, a key symptom of chronic pain. LPA is a pronociceptive ligand that sensitizes nociceptive neurons, causes long-lasting (>7 d) sensitization in vivo (including thermal hyperalgesia and mechanical allodynia), and is implicated in neuropathic pain mechanisms (Elmes et al., 2004; Inoue et al., 2004; Park and Vasko, 2005). In addition, LPA receptors are coupled to Gαq/11-proteins and signal through PLC in many cell types, including Rat1 fibroblasts (which endogenously express LPA receptors) (Mills and Moolenaar, 2003; Kelley et al., 2006).
Using Rat1 fibroblasts, we found that the amplitude and duration of LPA-evoked calcium responses were significantly reduced in cells expressing TM-PAP relative to untransfected cells in the same field of view (Fig. 4A,D). This “PAP effect” was species-conserved as cells transfected with TM-hPAP (human TM-PAP) were also less responsive to LPA stimulation (Fig. 4B,D). In contrast, the LPA-evoked calcium response was not reduced in cells transfected with the catalytically inactive TM-PAP(H12A) mutant (Fig. 4C,D).

TM-PAP reduces pronociceptive ligand-evoked calcium responses in Rat1 fibroblasts through activation of A1R. A–C, LPA (100 nm)-evoked calcium responses in untransfected cells and in cells transfected with the indicated constructs; n = 15 cells per condition. D, LPA-evoked calcium responses in cells expressing the indicated constructs and after incubation with PTX (500 ng/ml), caffeine (Caff) (1 mm), CPT (500 nm), CPX (5 μm), or U73122 (5 μm). Incubation time was 18 h for PTX and 3 h for all other compounds. Responses normalized to untransfected cells (Untrans); n = 70–100 cells per condition. E, Normalized calcium responses in Rat1 fibroblasts stimulated with ATP (10 μm), Thr (1 U/ml), or BK (1 μm). The indicated cells were incubated with CPX (5 μm) for 3 h before stimulation; n = 70–110 cells per condition. F, Model showing how PAP inhibits pronociceptive receptor signaling (see text for details). For A–C, two-way ANOVA was used to compare transfected and untransfected cells (p values indicated on graphs). For D and E, t tests were used to compare untransfected cells to transfected cells. ***p < 0.0005. All data are presented as means ± SEM.
To determine whether TM-PAP inhibited LPA-evoked signaling by generating adenosine and activating A1R, we assessed whether PTX (an inhibitor of Gαi/o-coupled receptors) or adenosine receptor antagonists could block the effect of TM-PAP on LPA-evoked signaling. We found that PTX completely blocked the PAP effect, as evidenced by no significant differences between untransfected cells and TM-PAP plus PTX-treated cells (Fig. 4D). Additionally, the PAP effect was blocked by the A1/A2B adenosine receptor antagonist caffeine (Caff) and by two different A1R-selective antagonists: CPT and CPX (Fig. 4D). In contrast, selective antagonists of all other adenosine receptors (A2AR, SCH 58261; A2BR, MRS 1754; A3R, MRS 1523) did not block the PAP effect (data not shown).
Next, we evaluated whether increasing or decreasing PIP2 affected LPA-evoked calcium responses in our cell-based assay. We found that both TM-PAP and PLCβ3 reduced LPA-evoked calcium responses to a similar extent (Fig. 4D), suggesting indirect or direct depletion of PIP2 was sufficient to reduce signaling. This is consistent with a previous study showing that LPA-evoked Ca2+ responses were reduced to baseline levels when PIP2 was depleted using an inducible PIP2 phosphatase (Várnai et al., 2006). Conversely, increasing PIP2 levels by overexpressing PIPK blocked the TM-PAP- and PLCβ3-mediated reduction in LPA-evoked calcium responses (Fig. 4D). Moreover, restoring PIP2 levels with the PIP2 shuttle blocked the inhibitory effect of TM-PAP (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). These experiments provide complementary support that TM-PAP inhibits LPA receptor signaling as a direct result of PIP2 depletion. The inhibitory effect of TM-PAP was also blocked with the PLC inhibitor U73122 (Fig. 4D), further indicating that TM-PAP acts via PLC to deplete PIP2. As expected, the magnitude of the LPA-evoked Ca2+ influx was smaller in all cells when PLC was inhibited. Last, TM-PAP did not reduce LPA signaling by acting through other pathways that are downstream of A1R, including Gαi/o-mediated inhibition of PKA or DAG-mediated PKC activation (data not shown).
