

Abstract
Endocannabinoids (eCBs) are retrograde lipid messengers that, by targeting presynaptic type 1 cannabinoid receptors (CB1Rs), mediate short- and long-term synaptic depression of neurotransmitter release throughout the brain. Short-term depression is typically triggered by postsynaptic, depolarization-induced calcium rises, whereas long-term depression is induced by synaptic activation of Gq/11 protein-coupled receptors. Here we report that a physiologically relevant pattern of postsynaptic activity, in the form of theta-burst firing (TBF) of hippocampal CA1 pyramidal neurons, can trigger long-term depression of inhibitory transmission (iLTD) in rat hippocampal slices. Paired recordings between CA1 interneurons and pyramidal cells, followed by post hoc morphological reconstructions of the interneurons' axon, revealed that somatic and dendritic inhibitory synaptic inputs equally expressed TBF-induced iLTD. Simultaneous recordings from neighboring pyramidal cells demonstrated that eCB signaling triggered by TBF was highly restricted to only a single, active cell. Furthermore, pairing submaximal endogenous activation of metabotropic glutamate or muscarinic acetylcholine receptors with submaximal TBF unmasked associative iLTD. Although CB1Rs are also expressed at Schaffer-collateral excitatory terminals, long-term plasticity under various recording conditions was spared at these synapses. Consistent with this observation, TBF also shifted the balance of excitation and inhibition in favor of excitatory throughput, thereby altering information flow through the CA1 circuit. Given the near ubiquity of burst-firing activity patterns and CB1R expression in the brain, the properties described here may be a general means by which neurons fine tune the strength of their inputs in a cell-wide and cell-specific manner.
Introduction
Endocannabinoids (eCBs) are fundamental retrograde regulators of short- and long-term plasticity at excitatory and inhibitory synapses (Chevaleyre et al., 2006; Kano et al., 2009; Regehr et al., 2009; Castillo et al., 2012). Mechanistically, eCB mobilization results from postsynaptic depolarization, metabotropic receptor activation, or a synergistic combination thereof (Hashimotodani et al., 2007b). Once produced, eCBs act presynaptically by engaging type-1 cannabinoid receptors (CB1Rs) to transiently or persistently depress neurotransmitter release. Short-term eCB-dependent plasticity is usually evoked postsynaptically by depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE; Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001) or brief postsynaptic metabotropic receptor activation (Maejima et al., 2001; Kim et al., 2002; Ohno-Shosaku et al., 2003). In contrast, eCB-mediated long-term depression (eCB-LTD) is usually triggered by synaptic activation of group I metabotropic glutamate receptors (I-mGluRs; Heifets and Castillo, 2009). It is unclear, however, whether postsynaptic activity is sufficient for eliciting eCB-LTD.
The functional impact that eCBs have on a neural circuit depends on the magnitude and duration of their effects at synapses, as well as their spatiotemporal signaling profile within and between neurons. In hippocampal area CA1, protocols eliciting strong DSI at eCB-sensitive inhibitory inputs only triggers weak DSE at Schaffer collateral (Sch)–CA1 pyramidal cell excitatory synapses (Ohno-Shosaku et al., 2002; Kawamura et al., 2006). Likewise, endogenous or pharmacological I-mGluR activation mobilizes eCBs to induce heterosynaptic LTD of inhibitory transmission (iLTD; Chevaleyre and Castillo, 2003, 2004; Edwards et al., 2006), with no lasting impact at Sch–CA1 inputs (Rouach and Nicoll, 2003; Chevaleyre and Castillo, 2004). Recent studies, however, reported that eCBs can mediate LTD at the Sch–CA1 synapse (Xu et al., 2010; Izumi and Zorumski, 2012; Péterfi et al., 2012), making the influence of eCB signaling on the excitatory/inhibitory balance difficult to predict. Evidence also indicates that somatic inhibitory inputs exhibit greater eCB sensitivity than dendritic inhibitory inputs (Lee et al., 2010), and that eCBs mobilized from an active CA1 pyramidal cell can spread to neighboring inactive neurons (Wilson and Nicoll, 2001; Yasuda et al., 2008). Whether a similar spread can occur for iLTD is unknown. Finally, while the duration over which CB1Rs are activated is thought to determine whether short-term or long-term eCB-mediated synaptic plasticity is observed (Chevaleyre et al., 2006), the precise patterns of activity that lead to short-term versus long-term synaptic plasticity are not well understood.
In this study, we report that a physiologically relevant pattern of activity, such as theta-burst firing (TBF) of CA1 pyramidal neurons, can trigger robust iLTD at both somatic and dendritic inputs. Notably, this form of plasticity was restricted to the single, activated pyramidal neuron. Moreover, excitatory synapses were completely spared by TBF under a variety of experimental conditions. As a result of TBF-induced disinhibition, the balance of excitation and inhibition was shifted, thereby facilitating excitatory throughput. In addition, a synergistic and associative component for postsynaptic TBF-induced iLTD (TBF-iLTD) was unmasked when submaximal postsynaptic activity was paired with submaximal endogenous Gq/11 protein-coupled receptor activation. Together, our results suggest that CA1 pyramidal cell TBF, alone or in combination with metabotropic receptor activation, triggers an eCB-dependent, long-lasting disinhibitory state capable of routing information through the hippocampus.
Materials and Methods
Hippocampal slice preparation.
Animal procedures were approved by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee and adhered to National Institutes of Health guidelines. Male and female Wistar rats (Charles River) aged postnatal day (P7) to P27 or C57BL/6 mice (P20–P25) were deeply anesthetized with isoflurane and then killed by decapitation. The brain was removed and quickly placed in ice-cold cutting solution containing the following (in mm): 215 sucrose, 20 glucose, 26 NaHCO3, 4 MgCl2, 4 MgSO4, 1.6 NaH2PO4, 1 CaCl2, and 2.5 KCl. Hippocampi were mounted on an agar block, and transverse slices 400 μm thick were prepared with a DTK-2000 microslicer (Dosaka Em) or VT1200 S microtome (Leica). Slices were placed, at room temperature, in a holding chamber containing 50% cutting solution and 50% artificial CSF (ACSF) recording solution containing the following (in mm): 124 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, and 1.3 MgSO4. After 30 min, the 1:1 solution was switched to ACSF at room temperature. Slices recovered in ACSF for at least 1 h, and then were transferred to a submersion-type, temperature-controlled recording chamber (TC-344B, Warner Instruments) and perfused with ACSF at 2 ml/min using a peristaltic pump (Dynamax RP-1, Rainin). Experiments were performed at 25.0 ± 0.1°C, unless otherwise indicated. All solutions were equilibrated for at least 30 min with 95% O2 and 5% CO2, pH 7.4.
Electrophysiology.
Conventional single, dual, and paired whole-cell voltage-clamp and current-clamp recordings were performed using a MultiClamp 700A or 700B amplifier (Molecular Devices). For single-cell voltage-clamp experiments (holding potential = −60 mV), CA1 pyramidal cells located ∼150 μm deep in the slice were blind patched with a micropipette (2.3–3.4 MΩ) fabricated on a two-step micropipette puller (PP-830 or PC-10, Narishige) and filled with an intracellular recording solution containing the following (in mm): 135 KMeSO4, 5 KCl, 1 CaCl2, 5 EGTA, 5 NaOH, 10 HEPES, and 10 glucose, pH 7.2 (280–290 mOsm). Stock ATP and GTP aliquots were added before experiments to give 5 and 0.4 mm, respectively. In some experiments, 20 mm 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) was included in the internal solution, and an equimolar amount of KMeSO4 removed. For experiments shown in Figure 2H, the internal solution was as follows (in mm): 130 K-gluconate, 4 NaCl, 10 HEPES, and 10 glucose, pH 7.2 (285 mOsm). KOH was used to adjust pH. To assess cell stability, series and input resistances were monitored with a 5 mV, 80 ms hyperpolarizing test pulse, and cells with >15% change in series resistance were excluded from analysis (>20% for cell-paired recordings). Input resistance was similarly monitored during current-clamp experiments, and the membrane potential was held constant near −60 mV, if needed, with DC injection. To elicit synaptic responses, paired, monopolar square-wave voltage or current pulses (100–200 μs pulse width) were delivered through a stimulus isolator (Isoflex, AMPI) connected to a broken tip (∼10–20 μm) stimulating patch-type micropipette filled with ACSF. Typically, stimulating pipettes were placed in CA1 stratum radiatum and/or stratum pyramidale (150–300 μm from the putative apical dendrite of the recorded pyramidal cell, 150–200 μm slice depth). Stimulus intensity was adjusted to give comparable magnitude synaptic responses across experiments (∼200–500 pA for whole-cell recordings and less than ∼0.6 mV for extracellular field recordings). Baseline stimulation was delivered at 0.2–0.25 or 0.05–0.1 Hz, respectively, before and after the induction of short-term or long-term plasticity. Short-term plasticity was induced after a 2 min stable baseline with a 5 s depolarization from −60 to 0 mV for DSI or with one to three bouts of TBF (see below) for burst firing-induced suppression of inhibition (BSI). iLTD was typically triggered after a 20 min baseline period with multiple postsynaptic DSIs (mDSIs; 5 s depolarization from −60 to 0 mV every 15 s) or TBF. A single TBF episode is defined as 10 bursts, with each burst containing five action potentials at 50 Hz, delivered at 5 Hz. For multiple episodes of TBF, this protocol was repeated every 5, 7.5, or 15 s (see Results). Action potentials were evoked from CA1 pyramidal cells in current-clamp by brief (2 ms) depolarizing pulses (1–2 nA). Heterosynaptic iLTD (Chevaleyre and Castillo, 2003) was triggered with a theta-burst stimulation protocol: 10 bursts of five stimuli (applied at 100 Hz) with a 200 ms interburst interval, repeated four times every 5 s.
