

Abstract
We previously identified SPARC-related protein-containing immunoglobulin domains 1 (SPIG1, also known as Follistatin-like protein 4) as one of the dorsal-retina-specific molecules expressed in the developing chick retina. We here demonstrated that the knockdown of SPIG1 in the retinal ganglion cells (RGCs) of developing chick embryos induced the robust ectopic branching of dorsal RGC axons and failed to form a tight terminal zone at the proper position on the tectum. The knockdown of SPIG1 in RGCs also led to enhanced axon branching in vitro. However, this was canceled by the addition of a neutralizing antibody against brain-derived neurotrophic factor (BDNF) to the culture medium. SPIG1 and BDNF were colocalized in vesicle-like structures in cells. SPIG1 bound with the proform of BDNF (proBDNF) but very weakly with mature BDNF in vitro. The expression and secretion of mature BDNF were significantly decreased when SPIG1 was exogenously expressed with BDNF in HEK293T or PC12 cells. The amount of mature BDNF proteins as well as the tyrosine phosphorylation level of the BDNF receptor, tropomyosin-related kinase B (TrkB), in the hippocampus were significantly higher in SPIG1-knockout mice than in wild-type mice. Here the spine density of CA1 pyramidal neurons was consistently increased. Together, these results suggest that SPIG1 negatively regulated BDNF maturation by binding to proBDNF, thereby suppressing axonal branching and spine formation.
Introduction
We and other groups have demonstrated that several morphogens and transcription factors play crucial roles in the formation (and maintenance) of regional specificity in the developing retina (Yuasa et al., 1996; Koshiba-Takeuchi et al., 2000; Schulte and Cepko, 2000; Sakuta et al., 2001, 2006; Takahashi et al., 2003, 2009). These molecules regulate the expression of downstream genes, including Eph-ephrin systems, directly involved in the axon guidance of retinal ganglion cells (RGCs) to the optic tectum/superior colliculus (SC) along the anteroposterior (AP) and dorsoventral (DV) axes (McLaughlin et al., 2003b; Noda et al., 2009). Topographically appropriate connections may be established by selective branching along the axon shaft with a bias at the AP location of their future appropriate terminal zone (TZ) (Simon and O'Leary, 1992a; Yates et al., 2001) and preferential extension toward the TZ along the DV axis in the tectum (Nakamura and O'Leary, 1989; Hindges et al., 2002; McLaughlin et al., 2003a). Refined topography has then been shown to develop through the large-scale elimination of overshoots against ectopic branches and arbors (Nakamura and O'Leary, 1989; Simon and O'Leary, 1992a, 1992b; Yates et al., 2001). However, the mechanisms underlying the formation and refinement of axonal branches, which are requisite steps for the establishment of the final pattern of connections between RGCs and its targets, have not yet been examined in detail, although previous studies proposed the involvement of Ephs/ephrins and TrkB/BDNF in branch formation (Cohen-Cory and Fraser, 1995; Yates et al., 2001; Hindges et al., 2002; Sakurai et al., 2002; Marler et al., 2008).
We previously identified a clone with dorsal-rich expression in the developing chick retina by using a cDNA display system (D/Bsp120I #1 in Shintani et al., 2004; GenBank accession number, EF692644), and later referred to this molecule as SPIG1 (Yonehara et al., 2008; AF374459). SPIG1, a member of the SPARC family, is composed of a signal peptide, a follistatin-like domain, an extracellular calcium-binding domain with two EF-hand motifs, and two immunoglobulin (Ig)-like domains (see Fig. 1A). SPARC family molecules are known to have various functions, such as regulating the assembly and deposition of the extracellular matrix, counter-adhesion between cells, and modulating the activity of extracellular proteases and growth factors/cytokines (Bradshaw, 2012). We demonstrated that SPIG1 was highly expressed in upward-motion-preferring ON direction-selective RGCs in the whole retinal region, and in a large population of RGCs in the dorsotemporal region of the retina in mice (Yonehara et al., 2008, 2009). However, the physiological functions of SPIG1 remain unknown.