TM-PAP reduces signaling through several pronociceptive GPCRs
Since many pronociceptive receptors are Gαq/11-coupled and signal via PLC, we hypothesized that TM-PAP might reduce signaling through additional pronociceptive receptors, including purinergic receptors (using the ligand ATP), protease-activated receptors (using the ligand Thr), and BK receptors. Importantly, activation of these receptors evokes transient calcium responses in Rat1 cells and causes nociceptive sensitization in vivo (Kelley et al., 2006; Wang et al., 2006; Burnstock, 2007; Sawynok, 2007; Dale and Vergnolle, 2008). Strikingly, calcium responses induced by all three ligands were reduced in TM-PAP-transfected cells relative to untransfected cells, and these reductions were blocked by the A1R antagonist CPX and by overexpressing PIPK (Fig. 4E).
Collectively, our data further support a mechanism (Fig. 4F) in which (1) PAP functions as an ectonucleotidase to generate adenosine over a sustained time period. (2) Adenosine then stimulates A1R followed by (3) PLC activation and (4) PIP2 hydrolysis. With less PIP2 available for (5) Gαq/11/PLC-mediated receptor signaling, less IP3 (and DAG) is generated after receptor stimulation, resulting in smaller pronociceptive ligand-evoked calcium responses.
PAP enduringly blocks LPA- and ATP-induced hyperalgesia and allodynia
Since PAP reduced pronociceptive receptor signaling in Rat1 cells by depleting PIP2, we hypothesized that PAP might also reduce signaling through pronociceptive receptors in vivo. To test this possibility, we took advantage of the fact that both LPA and ATP produce long-lasting (>7 d) thermal hyperalgesia and mechanical allodynia when injected intrathecally (Inoue et al., 2004; Nakagawa et al., 2007). This is longer than the 3 d antihyperalgesic and antiallodynic effects of S-hPAP (Zylka et al., 2008). Thus, by injecting S-hPAP 1 d before ATP or LPA, we could ascertain whether S-hPAP reduced initiation of LPA- or ATP-evoked signaling by quantifying hyperalgesia and allodynia on days 4 and 8 (i.e., after the 3 d antinociceptive effects of PAP wore off).
First, we measured baseline (BL) thermal and mechanical sensitivity in two groups of WT mice. Next, we injected S-hPAP (intrathecally) into one of the groups and heat-inactivated S-hPAP (control; catalytically dead) into the other group (Fig. 5) (a third group of S-hPAP-injected mice from a different experiment is shown, to provide a visual reference for how S-hPAP typically affects naive mice). One day later, S-hPAP increased paw withdrawal latency to the noxious thermal stimulus but had no effect on mechanical sensitivity, whereas inactive S-hPAP had no effects on thermal or mechanical sensitivity (Fig. 5). These results were expected (Zylka et al., 2008) and confirmed that mice received an active or inactive dose of S-hPAP. After taking these measurements, we injected (intrathecally) either 5 nmol of LPA (Fig. 5A,B) or 100 nmol of ATP (Fig. 5C,D). These doses produce maximal sensitization in animals (Inoue et al., 2004; Nakagawa et al., 2007). Both pronociceptive compounds produced long-lasting thermal hyperalgesia and mechanical allodynia in the control (inactive S-hPAP-injected) mice. In contrast, mice injected with S-hPAP and then LPA/ATP were significantly different from controls at all times after LPA/ATP injections and did not develop thermal hyperalgesia or mechanical allodynia, as evidenced by latencies and thresholds that were at or near baseline levels on days 4 and 8. These data suggest that PAP, an enzyme that reduces PIP2 levels in vivo (Fig. 3A), blocked physiologically relevant signaling and sensitization through two distinct pronociceptive receptors. Conversely, thermal hyperalgesia and mechanical allodynia were significantly and enduringly enhanced after LPA or ATP injection in Pap−/− mice (Fig. 6)—that is, mice that have elevated levels of PIP2 in lumbar DRGs (Fig. 3A).