Inhibitory synaptic transmission was monitored in the continuous presence of the NMDA receptor antagonist d-(-)-2-amino-5-phosphonopentanoic acid (d-APV; 25–50 μm) and the AMPA/kainate receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; 5–10 μm); for excitatory synaptic transmission, the GABAA receptor antagonist picrotoxin (100 μm) was present in the ACSF. For extracellular field recordings, a stimulating pipette filled with ACSF was placed in the middle third of stratum radiatum in area CA1. A recording pipette containing 1 m NaCl was placed ∼150 μm away at approximately the same slice depth (150–200 μm).
For dual recordings, neighboring CA1 pyramidal cells ∼50–100 μm deep in the slice were visually patched using an upright microscope (Eclipse E600FN, Nikon) equipped with infrared differential interference contrast optics. A stimulating pipette was placed 150–200 μm away from the recorded cells in stratum pyramidale. The recording pipette tips were used to measure the distance between the two somas. For associativity experiments combining submaximal presynaptic stimulation with submaximal postsynaptic TBF, a presynaptic train was paired with TBF. For submaximal group I-mGluR activation, three pulses at 10 Hz were delivered to stratum radiatum 0.5 or 7.5 s before each submaximal TBF episode (see Results). For submaximal activation of muscarinic acetylcholine receptors (mAChRs), 10 pulses at 20 Hz every 10 s, or 25 pulses at 50 Hz every 5 s, was delivered to stratum oriens/stratum alveus concomitantly with submaximal TBF (see Results). The stimulating pipette (broken tip ∼30–50 μm) was placed within 100 μm of the recorded CA1 pyramidal cell putative basal dendrites. Strong synaptic stimulation (∼30 V or ∼300 μA) is required to release endogenous acetylcholine in vitro (Cole and Nicoll, 1983).
For cell-paired recordings, interneuron somas located in stratum radiatum at least 100 μm deep in the slice were identified, visually patched, and recorded in current-clamp mode (initial resting membrane potentials ranged from −80 to −55 mV) using a KMeSO4-based recording solution similar to above but with 0.2% biocytin included for post hoc morphological reconstructions. A CA1 pyramidal cell, approximately the same depth in the slice, located directly across from the interneuron in stratum pyramidale, was blind patched with the original KMeSO4-based recording solution mentioned above or a relatively high chloride internal recording solution (Cl−reversal approximately −20 mV) containing the following (in mm): 54.5 KMeS04, 60 KCl, 1 CaCl2, 5 EGTA, 2 NaOH, 10 HEPES, 10 glucose, 5 ATP, and 0.4 GTP, pH 7.2 (280–290 mOsm). The probability of finding an interneuron–pyramidal cell pair was ∼15–20%. Action potentials were evoked from interneurons with a pair of 2 ms, 1.5–2.0 nA depolarizing square-wave current pulses spaced 100 ms apart, while unitary IPSCs (uIPSCs) were recorded from the pyramidal cell. Cell pairs were then tested for DSI at least twice. When observed (i.e., >20% suppression from baseline), DSI was quantified by averaging the peak amplitude of three to five traces before, immediately after the delivery of, and upon recovery from (usually <1 min) DSI. After a stable baseline (≥10 min), iLTD was induced. During the induction period, interneurons were slightly depolarized with somatic current injection to yield spontaneous firing frequencies of 2.0–2.5 Hz (Heifets et al., 2008). Four cell-paired recordings that were included in the DSI analysis were excluded from the iLTD analysis because the series resistance of the postsynaptic CA1 neuron increased significantly toward the end of the experiment.
Post hoc morphological reconstructions.
After completing the functional portion of cell-paired recordings, slices were fixed in 4% paraformaldehyde and 0.1% picric acid, and stored up to 3 d at 4°C. Slices were then washed with phosphate buffer and incubated with 1% H2O2 to reduce endogenous peroxidase activity, and biocytin was detected with an avidin–biotin–horseradish peroxidase complex using the Vectastain ABC kit (Vector Laboratories) containing 0.5% Triton X-100. The reaction was developed with the DAB substrate kit (Vector Laboratories) containing (NH4)2Ni(SO4)2. Slices were then washed with buffer, dehydrated with ethanol, cleared with CitriSolv (Fisher Scientific), mounted on glass slides, and coverslipped (Fisher Scientific) for subsequent morphological reconstructions using NeuroLucida (MBF Bioscience). Based on the predominant distribution of their axons in stratum pyramidale or stratum radiatum, interneurons were classified as basket cells (BCs) or Sch collateral-associated (SCA) interneurons, respectively (Vida et al., 1998; Cope et al., 2002).
Data acquisition and statistical analysis.
Stimulation and acquisition were controlled with custom software written in IgorPro (Wavemetrics). Output signals from whole-cell recordings were acquired at 5 kHz, filtered at 2.4 kHz, and stored online. Statistics were performed using OriginPro (Origin Laboratory). Statistical significance was set to p < 0.05 (*** indicates p < 0.001, ** indicates p < 0.01, and * indicates p < 0.05). Results are reported as the mean ± SEM. Student's paired and unpaired t tests were used to assess within-group and between-group differences, respectively. Typically, the magnitude of long-term plasticity was determined by comparing baseline-averaged responses before induction with responses 30–40 min after induction.
Reagents.
Reagents were bath applied following dilution into ACSF from stock solutions stored at −20°C prepared in water or DMSO, depending on the manufacturer's recommendation. The final DMSO concentration was <0.01% total volume. Reagents N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251), NBQX, d-APV, 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY 367385), and (S)-3,5-dihydroxyphenylglycine (DHPG) were purchased from Abcam Biochemicals. (2-Hydroxyethyl) trimethylammonium chloride carbamate [carbachol (CCh)], BAPTA, hyoscyamine tropine tropate endo-(±)-α-(hydroxymethyl)benzeneacetic acid 8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester (atropine), (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate (WIN 55212-2), N6-[5-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4-yl]-1-oxopentyl]-l-lysine (biocytin), 8-cyclopentyl-1,3-dipropylxanthine (DCPCX), 1,1-dimethyl-4-diphenylacetoxypiperidinium iodide (4-DAMP), 5,11-dihydro-11-[(4-methyl-1-piperazinyl)acetyl]-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one dihydrochloride (pirenzepine), and 3,6a,11,14-tetrahydro-9-methoxy-2-methyl-(12H)-isoquino[1,2-b]pyrrolo[3,2-f][1,3]benzoxazine-1-carboxylic acid, ethyl ester (PD 102807) were purchased from Tocris Bioscience. N-arachidonoyl maleimide (NAM) was purchased from Cayman. Picrotoxin, 1,4-dihydro-2,6-dimethyl-4-(2-nitrophenyl)-3,5-pyridinedicarboxylic acid dimethyl ester (nifedipine), gallamine triethiodide (gallamine), and salts for making ACSF and internal solutions were purchased from Sigma-Aldrich. 4-[Bis(1,3-benzodioxol-5-yl)hydroxymethyl]-1-piperidinecarboxylic acid 4-nitrophenyl ester (JZL-184) was a gift from Dr. Sachin Patel (Vanderbilt University, Nashville, TN). NBQX, dl-APV, and WIN 55212-2 also were obtained from the Chemical Synthesis and Drug Supply Program of the National Institute of Mental Health.
Results
Multiple postsynaptic depolarizations cause iLTD
Endocannabinoids are mobilized by brief postsynaptic depolarizations (Wilson and Nicoll, 2001), and iLTD induction is thought to require minutes-long CB1R activation (Chevaleyre and Castillo, 2003). We explored whether multiple depolarizations of CA1 pyramidal neurons per se, by controlling the duration of eCB release and CB1R activation over the course of minutes, were sufficient to induce iLTD at inhibitory synapses onto CA1 pyramidal neurons. To this end, IPSCs were elicited by extracellular stimulation in stratum radiatum while voltage-clamping CA1 pyramidal neurons (see Materials and Methods). This form of stimulation likely activates mostly dendritic inhibitory inputs (Chevaleyre and Castillo, 2003). After obtaining a stable baseline, multiple episodes of DSI (mDSI) were delivered to CA1 pyramidal cells through the patch pipette (see Materials and Methods). We found that mDSI delivered over the course of 2 min only triggered a transient depression of IPSCs, whereas 10 min mDSI triggered robust iLTD (Fig. 1A). On average, the magnitude of iLTD was proportional to the duration of the mDSI protocol (Fig. 1B; 2 min mDSI: 99.4 ± 3.6% of baseline, n = 6, p > 0.25; 5 min mDSI: 87.9 ± 1.2% of baseline, n = 5, p < 0.001; 10 min mDSI: 71.8 ± 2.9% of baseline, n = 9, p < 0.001). Consistent with a requirement for eCB signaling, the selective CB1R antagonist AM251 (2 μm) abolished mDSI-induced iLTD (Fig. 1C; control: 71.4 ± 0.7% of baseline, n = 6, p < 0.001; AM251: 98.9 ± 1.3% of baseline, n = 6, p > 0.5; control vs AM251, p < 0.001). In addition, mDSI-iLTD was associated with an increase in the paired-pulse ratio (PPR), a measure that is inversely proportional to changes in neurotransmitter release probability. The mDSI-driven increase in PPR was abolished in the presence of AM251 (Fig. 1D; control: 1.28 ± 0.07-fold change, n = 6, p < 0.001; AM251: 1.08 ± 0.03-fold change, n = 6, p < 0.05; control vs AM251, p < 0.02). To determine whether heterosynaptically induced iLTD (Chevaleyre and Castillo, 2003) and mDSI-induced iLTD occlude each other and share a common target, we first elicited mDSI and then repetitively activated presynaptic fibers. This manipulation failed to evoke further depression (101.9 ± 1.6% of baseline, n = 5, p > 0.5, data not shown). Collectively, these findings indicate that multiple CA1 pyramidal cell depolarizations, over the course of a few minutes, are sufficient to trigger eCB-mediated presynaptic iLTD.