In the present study, we performed knockdown (KD) experiments for SPIG1 to elucidate its function in the axonal projection of RGCs to the tectum. We found that manipulated RGC axons formed branches and synapses aberrantly in the tectum. SPIG1 and BDNF were colocalized in vesicle-like structures in cells. Moreover, SPIG1 bound with proBDNF but bound very weakly with mature BDNF. When SPIG1 was coexpressed with BDNF in HEK293T and PC12 cells, mature BDNF protein levels were significantly decreased not only in the cells, but also in the culture medium. Thus, SPIG1 likely plays an essential role in the control of BDNF maturation.
Materials and Methods
Animals.
Fertilized white leghorn eggs were incubated at 37.5°C under humidified conditions, and the embryos were staged by Hamburger and Hamiltons' criteria (Hamburger and Hamilton, 1951). SPIG1-knockout (KO) mice were generated by the insertion of the enhanced green fluorescent protein (EGFP) gene into the signal sequence region in exon 2 of the SPIG1 gene (Yonehara et al., 2008). SPIG1-KO mice backcrossed with the inbred C57BL/6 strain for >10 generations were used for experiments. Mice were given free access to food and water at a constant temperature (23°C) and humidity (50%) under a typical 12 h light-dark cycle. All animal experiments in this study were performed according to the guidelines of Animal Care with approval by the Committee for Animal Research, National Institutes of Natural Sciences.
Reagents and antibodies.
Merosin and poly-d-lysine were purchased from Merck Millipore and Sigma, respectively, and the other reagents for the tissue culture, including a neurobasal medium, were from Invitrogen. The antibodies used were commercially available: anti-BDNF (N-20, Santa Cruz Biotechnology), anti-proBDNF (mAb287, GeneCopoeia), anti-alkaline phosphatase (8B6, Sigma), HRP-conjugated anti-human IgG (NA933V, GE Healthcare), anti-Trk (#610101, BD Transduction Laboratories), anti-TrkB (SC-139, Santa Cruz Biotechnology), anti-phosphotyrosine (4G10, Merck Millipore), Alexa Fluor 488 conjugated DYKDDDDK Tag Antibody (#5407, Cell Signaling Technology), and Alexa Fluor 594 anti-mouse IgG (A11032, Invitrogen).
Constructs for knockdown of SPIG1 and BDNF, and the expression of SPIG1 and BDNF.
RCASDC-U6/SPIG1 (shRNA#1-shRNA#4), RCASDC-U6/BDNF, or RCASDC-U6/EGFP was constructed with the RCASDC vector (Sakuta et al., 2006), which expresses a short hairpin RNA (shRNA) under the control of the chick U6 promoter. The target sequences of shRNA for chick SPIG1 were as follows: 5′-GAGGTATCCGGAAGGTTTG-3′ (shRNA#1, nucleotide residues 127–145), 5′-GAAATACTGCGGCCGAGGG-3′ (shRNA#2, nucleotide residues 204–222), 5′-GACGATTCCCTCTACATCA-3′ (shRNA#3, nucleotide residues 904–922), and 5′-CACGTTACGCTGTGAGGTT-3′ (shRNA#4, nucleotide residues 2457–2475) (see Fig. 1A). The target sequence of shRNA for chick BDNF (NM_001031616) was 5′-GGTTCAAGAGGACTGACAT-3′ (nucleotide residues 154–172). The control shRNA for EGFP was 5′-GGAGTTGTCCCAATTCTTG-3′ (nucleotide residues 28–46).
The cDNA coding regions for SPIG1 and BDNF were obtained from chick or mouse retina total RNA by RT-PCR. The expression constructs for SPIG1 and BDNF were prepared by inserting a cDNA fragment containing the entire coding sequence of SPIG1 (EF692644 for chick; AF374459 for mouse) and BDNF (NM_001031616 for chick; NM_001048139 for mouse), respectively, into the pcDNA3.1 vector. The construct for FLAG-tagged SPIG1 (pcDNA-SPIG1-FLAG) was prepared by conjugating FLAG-tag to the C terminus of the entire coding sequence of SPIG1. The expression construct for the Fc region of human IgG (pcDNA-Fc) was generated by inserting the cDNA fragment of the human IgGγ1-Fc domain (Ellison et al., 1982) into the pcDNA3.1 vector. The construct for Fc-fused SPIG1 (pcDNA-SPIG1-Fc) was generated by inserting the cDNA fragment containing the entire cording region of chick SPIG1 fused in frame into pcDNA-Fc.