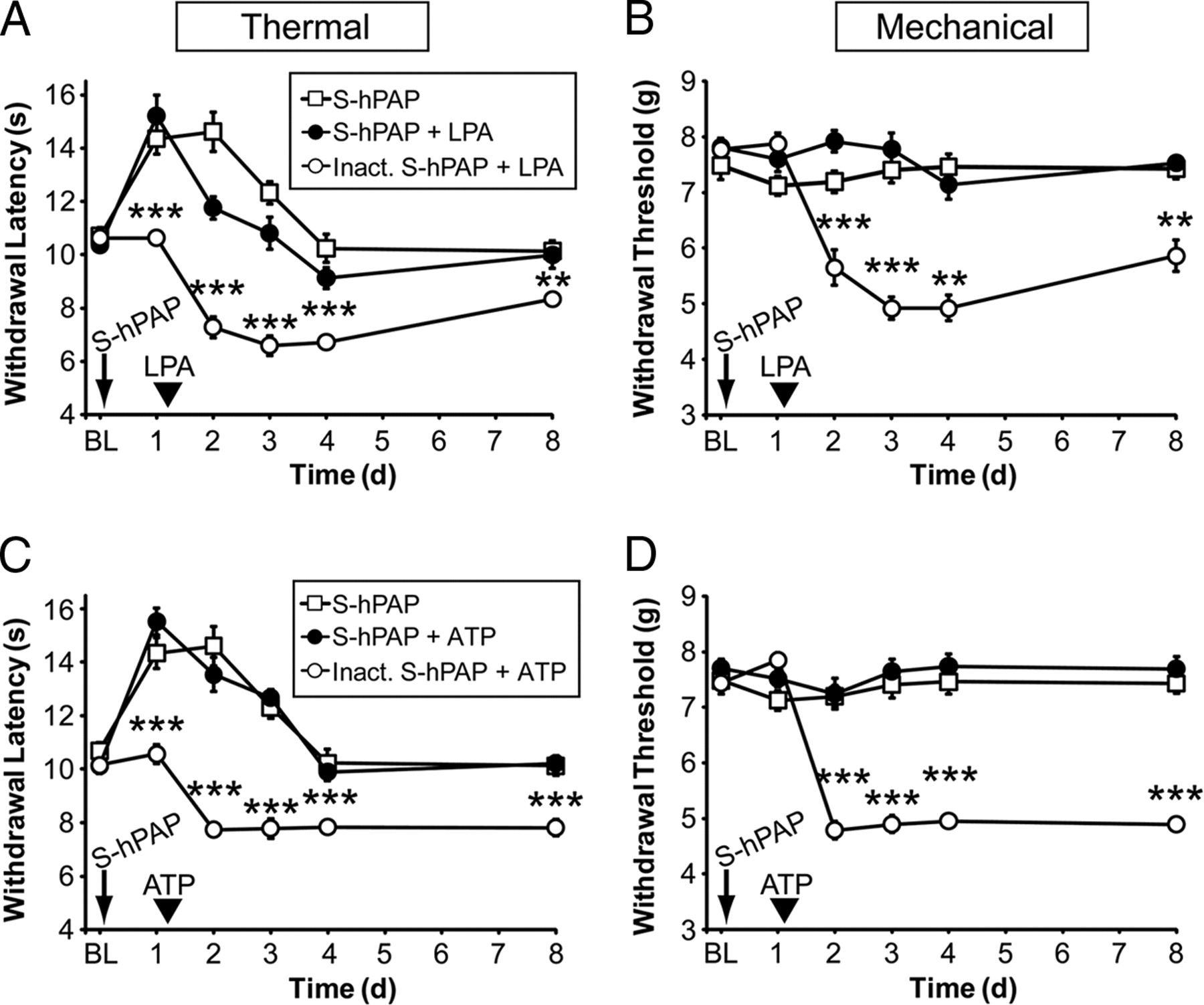
Secretory PAP inhibits pronociceptive receptor signaling in vivo. A–D, The hindpaws of WT mice (n = 10 per group) were tested for noxious thermal and mechanical sensitivity before [baseline (BL)] and after intrathecal injection of S-hPAP (250 mU) or heat-inactivated S-hPAP (0 mU). One day later, mice were injected intrathecally with 5 nmol of LPA (A, B) or 100 nmol of ATP (C, D). Paired t tests were used to compare responses at each time point between mice injected with S-hPAP plus LPA to mice injected with inactive S-hPAP plus LPA. The S-hPAP data (open squares) shown in A–D are from a different experiment with WT mice and are plotted here to provide a visual, not statistical, reference for comparison. **p < 0.005, ***p < 0.0005. All data are presented as means ± SEM.