Multiple postsynaptic depolarizations cause eCB-mediated iLTD. A, Representative experiment. A 2 min mDSI (black bar) only produced short-term suppression, whereas 10 min mDSI (white bar) triggered iLTD. IPSC traces, which correspond to the numbers in the time course plot below (for this and all remaining figures), are shown above for each condition. The mDSI induction protocol is also schematized. B, Group data showing 2, 5, and 10 min mDSI leading to progressively more depression of IPSCs. Schematic highlights basic experimental configuration in which IPSCs are recorded somatically from CA1 pyramidal cells (black) in response to extracellular stimulation of presynaptic interneuronal fibers (gray). Synaptic plasticity was triggered by postsynaptic activity. C, iLTD induced by 10 min mDSI was abolished when CB1Rs were blocked with 2 μm AM251. D, A 10 min mDSI caused a long-lasting increase in the PPR that was not observed in the presence of AM251 (same cells from C). Data are presented as the mean ± SEM, and “n” refers to the number of cells for this and all remaining figures.
Postsynaptic theta-burst firing converts short-term into long-term depression of inhibition
Although the mDSI protocol allowed us to tightly control eCB production in time and is thus a useful experimental tool, step depolarizations are rather nonphysiological manipulations. We therefore sought to determine whether postsynaptic burst firing can elicit eCB-mediated long-term synaptic plasticity. In vivo, during exploratory behaviors and sleep, CA1 pyramidal cells can repetitively discharge, over the course of minutes, high-frequency bursts of action potentials in organized theta rhythms (O'Keefe and Dostrovsky, 1971; Ranck, 1973; O'Keefe, 1976; Buzsáki, 2002; Harvey et al., 2009; Epsztein et al., 2011). To mimic this activity in vitro, CA1 pyramidal cells were directly activated by delivering a TBF protocol through the patch pipette (see Materials and Methods). Action potentials can back-propagate into apical dendrites of CA1 pyramidal cells, thereby elevating both somatic and dendritic Ca2+ levels (Callaway and Ross, 1995; Spruston et al., 1995) that are sufficient for eCB mobilization (Morishita and Alger, 2001; Wang and Zucker, 2001; Brenowitz et al., 2006; Rancz and Häusser, 2006). Consistent with these observations, just one TBF episode transiently (<30 s) suppressed IPSCs evoked by extracellular stimulation in stratum radiatum (Fig. 2A; 83.3 ± 1.6% of baseline, n = 10, p < 0.001). Similar to DSI, this plasticity was prevented by including BAPTA (20 mm) in the recording pipette (98.7 ± 1.1% of baseline, n = 4, p > 0.5), was accompanied by a transient increase in PPR (122.5 ± 4.5% of baseline, n = 8, p < 0.001), and was blocked by 2 μm AM251 (Fig. 2A; 100.6 ± 0.6% of baseline, n = 5, p > 0.5). These results indicate that postsynaptic TBF elicits short-term eCB-mediated inhibitory synaptic plasticity, and we refer to this phenomenon as burst firing-induced suppression of inhibition (BSI).

Postsynaptic TBF triggers LTD at inhibitory synapses. A, One bout of postsynaptic burst firing transiently suppressed IPSCs (BSI), which was not observed when CB1Rs were blocked with 2 μm AM251. The schematic illustrates one TBF (1×) episode. Inset, 50 Hz burst of action potentials elicited by somatic current injection. For this and all remaining figures, arrow marks the time at which TBF was initiated. B, Postsynaptic TBF (60×) elicited iLTD, which required intact CB1R signaling (2 μm AM251). The schematic illustrates a TBF-iLTD induction protocol consisting of 60 TBF episodes, delivered once every 5 s, over the course of 5 min. C, Intracellular loading of 20 mm BAPTA blocked TBF-iLTD. D, l-type voltage-gated Ca2+ channels (10 μm nifedipine) were necessary for TBF-iLTD induction, whereas I-mGluRs were not (4 μm MPEP/100 μm LY367385). Bar indicates drug application. E, Increasing the interval between TBF episodes from 5 to 15 s, while keeping the total number of episodes (60) the same, decreased the magnitude of TBF-iLTD. F, A submaximal TBF protocol (36 episodes, every 5 s, 3 min total) failed to elicit iLTD under control conditions but elicited robust iLTD when the eCB degradative enzyme MGL was inhibited with JZL 184 (0.2 μm). G, TBF-iLTD was observed at 37°C. Note the apparent high-temperature-induced rundown in IPSCs. H, Using a K-gluconate internal recording solution, TBF (60×) elicited iLTD, which was blocked in the presence of AM251 (2 μm).
Like mDSI, multiple episodes of postsynaptic TBF might convert short-term into long-term eCB-dependent plasticity. Simply increasing the number of TBF episodes (60 total, delivered every 5 s) to span 5 min caused iLTD (Fig. 2B; control, 83.6 ± 1.3% of baseline, n = 10, p < 0.001). This form of plasticity, which we term TBF-iLTD, was accompanied by an enhanced PPR (117.0 ± 3.0% of baseline, n = 10, p < 0.001) and was blocked by 2 μm AM251 (Fig. 2B; 100.1 ± 0.3% of baseline, n = 8, p > 0.5; control vs AM251, p < 0.001). In addition, TBF-iLTD was not observed when CA1 pyramidal cells were loaded with 20 mm BAPTA (Fig. 2C; control: 79.1 ± 5.7% of baseline, n = 4, p < 0.001; BAPTA: 106.4 ± 4.1% of baseline, n = 5, p > 0.25; control vs BAPTA, p < 0.001). Similarly, blocking l-type voltage-gated Ca2+ channels by adding 10 μm nifedipine to the bath prevented TBF-iLTD (Fig. 2D; 99.2 ± 2.0% of baseline, n = 6, p > 0.5). Unlike heterosynaptically triggered iLTD, whose induction relies on presynaptic glutamate release and postsynaptic activation of I-mGluRs for eCB production (Chevaleyre and Castillo, 2003), TBF-iLTD was still induced in the presence of the I-mGluR antagonists MPEP (4 μm) and LY 367385 (100 μm), which antagonize mGluR5 and mGluR1, respectively (Fig. 2D; 77.7 ± 3.3% of baseline, n = 8, p < 0.001). To determine whether heterosynaptically induced iLTD (Chevaleyre and Castillo, 2003) and TBF-induced iLTD occlude one another, we elicited heterosynaptic iLTD (see Materials and Methods; 61.7 ± 4.0% of baseline, n = 5, p < 0.001), renormalized these values to baseline, and then delivered the TBF-iLTD protocol. TBF-iLTD did not elicit further depression (95.7 ± 6.5% of baseline, n = 5, p > 0.3; data not shown). Together, these results indicate that postsynaptic TBF alone is sufficient to elicit long-term presynaptic depression of inhibition (i.e., TBF-iLTD) in an eCB-dependent manner.
BSI is a transient phenomenon, lasting ∼30 s (Fig. 2A). This time course likely reflects the duration of CB1R activation given that CB1R desensitization has not been observed within this time window (Jin et al., 1999; Kouznetsova et al., 2002). Thus, increasing the interburst interval between TBF episodes, which presumably allows eCB concentration to partially fall between stimulations, could decrease the magnitude of iLTD. IPSCs were again evoked by extracellular stimulation in stratum radiatum. While TBF every 5 s triggered iLTD (Fig. 2E; 81.4 ± 2.7% of baseline, n = 6, p < 0.001), the same number of episodes (60 total) applied every 15 s elicited weaker iLTD (Fig. 2E; 95.9 ± 1.4% of baseline, n = 7, p < 0.01; 5 vs 15 s, p < 0.01). This observation could help to explain, at least in part, why a previous report (Edwards et al., 2006) did not evoke iLTD with a protocol consisting of 2 s postsynaptic depolarizations delivered every 12 s for 10 min. Indeed, this stimulation protocol did not trigger iLTD in our study either (99.5 ± 1.5% of baseline, n = 3, p > 0.5). These results imply that to observe iLTD, eCB release intervals must be relatively short to allow increased or more continuous CB1R activation, and they also predict that extending the lifetime of eCB signaling would convert a submaximal TBF induction into iLTD. It is known that the presynaptically expressed enzyme monoacylglycerol lipase (MGL; Dinh et al., 2002; Blankman et al., 2007), which catabolizes the eCB 2-arachidonoylglycerol (2-AG), can control the duration and magnitude of eCB-mediated short-term and long-term plasticity (Makara et al., 2005; Szabo et al., 2006; Hashimotodani et al., 2007a; Pan et al., 2009, 2011). We found that a submaximal TBF protocol (e.g., 36 episodes, delivered every 5 s over 3 min) failed to elicit long-term plasticity under control conditions (Fig. 2F; 99.1 ± 1.9% of baseline, n = 5, p > 0.5), whereas iLTD was unmasked when slices were preincubated in (>30 min, 0.2 μm) and perfused with the potent and selective MGL inhibitor JZL 184 (Long et al., 2009; Fig. 2F; 77.0 ± 3.5% of baseline, n = 6, p < 0.001; control vs JZL, p < 0.001). Notably, TBF-iLTD was also detected at physiological temperatures (Fig. 2G; control: 77.6 ± 2.4% of baseline, n = 7, p < 0.001; AM251: 99.4 ± 2.0% of baseline, n = 8, p > 0.5; control vs AM251, p < 0.001; measured 20–25 min after induction due to apparent rundown) and with a K-gluconate internal solution (Fig. 2H; control: 78.0 ± 1.7% of baseline, n = 9, p < 0.001; AM251: 96.0 ± 3.4% of baseline, n = 8, p > 0.05; control vs AM251, p < 0.001). Together, these observations support the notion that iLTD induction requires eCB release for several minutes. Moreover, CA1 pyramidal cell TBF uncovers a possible physiologically relevant paradigm to evoke iLTD.