Northern blotting and in situ hybridization.
Northern blotting and section in situ hybridization were performed as described previously (Suzuki et al., 2000). Total RNA was isolated from the dorsal, ventral, temporal, or nasal one-third of chick retinas with TRIzol (Invitrogen) following the manufacturer's protocol. Templates used for probe preparation were the 1004 bp fragment of chick SPIG1 (nucleotide residues 1426–2429; EF692644) and the 577-bp fragment of chick glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (218–794; K01458).
In ovo electroporation.
In ovo electroporation was performed as described previously (Sakuta et al., 2001, 2008). Briefly, retroviral constructs (0.125–0.5 μg/μl) in TE buffer (10 mm Tris-HCl, 1 mm EDTA, pH 8.0) containing 0.05% fast green were injected into the optic vesicle or mesencephalon at Hamburger-Hamilton (HH) stage 9–10 with a glass micropipette. The embryos were continually incubated in a humidified incubator until the appropriate developmental stage.
DiI labeling of RGC axons.
RGC axons were labeled with a small crystal of DiI (Invitrogen) at embryonic day 10 (E10) to E16 as described previously (Yuasa et al., 1996). Embryos were incubated for an additional 2 d to allow DiI to label the axonal fibers of RGCs from the retina to the tectum. Tecta were cut into lateral and medial halves and were then observed using a fluorescent microscope (Axiophot 2, Zeiss).
Electron micrographs of DiI-labeled RGC axons by photoconversion.
The photoconversion of DiI was performed as described previously (von Bartheld et al., 1990). Tectal tissues were prefixed with 4% PFA and 0.4% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.4, for 10 min, and postfixed with 2% glutaraldehyde in the same buffer for 5 h. The sliced tissue (100 μm thickness) was preincubated with 1 mg/ml DAB tetrahydrochloride in 0.1 m Tris-HCl, pH 8.0, for 1 h, flattened onto a depression slide glass, and the region of interest was then irradiated using a fluorescent microscope (Axiophot 2; Zeiss) through a ×20 objective (NA, 0.5; Zeiss) with a rhodamine filter. DAB solution on the surface of the slice was changed with fresh solution every 15 min during the photoconversion. After washing with the cacodylate buffer (0.1 m), slices were then fixed with 2% osmium oxide in the sodium cacodylate buffer and embedded in Epon812 (TAAB Laboratories Equipment). Ultrathin sections were stained with uranyl acetate and lead citrate and observed with a JEM-1200 EX electron microscope (JEOL).
Golgi impregnation.
Golgi-Cox staining was performed as described by Gibb and Kolb (1998). Briefly, brains were immersed in the Golgi-Cox solution at room temperature for 14 d, transferred to a 30% sucrose solution for 2 d, and were then sagittally sectioned at 120 μm intervals using a vibratome. The sections were impregnated with a mixture of potassium dichromate and mercuric chloride and were mounted on glass slides.
Primary culture of RGCs and hippocampal neurons.
The dorsal one-third of the retina was dissociated by trituration after incubation with 0.1% trypsin (Worthington) in Ca2+ and Mg2+-free PBS for 10 min at 37°C. The dissociated cells collected by centrifugation (1,000 × g for 2 min) were plated on glass-bottom dishes coated with laminin (10 μg/ml) and merosin (2 μg/ml), and were then cultured in a neurobasal medium (Invitrogen) supplemented with 1× B27, 1 mm glutamine, and 5 μm forskolin (1.0 × 105 cells per 3.5 cm dish). Dissociated hippocampal neurons were prepared from E18.5 mice. The cells were electroporated with the expression constructs using Amaxa nucleofector (Amaxa) according to the manufacturer's protocol and were cultured in neurobasal medium supplemented with 1× B27 and 1× GlutaMAX1 (Invitrogen).