Pap−/− mice show enhanced LPA- and ATP-induced thermal hyperalgesia and mechanical allodynia. A–D, The hindpaws of WT and Pap−/− mice were tested for noxious thermal and mechanical sensitivity before [baseline (BL)] and after intrathecal injection of LPA (5 nmol) (A, B) or ATP (100 nmol) (C, D); n = 10 mice per genotype. Pap−/− mice developed significantly greater thermal hyperalgesia (p < 0.0001 by two-way ANOVA) and mechanical allodynia (p < 0.0001 by two-way ANOVA) in response to intrathecal LPA or ATP. Post hoc paired t tests were used to compare responses at each time point between genotypes. *p < 0.05, **p < 0.005, ***p < 0.0005. All data are presented as means ± SEM.
PAP preemptively inhibits sensitization caused by inflammation and nerve injury
Although LPA and ATP produce long-lasting sensitization in vivo, injection of these chemicals may not fully model the sensitization and pathology that is associated with chronic pain conditions. To determine whether reducing PIP2 levels with S-hPAP had a more generalized effect on the signals that initiate pain sensitization, we tested S-hPAP in the CFA model of inflammatory pain and in the SNI model of neuropathic pain. Strikingly, intrathecal injection of S-hPAP before CFA-induced inflammation nearly eliminated thermal hyperalgesia and significantly reduced mechanical allodynia for the duration of the experiment compared with controls injected with inactive S-hPAP (Fig. 7A,B). These preemptive antinociceptive effects of S-hPAP were dependent on A1R activation (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). In addition, intrathecal injection of S-hPAP before nerve injury eliminated thermal hyperalgesia and significantly reduced mechanical allodynia for the duration of the experiment compared with mice injected with inactive S-hPAP (Fig. 7C,D). Collectively, these findings indicate that S-hPAP enduringly blocks sensitization in two chronic pain models when injected before inflammation/injury.

Secretory PAP partially blocks the initiation of inflammatory and neuropathic pain. A–D, The hindpaws of WT mice (n = 10 per group) were tested for noxious thermal and mechanical sensitivity before [baseline (BL)] and after intrathecal injection of S-hPAP (250 mU) or heat-inactivated S-hPAP (0 mU). One day later, CFA was injected into one hindpaw (CFA arrowhead) (A, B) or peripheral nerves were injured using the SNI model of neuropathic pain (Injure arrowhead) (C, D). Paired t tests were used to compare responses at each time point between mice injected with S-hPAP (open square; injured paw) and those injected with inactive hPAP (open circles; injured paw). *p < 0.05, **p < 0.005, ***p < 0.0005. All data are presented as means ± SEM.
Direct elevation of PIP2 in DRGs enhances nociceptive sensitization
Since PIP2 levels in DRGs were indirectly elevated and sensitization was enhanced in Pap−/− mice [data above and in the study by Zylka et al. (2008)], we next sought to determine whether direct elevation of PIP2 in DRGs could enhance sensitization. To accomplish this, we coinjected (intrathecally) WT mice with LPA plus Car (the control) or LPA plus PIP2 plus Car, and then measured noxious thermal and mechanical sensitivity for several days. Importantly, there is a critical window of 3 h over which LPA (injected intrathecally) signals to produce nociceptive sensitization in mice (Ma et al., 2009). Since PIP2 levels are only elevated in DRGs for 2 h after injection (Fig. 3D), these coinjection experiments allowed us to elevate PIP2 levels only when LPA receptors were active in vivo. Strikingly, when PIP2 levels were elevated coincident with LPA receptor activation, thermal hyperalgesia and mechanical allodynia were significantly and reproducibly enhanced for the duration of the experiment (Fig. 8A,B). In contrast, injection of Car alone or PIP2 plus Car in the absence of a pronociceptive stimulus had no long-term effects on thermal or mechanical sensitivity (Fig. 8C,D). And injection of Car alone or PIP2 plus Car did not sensitize mice when injected 3 d after LPA was injected (i.e., well past the 3 h critical window for LPA receptor signaling) (Fig. 8E,F). In this same experiment, PIP2 plus Car (but not Car alone) caused a transient (2 h) enhancement in thermal sensitivity (data not shown), thus reproducing our findings above (Fig. 3E,F) and confirming that these PIP2 injections were successful and had the capacity to affect behavior. Together, these experiments strongly argue that PIP2 levels must be elevated in DRGs at the time of pronociceptive receptor activation to enhance thermal and mechanical sensitization.