Postsynaptic TBF does not lead to any detectable changes in long-term plasticity at Schaffer collateral excitatory synapses
Glutamatergic Sch–CA1 synapses express functional CB1Rs (Domenici et al., 2006; Katona et al., 2006; Kawamura et al., 2006; Takahashi and Castillo, 2006), and eCB-dependent LTD has been observed at this synapse (Yasuda et al., 2008; Xu et al., 2010; Izumi and Zorumski, 2012; Péterfi et al., 2012). To investigate whether TBF might also trigger long-term eCB-mediated plasticity at Sch–CA1 synapses, we monitored EPSCs from CA1 pyramidal cells while blocking inhibitory synaptic transmission (see Materials and Methods). Postsynaptic TBF, which reliably released eCBs to trigger iLTD (Fig. 2B), failed to elicit long-term plasticity at Sch–CA1 synapses in juvenile (P16–P26) rat slices (Fig. 3A,B; control, 107.8 ± 9.2% of baseline, n = 10, p > 0.3). A number of additional manipulations were performed to maximize the likelihood of detecting TBF-mediated LTD at this synapse. First, CB1Rs reportedly mediate stronger effects at hippocampal glutamatergic synapses in young animals (<P13) (Kang-Park et al., 2007; Yasuda et al., 2008; but see Zhu and Lovinger, 2010; Caiati et al., 2012). However, TBF did not trigger any Sch–CA1 LTD in young animals (Fig. 3B; P7–P10, 101.8 ± 6.7% of baseline, n = 8, p > 0.5). Second, activation of I-mGluRs or mAChRs facilitates eCB release (Maejima et al., 2001, 2005; Kim et al., 2002; Hashimotodani et al., 2005). TBF paired with 3 μm DHPG, a specific group I-mGluR agonist, or 0.5 μm CCh, a potent mAChR agonist, both failed to produce long-term plasticity at excitatory synapses (Fig. 3B; DHPG: 98.8 ± 5.3% of baseline, n = 6, p > 0.5; CCh: 103.1 ± 5.6% of baseline, n = 3, p > 0.5). Third, concomitant presynaptic activity and CB1R activation are known to be required for the induction of eCB-LTD (Singla et al., 2007; Heifets et al., 2008; Adermark et al., 2009). However, pairing presynaptic activity (10 pulses at 5 Hz every 5 s for 5 min; see Heifets et al., 2008) with postsynaptic TBF did not uncover eCB-LTD (Fig. 3B; 98.2 ± 7.5% of baseline, n = 7, p > 0.5). Fourth, while CB1R signaling at Sch collaterals may be affected by tonic adenosine acting at adenosine type 1 receptors (Hoffman et al., 2010) or indirectly by gliotransmitters acting on presynaptic mGluR1 (Navarrete and Araque, 2010), we found that TBF, in combination with the selective A1 receptor antagonist DPCPX (0.2 μm) or the mGluR1 antagonist LY 367385 (100 μm), unsuccessfully evoked long-term plasticity (Fig. 3B; DPCPX: 108.8 ± 5.0% of baseline n = 5, p > 0.1; LY 367385: 95.1 ± 5.2% of baseline, n = 5, p > 0.3). Fifth, we reasoned that inhibiting MGL might facilitate the spread of eCB signaling to activate presynaptic CB1Rs. In the presence of JZL 184 (0.2 μm), TBF did not elicit LTD at Sch–CA1 synapses (Fig. 3B; 101.3 ± 5.1% of baseline, n = 5, p < 0.5). Sixth, it was shown that DHPG can evoke a CB1R-sensitive component of LTD at Sch–CA1 synapses (Xu et al., 2010; Péterfi et al., 2012). While we and others have not yet observed this phenomenon (Chevaleyre and Castillo, 2003; Rouach and Nicoll, 2003; Izumi and Zorumski, 2012), we re-examined whether Sch–CA1 DHPG-induced LTD involves eCB signaling. Extracellular field recordings were used to monitor EPSPs in stratum radiatum of CA1 (see Materials and Methods). After a 10 min baseline period, DPHG (50 μm, 10 min) was bath applied in the absence and presence of AM251 (2 μm). There was no significant difference in DHPG-induced LTD at Sch–CA1 synapses between control and AM251 conditions (control: 63.9 ± 2.4% of baseline, n = 3, p < 0.01; AM251: 57.0 ± 6.9% of baseline, n = 3, p < 0.001; control vs AM251, p > 0.05; data not shown). Seventh, a spike-timing-dependent plasticity (STDP) induction protocol was recently reported to elicit robust eCB-LTD at these synapses in mouse hippocampus (Péterfi et al., 2012). However, such a protocol in our hands (six blocks of 100 pairings, 10 s interblock interval, pairings at 5 or 10 Hz, 10 ms post/pre delay) did not elicit eCB-LTD in rat (Fig. 3B; 99.5 ± 7.0% of baseline, n = 10, six cells with 5 Hz induction and four cells with 10 Hz induction were pooled because they were not statistically different, p > 0.5) or mice (Fig. 3B; 114.8 ± 14.2% of baseline, n = 4 cells with 10 Hz induction, p > 0.25). Finally, we tested in mouse slices the 10 Hz STDP protocol at 32°C in the presence of 0.2 μm JZL 184 (d-APV was omitted from the bath for these experiments). Still, no long-lasting plasticity was detected at Sch–CA1 synapses (Fig. 3B; 110.5 ± 9.2% of baseline, n = 6, p > 0.3). Collectively, our results indicate that in hippocampal CA1, TBF triggers LTD exclusively at eCB-sensitive inhibitory inputs, completely sparing excitatory synapses.

Postsynaptic TBF does not trigger long-term plasticity at excitatory synapses. A, TBF does not elicit long-term plasticity at Schaffer collateral excitatory synapses in juvenile animals aged P16–P26. Traces taken from a single representative experiment are shown at right. B, Long-term plasticity was not observed at CA1 excitatory synapses under various recording conditions. TBF in P16–P26 rats (data from A replotted); TBF in P7–P10 rats; TBF + I-mGluR agonist DHPG (3 μm); TBF + mAChR agonist CCh (0.5 μm); TBF + presynaptic activity (10 pulses at 5 Hz every 5 s for 5 min); TBF + A1R antagonist DPCPX (0.2 μm); TBF + mGluR1 antagonist LY 367385 (100 μm); TBF + MGL inhibitor JZL 184 (0.2 μm); all of the former experiments were performed in rat slices. STDP protocol (six blocks of 100 pairings, 10 s interblock interval, pairings at 5 or 10 Hz, 10 ms post/pre delay) in rat and mice; and STDP protocol + JZL 184 (0.2 μm) at 32°C in mouse. Numbers in parentheses indicate the number of cells. Data were expressed as the percentage change from baseline (dotted line) for each respective condition. n.s., Not significant.
TBF-iLTD targets both somatic and dendritic inhibitory inputs
Heterosynaptically induced iLTD in the CA1 area is a local phenomenon driven by glutamatergic inputs that impinge on the dendritic tree of pyramidal neurons (Chevaleyre and Castillo, 2003). Only nearby dendritic inhibitory synapses that are close to Sch inputs (<10 μm) express heterosynaptic eCB-LTD (Chevaleyre and Castillo, 2004). Given that TBF would be expected to mobilize eCBs throughout the somatodendritic compartment, we hypothesized that, unlike synaptically induced iLTD, TBF-iLTD targets both dendritic and somatic inhibitory inputs. To test this possibility, we performed two sets of experiments. We first monitored pharmacologically isolated IPSCs from a given CA1 pyramidal neuron by delivering focal stimulation in stratum pyramidale and stratum radiatum. We found robust TBF-iLTD at both sets of inputs (stratum pyramidale: 74.9 ± 2.5% of baseline, n = 5, p < 0.001; stratum radiatum: 76.7 ± 5.8% of baseline, n = 5, p < 0.001). Although useful, extracellular stimulation may not clearly distinguish between somatic and dendritic inputs. We therefore performed cell-paired recordings between single interneurons and CA1 pyramidal neurons (in the absence of blockers for excitatory and inhibitory synaptic transmission) followed by post hoc morphological reconstructions of the interneuron (Fig. 4A). In the hippocampus, CB1Rs are highly expressed on the axon terminals of somatic [i.e., basket cells (BCs)] and dendritic [i.e., Schaffer-collateral associated (SCA)] projecting regular-spiking interneurons (Katona et al., 1999; Tsou et al., 1999; Cope et al., 2002; Pawelzik et al., 2002; Nyíri et al., 2005; Glickfeld and Scanziani, 2006). Interneurons were first characterized electrophysiologically for their spiking behavior (i.e., fast spiking vs regular spiking), and then cell pairs were tested for DSI to assess their eCB sensitivity. Only DSI-sensitive connections (i.e., eCB-sensitive connections) were analyzed (see Materials and Methods).