DNA transfection of HEK293T cells and PC12 cells.
Human embryonic kidney epithelial cells (HEK293T cells) were grown and maintained on dishes in DMEM supplemented with 10% FBS. DNA transfection was performed using a Lipofectamine plus reagent (Invitrogen) according to the manufacturer's protocol. The medium was changed to a fresh serum-free medium 24 h after transfection.
The pheochromocytoma cell line, PC12, was grown and maintained on dishes in DMEM supplemented with 10% FBS and 5% horse serum. The medium was changed 12 h after transfection to DMEM supplemented with 1% horse serum and 50 ng/ml nerve growth factor (NGF-2.5S from murine submaxillary glands, Sigma) for 48 h to differentiate the cells into a neuronal phenotype.
BDNF-releasing assays.
Differentiated PC12 cells (2.0 × 105 cells on a 3.5 cm dish) were washed with normal artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 4.75 KCl, 26 NaHCO3, 10 glucose, 1.25 KH2PO4, 2 CaCl2, and 1 MgCl2, pH 7.4, with 95% O2 and 5% CO2, and incubated in 1 ml of normal aCSF or high-K+ aCSF (82 NaCl, 58.75 KCl) for 1 h at 30°C. The supernatants and cells were then collected for Western blot analyses: protein precipitation with acetone was performed for the supernatants to concentrate the proteins.
Pull-down, immunoprecipitation assays, Western blotting.
Tissues or cultured cells were lysed with 1% NP-40 in 20 mm Tris-HCl, pH 8.0, 137 mm NaCl, 1 mm vanadate along with an EDTA-free protease inhibitor mixture (cOmplete, EDTA-free; Roche), and the supernatant was collected by centrifugation at 15,000 × g for 15 min.
Cell extracts or culture media were precleaned by incubation with Sepharose beads (GE Healthcare) for the pull-down assay. Fc-fused proteins were incubated with Protein G-coated magnetic beads (Dynabeads protein G, Invitrogen) for 1 h at 4°C for pull-down experiments. To analyze the phosphorylation of TrkB, immunoprecipitation was performed with an anti-Trk antibody and protein G magnetic beads. The pulled-down proteins were eluted from the beads by boiling in sample buffer and were then analyzed by Western blotting as below.
Proteins were separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Merck Millipore) by a conventional semidry blotting method. Membranes blocked with 4% nonfat dry milk and 0.1% Triton X-100 in 10 mm Tris-HCl, pH 7.4, 150 mm NaCl were incubated overnight with the indicated antibodies. The binding of antibodies was detected with an ECL Western blotting system (GE Healthcare).
In vitro binding assay with purified proteins.
SPIG1-Fc or control Fc proteins expressed in HEK293T cells were purified from the culture medium using a HiTrap protein G column attached to a chromatography apparatus (AKTA prime plus, GE Healthcare). A 96-well polystyrene ELISA plate (#9018, Costar) was coated with 4 pmol of purified recombinant human proBDNF (B-257, Alomone Labs) or mature BDNF (GF029, Millipore). After blocking with 1% BSA and 0.1% Tween 20 in 10 mm phosphate buffer, pH 7.2, containing 150 mm NaCl (PBS), the wells were incubated overnight at 4°C with purified SPIG1-Fc proteins or Fc proteins (1 ng protein per well) in 10 mm acetate buffer, pH 5.5 containing 150 mm NaCl. After washing, the wells were incubated with 10% formalin in PBS for 30 min to prevent the secretion of the binding proteins. After blocking again, binding was detected with HRP-conjugated anti-human IgG and 3,3′,5,5′-tetramethyl-benzidine substrate (SureBlue Reserve TMB Microwell Peroxidase Substrate, KPL) according to the manufacturer's instructions. After color development, the optical density at 450 nm was measured with a microplate reader (SH-9000Lab, Corona Electric). The specific binding of SPIG1-Fc was determined by subtracting the nonspecific binding activity for the control Fc proteins.