Increasing PIP2 levels at the time of LPA injection or inflammation enhances sensitization. A–D, One hindpaw of WT mice was tested for noxious thermal and mechanical sensitivity (n = 10 mice per group). After taking baseline (BL) measurements, mice were injected intrathecally with LPA (5 nmol) plus Car or LPA (5 nmol) plus PIP2 (3 nmol) plus Car (A, B); Car or PIP2 (3 nmol) plus Car (C, D). E, F, One hindpaw of WT mice was tested for noxious thermal and mechanical sensitivity (n = 10 mice per group). After taking BL measurements, mice were injected intrathecally with LPA (arrowhead; 5 nmol). Three days later, mice were injected intrathecally with Car or PIP2 (3 nmol) plus Car (arrow). G, H, The hindpaws of WT mice were tested for noxious thermal and mechanical sensitivity. After taking BL measurements, mice were injected intrathecally with Car or PIP2 (3 nmol) plus Car, and then were immediately injected with CFA into one hindpaw (arrowhead). Two hours after CFA injection, mice were reinjected intrathecally with Car or PIP2 (3 nmol) plus Car (arrow). Inflamed and noninflamed (control) hindpaws were tested. For A–F, paired t tests were used to compare groups at each time point. For G and H, paired t tests were used to compare responses at each time point between mice injected with Car (open circles; injured paw) and those injected with PIP2 plus Car (open squares; injured paw). *p < 0.05, **p < 0.005, ***p < 0.0005. All data are presented as means ± SEM.
Last, we sought to determine whether direct elevation of PIP2 could enhance thermal and mechanical sensitization caused by CFA. For this experiment, all mice were injected intrathecally with Car or PIP2 plus Car immediately before injecting CFA into one hindpaw. CFA sensitizes nociceptors through the release of an “inflammatory soup” composed of diverse pronociceptive ligands (Basbaum et al., 2009). Since this soup could activate pronociceptive receptors for an extended period of time (and since PIP2 is only elevated for 2 h after a single injection), we reinjected (intrathecally) all mice 2 h later with PIP2 plus Car (or Car alone). This ensured that PIP2 levels remained elevated while CFA “initiated” sensitization. Strikingly, CFA-induced thermal hyperalgesia and mechanical allodynia were significantly enhanced for the duration of the experiment when PIP2 levels were transiently elevated (Fig. 8G,H) [compare the inflamed paw of PIP2 plus Car-injected mice to the inflamed paw of Car alone (control) mice]. In contrast, thermal and mechanical responses were not altered in the contralateral (uninflamed) paws of mice injected with PIP2 plus Car (Fig. 8G,H), further demonstrating that acute elevation of PIP2 does not sensitize mice in the absence of a pronociceptive stimulus.
Discussion
In our effort to determine how PAP regulated nociception at a mechanistic level, we found that sustained A1R activation reduced the levels of PIP2 in cells and in DRGs. This reduction in PIP2 reduced noxious thermal sensitivity, in part through inhibition of TRPV1. And this reduction in PIP2 enduringly reduced nociceptive sensitization to thermal and mechanical stimuli.
PAP regulates thermosensation through TRPV1
Numerous studies found that TRPV1 can be modulated by PIP2 in vitro (for review, see Rohacs et al., 2008). At low capsaicin concentrations and in the absence of extracellular calcium, PIP2 partially inhibits TRPV1 (Prescott and Julius, 2003). In contrast, at high capsaicin concentrations and in the presence of extracellular calcium, PIP2 is required for TRPV1 activity (Liu et al., 2005; Stein et al., 2006; Lishko et al., 2007; Lukacs et al., 2007; Klein et al., 2008; Yao and Qin, 2009). These contrasting in vitro results made it difficult to predict how alterations in the levels of PIP2 might affect thermosensation in animals. In our present study, we found that PAP inhibited TRPV1 in cultured cells through sustained A1R activation and PLC-mediated PIP2 depletion. Likewise, PAP inhibited thermosensation in mice through A1R activation (Zylka et al., 2008), PLC activation (Fig. 3B,C), and A1R-dependent PIP2 depletion (Fig. 3A and data above). Moreover, the inhibitory effect of PAP on thermosensation was partially dependent on TRPV1 activation (Fig. 1D). Conversely, thermosensation was modestly enhanced when PIP2 levels were elevated in vivo (Fig. 3E,F). Together, our findings suggest that PIP2 facilitates thermosensation in vivo through TRPV1-dependent and independent mechanisms. Our findings are consistent with a study showing that TRPV1-dependent thermosensation was enhanced through interactions with PIRT, a PIP2-binding protein (Kim et al., 2008).