Paired recordings between CA1 interneurons and pyramidal cells revealed that TBF-iLTD is equally expressed at somatic and dendritic inhibitory inputs. A, Example post hoc morphological reconstruction of a somatically projecting BC interneuron. Dendrites are highlighted in black, and axons in gray. Inset illustrates regular spiking action potentials distinctive of DSI-sensitive (i.e., eCB-sensitive) interneurons. SLM, Stratum lacunosum moleculare; SR, stratum radiatum; SP, stratum pyramidale. B, Similar to A, but for a dendritically projecting SCA interneuron. C, DSI in somatically projecting interneurons. Representative time course plot (left) and summary data (right). Averaged unitary IPSC (uIPSC) responses and failures (i.e., efficacy) from the time points indicated are shown on top, along with temporally aligned presynaptic action potentials that elicited the uIPSCs. Note the increased failure rate (decreased release probability) in response to DSI elicited in the pyramidal cell. D, Similar to C, but for dendritically projecting interneurons. Note different y-axis scale in C and D. E, Representative experiment depicting uIPSC time course plotted for a different somatically projecting interneuron before and after iLTD induction. Individual (gray) and averaged (black) uIPSC traces are shown on top (along with temporally aligned presynaptic action potentials) for responses (potency) and failures. Primed numbers indicate averaged uIPSCs (efficacy). F, Similar to E, but for a dendritically projecting interneuron. Note different y-axis scale in E and F. Representative experiments shown in C–F from different paired recordings. G, Group data for somatically projecting interneurons showing success rate, efficacy, and potency before and after iLTD induction. Success rate before, 0.70 ± 0.09 vs after: 0.46 ± 0.08, p < 0.01; efficacy before, −25.5 ± 4.7 pA vs after, −12.4 ± 1.6 pA, p < 0.035; potency before, −32.8 ± 4.9 pA vs after, −25.3 ± 3.6 pA, p > 0.1. Note that four cells included in the DSI analysis were discarded from the iLTD analysis (see Materials and Methods). H, Group data for dendritically projecting SCA interneurons. Success rate before, 0.47 ± 0.06 vs after, 0.27 ± 0.06, p < 0.01; efficacy before, −6.7 ± 1.8 pA vs after, −3.0 ± 0.9 pA, p < 0.035; potency before: −11.9 ± 2.7 pA vs after, −9.3 ± 1.5 pA, p > 0.2. n.s., Not significant. **p < 0.01; *p < 0.05.
Similar to a recent report (Lee et al., 2010), DSI was more pronounced in BCs versus SCA interneurons (Fig. 4A–D; somatic: 11.2 ± 4.1% of baseline, n = 9, p < 0.001; dendritic: 48.1 ± 3.6% of baseline, n = 9, p < 0.001; somatic vs dendritic, p < 0.001). In contrast, the magnitude of the TBF-iLTD of uIPSCs was virtually identical between BC and SCA interneuron synapses as indicated by a comparable decrease in success rate and synaptic efficacy (Fig. 4E–H). Despite the fact that the starting success rate, on average, was different between somatic and dendritic projecting interneurons (Fig. 4G,H; somatic, 0.70 ± 0.09 vs dendritic, 0.47 ± 0.06; p < 0.05), the success rate of synaptic responses was similarly reduced between compartments in response to postsynaptic TBF (Fig. 4G,H; somatic, 65.0 ± 5.0% of baseline vs dendritic, 54.0 ± 10.1% of baseline; p > 0.25). This observation suggests that inhibitory synaptic release probability was equally depressed along the somatodendritic axis by postsynaptic TBF. In addition, synaptic efficacy, which is the mean uIPSC amplitude of all events (i.e., successes and failures), was comparably depressed (Fig. 4G,H; somatic, 52.7 ± 6.8% of baseline vs dendritic, 44.7 ± 7.9% of baseline; p > 0.5). Even though synaptic potency (i.e., mean uIPSC amplitude of successful events only) was initially different between groups (Fig. 4G,H; somatic, −32.8 ± 4.9 pA vs dendritic, −11.9 ± 2.7 pA; p < 0.01), we found that the effect of TBF did not cause any differences in synaptic potency in either compartment (Fig. 4G,H; somatic, 81.7 ± 10.6% of baseline vs dendritic, 87.9 ± 10.6% of baseline; p > 0.5), making it unlikely that postsynaptic changes in GABAA receptor (GABAAR) number and/or function underlie TBF-iLTD. Altogether, these results indicate that CB1R-expressing inhibitory synapses are equally sensitive to eCB signaling along the somatodendritic axis, at least in response to sustained activity. Moreover, TBF reveals how endogenous CA1 pyramidal cell activity can effectively recruit eCB signaling to evoke somatic and dendritic iLTD.
Endocannabinoid signaling is restricted to single CA1 pyramidal cells
The distance over which eCBs can diffuse is still unresolved (Alger, 2012). Recordings from two neighboring CA1 pyramidal cells have shown that DSI triggered in one neuron can elicit DSI in a neighboring, non-depolarized cell, provided their somas are within ∼20 μm (Wilson and Nicoll, 2001). Similarly, there is evidence for eCB-mediated DSE spread between neighboring CA1 pyramidal neurons, and, indirectly, short-term potentiation via astrocytes (Navarrete and Araque, 2010). Spread of eCB signaling across CA1 pyramidal neurons has also been reported for the induction of heterosynaptic eCB-mediated Sch–CA1 LTD in neonatal rats (Yasuda et al., 2008). Thus, the multiple bouts of postsynaptic activity required for the induction of TBF-iLTD suggests that eCBs could build up and spread from active to neighboring naive neurons. To directly test this possibility, we performed dual recordings between closely spaced pyramidal neurons. Inhibitory fibers in stratum pyramidale were stimulated, and excitatory synaptic transmission was blocked (see Materials and Methods). After obtaining stable baselines in both cells, TBF-iLTD was initiated in one of these cells (Fig. 5A). As expected, 60× TBF (every 5 s for 5 min) elicited robust iLTD in the activated cell (71.7 ± 4.1% of baseline, n = 9, p < 0.001). Surprisingly, the naive neighbor showed no long-lasting plasticity (Fig. 5B; 99.0 ± 1.8% of baseline, n = 9, p > 0.5; control vs naive, p < 0.001). This was true for each distance probed (range, 5–30 μm), suggesting very limited eCB diffusion following TBF of a single CA1 neuron.

Dual recordings demonstrate that TBF-iLTD is restricted to only a single, active pyramidal cell. A, Representative experiment from neighboring CA1 pyramidal cells ∼5 μm apart as assessed by the recording interpipette distance. TBF 60× (every 5 s, 5 min) elicited iLTD in the activated cell, but not in its neighbor. Differential interference contrast image highlights proximity of the recording pipettes. Image contrast was adjusted. SO, stratum oriens. B, Summary data for iLTD magnitude observed in the nonactive naive cell as a function of interpipette distance from the active test cell. Horizontal line highlights averaged magnitude of plasticity in the naive cell, which showed virtually no difference relative to baseline. C, Representative control experiment showing that both pyramidal cells respond to eCB signaling as indicated by DSI sensitivity. Note that DSI triggered in the test cell did not affect inhibitory transmission in the naive cell. Same cells from A. D, Summary data for DSI magnitude observed in the nonactive naive cell as a function of interpipette distance from the active test cell. Under control conditions, in the presence of 5 μm CCh (mAChR agonist), or at 34°C, DSI was not observed in the naive cell. Horizontal line highlights the averaged magnitude of plasticity in control naive cells. As a positive control, 0.2 μm JZL 184 and 10 μm NAM (MGL inhibitors) were used to unmask eCB cross talk at close interpipette distances. The best-fit line corresponds to data collected in JZL 184 and NAM, suggesting eCBs diffused <13 μm, even when MGL was inhibited.
The cell-limited nature of TBF-iLTD seems at odds with previous studies showing spread of eCBs between CA1 pyramidal neurons (Wilson and Nicoll, 2001; Yasuda et al., 2008; Navarrete and Araque, 2010). In all our experiments, both test and naive pyramidal neurons were assayed for DSI sensitivity before inducing TBF-iLTD (Fig. 5C; average DSI magnitude, 65.4 ± 2.1% of baseline, n = 18, p < 0.001). Unexpectedly, DSI elicited in one neuron had no effect on naive neighboring neurons (Fig. 5D; 100.6 ± 1.1% of baseline, n = 18, p > 0.5). Moreover, adding CCh (5 μm) to the extracellular solution, a manipulation expected to facilitate eCB mobilization and hippocampal DSI (Kim et al., 2002), did not unmask DSI in naive neurons (Fig. 5D; naive 100.1 ± 1.6% of baseline, n = 8, p > 0.5). Recordings at 34°C, a condition that affects eCB production, diffusion, and uptake, also did not uncover any cross talk (Fig. 5D; 99.9 ± 2.2% of baseline, n = 14, p > 0.5). Finally, we tested whether interfering with the catabolism of 2-AG, the eCB that mediates DSI (Gao et al., 2010; Tanimura et al., 2010; Yoshino et al., 2011), could unmask DSI in naive neurons. To this end, we blocked the enzyme MGL, which regulates ∼85% of all 2-AG hydrolysis in the brain (Blankman et al., 2007). Indeed, it was only in the presence of JZL 184 (0.2 μm) that some DSI could be observed in nearby naive neurons (Fig. 5D). This finding was reproduced using a structurally unrelated, but equally potent inhibitor of MGL (NAM, 10 μm; Saario et al., 2005; Blankman et al., 2007; Fig. 5D; near: <15 μm, 92.1 ± 2.0% of baseline, n = 16, p < 0.002; far: >15 μm, 100.8 ± 1.1% of baseline, n = 12, p > 0.5; nine dual recordings with JZL 184 and five dual recordings with NAM were pooled because they were not statistically different, p > 0.5). These results indicate that, under normal signaling conditions in the hippocampus, 2-AG does not spread between neighboring neurons, and that the effects of both DSI and TBF-iLTD are restricted by MGL to the activated CA1 pyramidal cell.
Submaximal TBF paired with submaximal endogenous Gq/11 protein-coupled receptor activation reveals associative TBF-iLTD
eCB mobilization can result from two independent but synergistic pathways: postsynaptic Ca2+ influx via voltage-gated calcium channels and activation of Gq/11 protein-coupled receptors, such as I-mGluRs and mAChRs (Hashimotodani et al., 2007b). We therefore explored whether activation of these receptors could facilitate TBF-iLTD. To investigate the role of I-mGluRs, IPSCs were elicited by stimulating stratum radiatum. In the presence of a low dose of DHPG (3 μm), the magnitude of BSI was significantly enhanced (Fig. 6A; control: 88.6 ± 1.4% of baseline, n = 5, p < 0.001; DHPG: 75.3 ± 3.8% of baseline, n = 5, p < 0.001; control vs DHPG, p < 0.001). Likewise, DHPG augmented TBF-iLTD triggered by a submaximal TBF induction protocol consisting of 60 episodes delivered every 15 s (Fig. 6B; control: 95.9 ± 1.4% of baseline, n = 7, p < 0.05; DHPG: 85.2 ± 1.8% of baseline, n = 5, p < 0.001; control vs DHPG, p < 0.001). These findings indicate that exogenous activation of I-mGluRs enhanced the effect of TBF on iLTD.

Pairing submaximal postsynaptic TBF with submaximal endogenous I-mGluR activation revealed an associative and synergistic component to iLTD. A, BSI (elicited with 1× TBF) triggered short-term suppression of inhibition, the magnitude of which was enhanced by 3 μm DHPG (I-mGluR agonist). B, Submaximal TBF episodes delivered once every 15 s (60 total) in the absence and presence of 3 μm DHPG. C, Pairing submaximal synaptic stimulation in stratum radiatum (three stimuli, 10 Hz) followed 0.5 s later by submaximal postsynaptic TBF (30 episodes total, every 15 s) unmasked associative iLTD that was blocked by 4 μm MPEP/100 μm LY367385 (mGluR5 and mGluR1 antagonists, respectively). D, Increasing the delay between submaximal synaptic stimulation and submaximal TBF to 7.5 s limited associative iLTD compared with submaximal TBF alone. E, Two-pathway experiment highlighting input specificity of associative iLTD when pairing submaximal presynaptic stimulation of only one of the inputs with submaximal postsynaptic TBF (30 episodes total, every 15 s). F, Neither submaximal presynaptic stimulation nor submaximal postsynaptic TBF had a long-lasting impact on inhibitory synaptic transmission.
We next investigated whether a synergy could be achieved with endogenous activation of I-mGluRs by activating Schaffer collaterals with brief bursts of stimuli (three stimuli, 10 Hz). Repetitive pairing of presynaptic bursts 0.5 s before each submaximal TBF episode (30 episodes, every 15 s) triggered robust associative iLTD. Importantly, this synergy was not observed in the presence of 4 μm MPEP and 100 μm LY 367385 (Fig. 6C; control: 83.1 ± 1.5% of baseline, n = 11, p < 0.001; MPEP/LY 367385: 96.1 ± 1.3% of baseline, n = 4, p > 0.05; control vs MPEP/LY 367385, p < 0.001). Presynaptic stimulation followed 7.5 s later by each submaximal TBF episode did not facilitate iLTD induction (Fig. 6D; control: 92.7 ± 1.8% of baseline, n = 4 vs TBF alone: 96.2 ± 2.3% of baseline, n = 3; p > 0.25), indicating that some temporal coincidence was required for this form of associative plasticity.
Input specificity of associative iLTD was tested by monitoring two independent inputs impinging on the same pyramidal cell. Two stimulating pipettes were placed in stratum radiatum 50 μm apart along the main axis of the apical dendrite, and a presynaptic train (three stimuli, 10 Hz) was delivered to one input 0.5 s before each postsynaptic submaximal TBF episode. Associative iLTD was only observed when both presynaptic and postsynaptic activities were paired (Fig. 6E; paired input: 82.5 ± 1.9% of baseline, n = 8, p < 0.001; unpaired input: 98.1 ± 3.3% of baseline, n = 8, p > 0.25; paired vs unpaired input, p < 0.001). Neither presynaptic stimulation nor submaximal TBF, when applied independently, induced long-term synaptic plasticity (Fig. 6F; synaptic stimulation: 97.6 ± 1.9% of baseline, n = 4, p > 0.25; submaximal TBF: 98.7 ± 1.6% of baseline, n = 4, p > 0.25). These observations indicate that two submaximal events, namely, presynaptic glutamatergic activity and postsynaptic TBF, can synergistically interact within a relatively narrow time window to induce associative iLTD that is localized to specific dendritic inhibitory inputs.
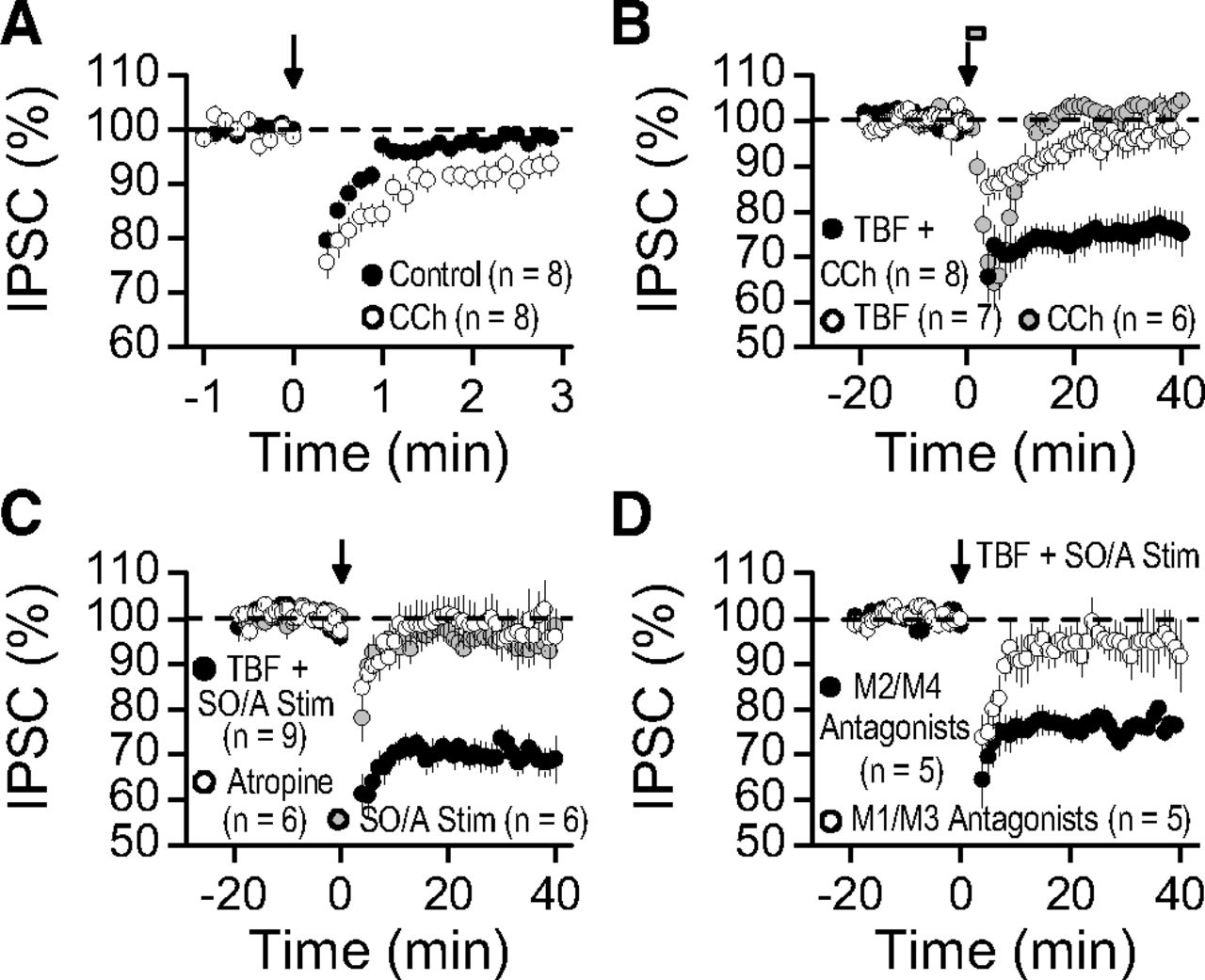
We also examined whether activation of mAChRs could facilitate BSI and the induction of TBF-iLTD. In these experiments, we activated somatic inhibitory inputs. The duration of BSI was significantly increased by 0.5 μm CCh (Fig. 7A; control recovery time constant tau, 31.1 ± 4.0 s vs CCh tau, 68.1 ± 13.9 s; n = 8, p < 0.02). Moreover, pairing a brief application of CCh (0.5 μm, 3 min) with a submaximal TBF protocol (36×, every 5 s for 3 min) induced TBF-iLTD (Fig. 7B; 76.2 ± 4.0%, n = 8, p < 0.001). Neither CCh application alone (Fig. 7B; 102.2 ± 2.5% of baseline, n = 6, p > 0.5) nor submaximal TBF alone (Fig. 7B; 96.3 ± 3.0% of baseline, n = 7, p > 0.25) triggered long-lasting changes in inhibitory synaptic transmission. To investigate whether release of endogenous ACh might synergize with postsynaptic TBF to cause iLTD, we stimulated stratum oriens/stratum alveus (see Materials and Methods) to activate cholinergic fibers (Cole and Nicoll, 1983). This stimulation alone did not induce long-term inhibitory synaptic plasticity (Fig. 7C; 94.1 ± 4.4% of baseline, n = 6, p > 0.2). In contrast, pairing submaximal stratum oriens/stratum alveus stimulation with submaximal TBF elicited robust iLTD (Fig. 7C; 66.7 ± 2.6% of baseline, n = 9, p < 0.001). This plasticity was blocked by 1 μm of the mAChR antagonist atropine (Fig. 7C; 95.9 ± 4.5% of baseline, n = 6, p > 0.25). Activation of the mAChR subtypes M1 and M3 that positively couple to phospholipase C (PLC) via Gq/11—but not M2 and M4, which negatively couple to adenylyl cyclase via Gαi/o—facilitates eCB mobilization from postsynaptic compartments (Ohno-Shosaku et al., 2003). To determine whether the M1/M3 receptor subtypes were important for this form of associative iLTD, we performed the submaximal stratum oriens/stratum alveus/postsynaptic TBF pairing protocol in the presence of selective M1 (pirenzepine, 10 μm) and M3 (4-DAMP, 0.2 μm) receptor antagonists, as well as the selective antagonists for M2 (gallamine, 100 μm) and M4 (PD 102807, 0.5 μm) receptors. Associative iLTD was not observed when M1/M3 receptors were blocked, whereas it remained intact in the presence of M2/M4 receptor antagonists (Fig. 7D; M1/M3: 94.2 ± 6.0% of baseline, n = 5, p > 0.3; M2/M4: 77.9 ± 1.7% of baseline, n = 5, p < 0.001; M1/M3 vs M2/M4, p < 0.02). Together, these results show that activation of two different Gq/11 protein-coupled receptors, namely, I-mGluRs and mAChRs, by endogenous ligands, can facilitate the induction of TBF-iLTD, thus revealing an associative and synergistic component for this form of plasticity.