Pronociceptive GPCR activation can sensitize TRPV1 via PKC/PLC (Bhave et al., 2003; Huang et al., 2006). Since A1R is also coupled to PLC (Murthy and Makhlouf, 1995; Jacobson and Gao, 2006), this raises the question of why PAP inhibits TRPV1 following A1R activation, as we observed, instead of sensitizing TRPV1? This likely reflects differences in how pronociceptive GPCRs and A1R couple to downstream signaling pathways. Pronociceptive receptors, including LPA receptors, are coupled to PLC isoforms via Gαq/11- and Gβγ-proteins. Stimulation of these receptors evokes transient PKC activation and large PLC-dependent calcium responses that desensitize rapidly (Mills and Moolenaar, 2003; Kelley et al., 2006). In contrast, A1R is coupled to PLC isoforms exclusively via Gβγ-proteins and A1R does not desensitize, at least when activated by ectonucleotidase-generated adenosine. Specifically, we found that A1R does not desensitize when activated for a sustained 3 d time period by PAP (Zylka et al., 2008; Sowa et al., 2009) or on repeated injection of S-hPAP (data not shown). And importantly, the PLC inhibitor U73122 transiently inhibited PAP antinociception (Fig. 3B,C), arguing that PLC is active over this 3 d period. These data suggest that ectonucleotidase-dependent activation of A1R is sufficient to deplete PIP2 and inhibit TRPV1 but is not sufficient to activate PKC, sensitize TRPV1, or detectably elevate calcium levels. Indeed, we found that PAP did not inhibit TRPV1 through PKC (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) nor did PAP reduce intracellular (IP3-sensitive) calcium stores (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material). And acute stimulation with the A1R agonist N6-cyclopentyladenosine (300 nm) did not evoke Ca2+ influx in cultured small-diameter DRG neurons from adult mice (E. McCoy and M. J. Zylka, unpublished data).
We found that most of the thermal antinociceptive effects of PAP were lost in Trpv1−/− mice, whereas the mechanical antinociceptive effects of PAP were preserved (Fig. 1D–F). This dissociation suggests that most of the thermal antinociceptive effects of PAP were mediated through TRPV1, whereas the remaining thermal and mechanical antinociceptive effects of PAP were likely mediated by other PIP2-sensitive channels or proteins. Depletion of PIP2 generally reduces ion channel activity, including KCNQ, P2X4, and N-type calcium channels (Gamper et al., 2004; Suh and Hille, 2005; Bernier et al., 2008), all of which could affect nociception (Basbaum et al., 2009). In addition, PIP2 depletion inhibits synaptic vesicle exocytosis (Di Paolo and De Camilli, 2006). Additional studies will be needed to determine whether additional antinociceptive effects of PAP are attributable to inhibition or modulation of other PIP2-sensitive channels, proteins, or mechanisms.
PIP2 levels at the time of pronociceptive receptor activation enduringly regulate the magnitude of pain sensitization
We unexpectedly found that nociceptive sensitization could be enduringly altered by manipulating the levels of PIP2 at the time of stimulation/injury. This suggests that any manipulation, be it genetic or environmental, which alters PIP2 levels could have a lasting impact on nociceptive sensitization. Indeed, we found that thermal hyperalgesia and mechanical allodynia were enduringly enhanced only when PIP2 levels were elevated coincident with LPA receptor activation. PIP2 injection alone or PIP2 injection several days after LPA injection did not produce, enhance, or prevent sensitization. Direct elevation of PIP2 in DRGs also enhanced sensitization caused by peripheral injection of CFA, further supporting the physiological importance of PIP2 in nociceptive sensitization. Intriguingly, these findings also hint that PIP2 levels in DRGs regulate sensitization regardless of whether pain-producing stimuli are administered centrally (i.e., LPA) or peripherally (i.e., CFA).