Neuromodulation of TBF-iLTD by endogenous mAChR activation. A, BSI (elicited with 3× TBF) triggered short-term suppression of inhibition, the duration of which was enhanced by the mAChR agonist CCh (0.5 μm). B, Pairing a submaximal 3 min postsynaptic TBF protocol (36 episodes total, every 5 s) with acute CCh application (0.5 μm, gray bar) converted short-term into long-term depression of inhibition, whereas CCh and submaximal TBF alone triggered only a transient suppression. C, Pairing submaximal synaptic stimulation (Stim; 10 pulses at 20 Hz every 10 s or 25 pulses at 50 Hz every 5 s) in stratum oriens/stratum alveolus (SO/A) with submaximal TBF (as in B) triggered iLTD that was blocked by 1 μm atropine, a mAChR antagonist. No long-lasting plasticity was seen when submaximal SO/A stimulation was applied alone. D, Associative iLTD was elicited in the presence of mAChR subtype-selective antagonists for M1 (pirenzepine, 10 μm) and M3 (4-DAMP, 0.2 μm) receptors as well as M2 (gallamine, 100 μm) and M4 (PD 102807, 0.5 μm) receptors.
Postsynaptic TBF persistently gates excitatory throughput via CB1R-mediated disinhibition
Previous studies found that repetitive postsynaptic CA1 pyramidal cell activity can transiently or persistently potentiate or depress Sch collateral synapses (Christofi et al., 1993; Huang and Malenka, 1993; Kuhnt and Voronin, 1994; Kato et al., 2009), thus making it challenging to predict how TBF-induced eCB signaling actually affects the balance of excitation and inhibition in the intact CA1 network. To examine this issue, we tested the long-term impact that TBF might have on EPSP–spike (E-S) coupling (i.e., the ability of a given synaptic input to trigger an action potential). Fibers located in stratum radiatum were stimulated, while CA1 pyramidal cells were maintained at −60 mV in current-clamp mode. Excitatory and inhibitory synaptic transmission was left intact. Five baseline test pulses (25 Hz intraburst interval, 0.1 Hz interburst interval) were elicited during the baseline period and after TBF induction. The stimulation intensity was adjusted such that, on average, one or two action potentials were elicited during baseline test pulses (Fig. 8). Under control conditions, TBF (60×, every 5 s for 5 min) led to a long-lasting increase in the number of action potentials per test pulse (Fig. 8A,E; control, 198.3 ± 34.7% of baseline, n = 6, p < 0.001). Except for a transient (a few minutes) ∼25% reduction right after TBF, the membrane input resistance remained unchanged throughout the experiments (105.4 ± 6.1% of baseline, n = 6, p > 0.5), suggesting that changes in the intrinsic membrane properties of CA1 pyramidal neurons did not contribute to this form of plasticity. Altogether, these observations indicate that postsynaptic TBF leads to E-S coupling potentiation.

Enduring E-S coupling potentiation relies on TBF-induced eCB-mediated disinhibition. A, Top, Traces depicting EPSPs and action potentials elicited by extracellular stimulation of Schaffer collaterals. Excitation and inhibition were intact for these current-clamp recordings. Bottom, Example time-course plot (single experiment) highlighting the change in the number of action potentials per burst (APs/Burst) before and after postsynaptic 60× TBF (every 5 s for 5 min) applied at time 0 (vertical arrow). This manipulation led to E-S coupling potentiation. B, Single representative experiment demonstrating that postsynaptic TBF does not trigger E-S coupling potentiation when CB1Rs were blocked with 2 μm AM251. C, Example experiment illustrating E-S coupling potentiation produced by 5 μm WIN 55212-2, a CB1R agonist (black bar indicates drug application). D, Single experiment representing E-S coupling potentiation observed by an acute application of the GABAAR antagonist picrotoxin (100 μm, black bar). Extracellular stimulation intensity was reduced to evoke two to three action potentials per burst to avoid a ceiling affect. TBF was then delivered in the continuous presence of picrotoxin, which did not elicit E-S coupling potentiation with inhibition blocked. E, Summary data for E-S coupling experiments. TBF alone (control); TBF in the presence (2 μm) of AM251; acute bath application of WIN 55212-2 (5 μm); acute bath application of picrotoxin (100 μm); and TBF in the continuous presence of picrotoxin (100 μm). Note the break in the y-axis. PiTX cont, Picrotoxin continuous; n.s., not significant. ***p < 0.001; **p < 0.01.
Consistent with a requirement for retrograde eCB signaling, E-S coupling potentiation was no longer observed in the presence of 2 μm AM251 (Fig. 8B,E; AM251, 107.3 ± 15.8% of baseline, n = 8, p > 0.5; control vs AM251, p < 0.001). In addition, E-S coupling potentiation was produced by bath application of the CB1R agonist WIN 55212-2 (5 μm) (Fig. 8C,E; WIN 55212-2, 357.6 ± 34.8% of baseline, n = 4, p < 0.001; control vs WIN 55212-2, p < 0.001). Given the functional expression of CB1Rs on Sch collaterals (Kawamura et al., 2006; Takahashi and Castillo, 2006), this finding indicates that CB1R-sensitive inhibitory synapses override excitatory inputs to gate action potential generation, and it also suggests that TBF-induced E-S coupling potentiation is likely due to long-lasting disinhibition. If so, blocking inhibition should mimic and occlude TBF-induced E-S coupling potentiation. In agreement with this prediction, acutely adding the GABAAR antagonist picrotoxin (100 μm) caused massive E-S coupling potentiation (Fig. 8D,E; 619.2 ± 86.8% of baseline, n = 3, p < 0.001). After reducing the stimulation strength and obtaining a new baseline to avoid a ceiling effect, TBF failed to elicit long-term E-S coupling potentiation (Fig. 8D,E; 87.5 ± 12.9% of baseline, n = 3, p > 0.2). These findings strongly suggest that TBF, by triggering CB1R-mediated disinhibition (i.e., iLTD), controls excitatory throughput in a long-lasting manner.
Discussion
The key finding from our study is that postsynaptic activity is sufficient to engage presynaptic long-term plasticity via retrograde eCB signaling. In hippocampal CA1 pyramidal cells, this activity effectively, persistently, and equally depresses somatic and dendritic inhibitory synaptic inputs while sparing excitatory synapses. As a result, eCB-mediated disinhibition can alter information flow through the CA1 circuit by shifting the synaptic balance in favor of excitation. Notably, eCB signaling seems to be tightly restricted to only a single, active neuron. When pairing postsynaptic activity with endogenous metabotropic receptor activation, two manipulations that support a synergy in eCB production, an associative component for iLTD was unmasked. Together, our results support a model in which single CA1 pyramidal cells achieve a new state of excitability in response to postsynaptic activity.
Neural activity can induce long-term eCB-LTD at both excitatory and inhibitory synapses (Heifets and Castillo, 2009). In the hippocampus, repetitive stimulation of Sch–CA1 synapses triggers heterosynaptic iLTD at CB1R-expresing inhibitory synapses (Chevaleyre and Castillo, 2003; Edwards et al., 2006; Zhu and Lovinger, 2007). Our results reveal that heterosynaptic iLTD and TBF-iLTD share a common presynaptic expression mechanism via retrograde eCB signaling. One conspicuous difference between these two forms of plasticity, however, is that heterosynaptic iLTD requires I-mGluR activation for induction, whereas TBF-iLTD relies on postsynaptic Ca2+ influx through l-type voltage-gated Ca2+ channels. Additional studies have used postsynaptic activity alone to probe long-term synaptic plasticity, but these were associated with changes in postsynaptic GABAAR number and/or function (Morishita and Sastry, 1996; Aizenman et al., 1998; Wang et al., 2006; Kurotani et al., 2008). Our paired recordings exclude this expression mechanism for TBF-iLTD. While certain neocortical cell types exhibit an activity-dependent and postsynaptic CB1R-dependent increase in intrinsic membrane properties (Bacci et al., 2004; Marinelli et al., 2009), we did not detect any long-term changes in these properties after CA1 pyramidal cell TBF, thus ruling out this expression mechanism.
Associative synaptic activity by means of coincident spike/burst-timing is thought to lead to long-term changes in synaptic efficacy in vivo (Caporale and Dan, 2008; Feldman, 2012). Here, induction relies on the relative timing between repetitive presynaptic and postsynaptic pairings, with eCB signaling playing an important role and typically dominating the LTD component (Sjöström et al., 2003; Bender et al., 2006; Nevian and Sakmann, 2006; Zhao and Tzounopoulos, 2011; Min and Nevian, 2012). Although timing-dependent eCB-LTD was recently reported at Sch–CA1 excitatory synapses (Péterfi et al., 2012), we were unable to detect eCB-LTD at this synapse under a variety of experimental conditions. Other studies also provided evidence for eCB-LTD at the Sch–CA1 synapse (Xu et al., 2010; Izumi and Zorumski, 2012), findings that contrast with current and previous results (Rouach and Nicoll, 2003; Chevaleyre and Castillo, 2004). While differences in experimental conditions between laboratories may explain the discrepancy, our findings are consistent with the idea that, at least in hippocampal area CA1, inhibitory inputs are vastly more sensitive to eCB signaling than excitatory inputs (Ohno-Shosaku et al., 2002; Kawamura et al., 2006). We also uncovered a synergistic and associative component to iLTD when pairing two submaximal stimuli that, when applied alone, had little impact on synaptic transmission. The mechanism for this form of associativity likely relies on PLC as a coincidence detector (Hashimotodani et al., 2005, 2007b) and suggests that other Gq/11 protein-coupled receptors could also participate in associative TBF-iLTD.
Our study highlights that under conditions designed to reliably detect normal eCB signaling, virtually no spread of eCB effect was observed beyond a single, active neuron. This observation is consistent with eCBs having limited diffusive properties and is strongly supported by a number of reports indirectly showing local actions of retrograde eCB signaling (for review, see Alger, 2012). Some studies, however, have implicated eCB diffusion under normal conditions over seemingly great distances (Vincent and Marty, 1993; Wilson and Nicoll, 2001; Kreitzer et al., 2002; Yasuda et al., 2008; Navarrete and Araque, 2010). Several possibilities could explain these disparate findings. Upon release, eCBs might integrate into lipid bilayers of adjacent neurons and diffuse laterally. Alternatively, eCBs could indirectly affect inhibitory synaptic transmission via an intermediary astrocyte (for review, see Castillo et al., 2012). Neither of these possibilities, however, likely contributes to normal eCB signaling in the hippocampus in response to TBF or DSI. Another possibility is that the activity level of the presynaptic eCB degradative enzyme MGL helps regulate the distance over which 2-AG is capable of diffusing (Tanimura et al., 2012). While MGL expression levels are heterogeneous across different synapses in the brain (Wu et al., 2008; Yoshida et al., 2011; Tanimura et al., 2012), it remains to be tested whether the intrinsic enzymatic activity levels of MGL are also different, or whether the recent history of a given synapse can modify MGL activity.
One important property of heterosynaptic iLTD is that only inhibitory inputs close enough to Sch–CA1 synapses (<10 μm) will detect the eCB signal required for induction (Chevaleyre and Castillo, 2004). As expected for the dendritic but not somatic localization of Sch–CA1 synapses (Megías et al., 2001), only dendritic inhibitory inputs undergo heterosynaptic iLTD (Chevaleyre and Castillo, 2003). In contrast, we now show that a physiologically relevant pattern of CA1 pyramidal cell activity (e.g., TBF) can trigger iLTD at both somatic and dendritic CB1R-expressing inputs. Although somatic iLTD has not yet been reported in the hippocampus, our observation is in line with previous work showing that action potentials can engage eCB-mediated short-term plasticity such as DSI/DSE (Beau and Alger, 1998; Fortin et al., 2004; Brenowitz et al., 2006; Romo-Parra et al., 2009; Sedlacek et al., 2011) that is distributed across somatic and dendritic synapses (Morishita and Alger, 2001; Brenowitz et al., 2006). Our findings confirm that DSI is more pronounced in somatic rather than dendritic inhibitory domains (Lee et al., 2010). TBF-iLTD, on the other hand, is expressed equally in magnitude along the somatodendritic axis, a result supporting immunoelectron microscopy findings that no significant difference in CB1R density exists between these compartments (Nyíri et al., 2005).
Plasticity of inhibitory synapses is an important determinant of many cellular properties that contributes to local circuit function (Castillo et al., 2011; Isaacson and Scanziani, 2011; Kullmann et al., 2012). At the network level, our data further support a model in which active pyramidal cells selectively break free from a subset of inhibitory inputs (i.e., CB1R-expressing inputs), presumably increasing the “contrast” between inactive pyramidal cells still under inhibitory control (Freund and Katona, 2007). This type of neural behavior is thought to help route excitation through dynamic circuits, in line with several studies showing a role for inhibitory synaptic transmission in E-S coupling potentiation (Abraham et al., 1987; Chavez-Noriega et al., 1989; Lu et al., 2000; Chevaleyre and Castillo, 2003; Marder and Buonomano, 2003; Staff and Spruston, 2003; Carvalho and Buonomano, 2009). For instance, DSI, or short trains of action potentials, can transiently increase the sensitivity of pyramidal cells to excitatory inputs by suppressing inhibition (Wagner and Alger, 1996; Fortin et al., 2004), and DSI and tetanus-induced iLTD support long-term plasticity by lowering the induction threshold for excitatory Sch–CA1 LTP (Carlson et al., 2002; Chevaleyre and Castillo, 2004; Zhu and Lovinger, 2007). Thus, CA1 pyramidal cell TBF can act as a disinhibitory gate that transiently or permanently alters the way in which information is transmitted through the circuit.
We show that postsynaptic TBF depresses inhibitory inputs along the somatodendritic axis of hippocampal CA1 pyramidal cells, which is predicted to enhance somatic output by making it easier to generate action potentials (Miles et al., 1996). Previous studies found that inhibitory synaptic transmission limits the spread of back-propagating action potentials and Ca2+ signaling in dendrites (Buzsáki et al., 1996; Tsubokawa and Ross, 1996; Hsieh and Levine, 2013). Our results predict TBF-iLTD enhances the efficacy of back-propagating action potentials into more distal dendritic domains. By this means, TBF-iLTD could regulate the generation and duration of dendritic Ca2+ signals critical for inducing dendritic spikes and long-term plasticity of excitatory synaptic transmission (Golding et al., 2002; Takahashi and Magee, 2009; Müller et al., 2012).
Our study reveals that postsynaptic CA1 pyramidal cell TBF, mimicking in vivo patterns of activity that normally occur during exploratory behaviors and sleep, can engage retrograde eCB signaling that diminishes presynaptic inhibition and indirectly enhances excitation. These activity-dependent changes in hippocampal synaptic transmission may contribute to place cell formation and maintenance. Intriguingly, place cell formation is thought to occur in an activity-dependent manner over the course of several minutes with refinement requiring tens of minutes (Wilson and McNaughton, 1993). The time course over which TBF leads to iLTD (i.e., minutes), especially when paired with endogenous mGluR and mAChR activation to achieve a synergy in eCB production, raises the possibility that eCB-mediated disinhibition might contribute to this process. Finally, although we favor the physiological consequences of TBF on long-term inhibitory synaptic plasticity in the hippocampus, it is possible that this form of plasticity, in extreme circumstances like that of epileptic activity, could be maladaptive and lead to hyper-network excitability (Alger, 2004; Lutz, 2004; Katona and Freund, 2008).
Footnotes
This work supported by National Institutes of Health Grants R01-MH081935 and R01-DA17392, and the Irma T. Hirschl Career Scientist Award to P.E.C. We thank all the members of the Castillo laboratory, in particular Drs. Sachin Makani and Yuki Hashimotodani, for critical review of early versions of the manuscript and for invaluable discussions. We also thank Dr. Csaba Földy for providing the morphological reconstruction protocol; Kevin Fisher and the Imaging Core Facility at the Rose F. Kennedy Center (supported by NIH grant P30-HD071593) for NeuroLucida training; and Catherine Castillo for technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Pablo E. Castillo, Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Rose F. Kennedy Center, Room 703, 1410 Pelham Parkway South, Bronx, NY 10461. pablo.castillo{at}einstein.yu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}