Likewise, we found that sensitization could be enduringly altered by increasing or decreasing PIP2 levels through manipulation of PAP activity. Pap−/− mice have elevated PIP2 levels in DRGs and enhanced LPA-, ATP-, CFA-, and nerve injury-induced nociceptive sensitization [data above and in the study by Zylka et al. (2008)]. Conversely, S-hPAP injections reduced PIP2 levels in DRGs and enduringly inhibited LPA-, ATP-, CFA-, and nerve injury-induced nociceptive sensitization. S-hPAP acted exclusively through A1R to mediate these enduring effects (supplemental Fig. S6, available at www.jneurosci.org as supplemental material), ruling out the possibility that PAP acted through other adenosine receptor subtypes, including A2A (Loram et al., 2009). Importantly, these alterations in nociceptive sensitization outlasted the 3 d antinociceptive effects of S-hPAP and the acute (2 h) elevation of PIP2, arguing that neither maintained activity of PAP nor long-term elevations of PIP2 contributed to these enduring effects. Instead, these long-term changes in nociceptive sensitization could be attributable to reduced or enhanced engagement of transcriptional and nontranscriptional mechanisms that are downstream of pronociceptive receptor/PLC stimulation (Ji et al., 2009).
Intrathecal injections target DRGs and spinal cord (Luo et al., 2005). This raises the question of precisely where S-hPAP and PIP2 act to regulate nociception. Since S-hPAP (intrathecally) regulates PIP2 levels and nociception through A1R (data above) (Zylka et al., 2008), S-hPAP has the potential to act on any cell that expresses A1R. This includes peptidergic and nonpeptidergic nociceptive neurons as well as postsynaptic neurons in the spinal cord (Reppert et al., 1991; Li and Perl, 1994; Lao et al., 2001; Schulte et al., 2003) but excludes microglial cells because they do not express A1R (Orr et al., 2009). Despite repeated attempts, we were unable to determine at a cellular level which DRG neurons incorporated PIP2 (by studying the distribution of PIP2 conjugated to fluorochromes) (data not shown). However, nociception was only affected over the time period in which PIP2 levels were elevated in DRGs, arguing that nociceptive neurons in DRGs were targeted. Furthermore, the elevated levels of PIP2 in DRGs from Pap−/− mice are likely to be restricted to the subset of peptidergic and nonpeptidergic nociceptive neurons that normally express PAP (Zylka et al., 2008). We cannot further pinpoint at the cellular level where S-hPAP and PIP2 act to regulate nociception with existing technologies.
Last, PIP2 levels can be increased or decreased relative to an intermediate level in DRGs (Fig. 3A,D), suggesting nociception may be influenced by an underlying “phosphoinositide tone.” This tone may be coordinated with the “adenosine tone” that is present in diverse tissues, including the nervous system (Boison, 2008). Our data indicate that adenosine-generating ectonucleotidases like PAP contribute to this underlying phosphoinositide tone both positively and negatively. It may thus be possible to harness any molecule or mechanism that causes a sustained reduction in this tone to enduringly prevent or treat symptoms associated with chronic pain.
Footnotes
This work was supported by grants to M.J.Z. from The Sloan Foundation, The Searle Scholars Program, The Klingenstein Foundation, The Whitehall Foundation, and Rita Allen Foundation, and National Institute of Neurological Disorders and Stroke (NINDS) Grant R01NS060725, and grants to P.V. from The Sigrid Juselius Foundation, The Finnish Cancer Foundation, and The Research Council for Medicine of the Academy of Finland. N.A.S. was supported by NINDS Grant F30NS063507 and Medical Scientist Training Program Grant T32GM008719. Confocal imaging was performed at the University of North Carolina–Chapel Hill Confocal Imaging Facility, which is cofunded by NINDS and National Institute of Child Health and Human Development Grant P30NS045892. Calcium imaging was performed at the Michael Hooker Microscopy Facility, which is funded by an anonymous private donor. M.J.Z. is a Rita Allen Foundation Milton E. Cassel Scholar. We thank Sang-Kyou Han for PLCβ3 (EGFP- and FLAG-tagged versions), Tamas Balla for PLCδ-PH-GFP, Ira Milosevic for PIPK (mRFP-PIPK1-γ), David Julius for TRPV1-GFP, and Xinzhong Dong for TRPV1Δ42 and HEK293-TRPV1 stable cells; Yvette Chuang, Bonnie Taylor-Blake, and Eric McCoy for expert technical assistance; and Paul Farel, Ben Philpot, and Anthony LaMantia for comments on this manuscript.
- Correspondence should be addressed to Mark J. Zylka, Department of Cell and Molecular Physiology, University of North Carolina Neuroscience Center, University of North Carolina, CB #7545, Chapel Hill, NC 27599. zylka{at}med.unc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}