Results
The expression of SPIG1 is asymmetric in the developing chick retina
We previously identified SPIG1 as one of the dorsal-rich molecules in RGCs in the chick retina by RLCS screening at E8 (Shintani et al., 2004; Yonehara et al., 2008). We performed Northern blotting (Fig. 1B) and in situ hybridization (Fig. 1C) on the developing chick retina to examine the developmental and regional expression of SPIG1 transcripts in detail.

SPIG1 expression in the developing chick retina. A, Schematic representation of the domain structure of SPIG1. The probe region for Northern blotting and in situ hybridization is indicated below. The target sites of SPIG1 shRNA#1 to shRNA#4 examined are shown. B, Northern blot analyses of SPIG1 mRNA expression in the dorsal (D), ventral (V), temporal (T), or nasal (N) retina at embryonic day 6–18 (E6–E18). The upper four blots were hybridized with the SPIG1 cDNA probe in A; the lower four were reprobed with the GAPDH probe as a control for RNA loading. C, In situ hybridization for SPIG1 expression in the developing chick retina. Boxed areas in the retina (D, V, T, and N) are shown developmentally. Scale bars, 50 μm. D, shRNA knockdown of SPIG1. The retroviral construct expressing shRNA directed against chick SPIG1 mRNA or a control shRNA for EGFP (Con) was electroporated into the optic vesicle at HH stage 9–10 (at E1.5), and SPIG1 expression in the retina at E18 was analyzed by Northern blotting. The results with shRNA#3 were shown because it was the most effective. The amount of SPIG1 mRNA quantified by densitometry was normalized to the value for the control. Data are mean ± SE (n = 4). *p < 0.001 (Student's t test). E, Hematoxylin and eosin staining of E12 retina following electroporation as described above. Scale bars, 50 μm.
SPIG1 mRNA was first detected at E8 in the dorsal and temporal regions of the chick retina and markedly increased from E10 while maintaining asymmetric expression along the DV and nasotemporal (NT) axes by ∼E12-E14 (Fig. 1B). SPIG1 mRNA signals were initially localized in the ganglion cell layer (GCL), and then the inner nuclear layer (INL) also became positive from E12 (Fig. 1C): Signals in the nasal retina and ventral retina were faint at E8, which was consistent with the results of Northern blotting. Positive cells were present in both the GCL and INL in the whole retinal region from E12. The intensity of the SPIG1 signal in the GCL was higher in the dorsotemporal region than in the ventronasal region at every developmental stage, whereas the signal in the INL was not asymmetric.
SPIG1 knockdown in the developing retina causes dorsal RGC axons to form aberrant branches and ectopic terminal arbors in the tectum
We investigated whether SPIG1 was implicated in the topographic retinotectal projection by the KD of SPIG1 expression. The in ovo electroporation of SPIG1-specific shRNA construct #3 into the optic vesicle at HH stage 9 (E1.5) resulted in SPIG1 mRNA expression in the E18 retina being 70% lower than control levels (Fig. 1D). No histological alterations, including the total number of RGCs or organization of the retina, were observed in the manipulated retina (Fig. 1E). We then performed the anterograde tracing of RGC axons (48 h before the observation) by placing a DiI crystal in the dorsal retina in which SPIG1 was highly expressed.
Dorsal RGC axons entered orderly into the ventral tectum and developed axonal branches at E12 in control embryos (Con) (10 of 10 embryos) (Fig. 2A, left). These branches exhibited a topographic bias in their location at E14 (6 of 6 embryos) and finally formed a single dense TZ in an appropriate place at E18 (10 of 10 embryos) (Fig. 2D,G, left). However, dorsal RGC axons in SPIG1-KD embryos entered broadly into the ventral tectum at E12 (9 of 10 embryos) (Fig. 2A, right). Distribution of branches shifted toward the anterior and dorsal directions more than in the control embryos (Fig. 2B,C). In addition, the branch number of the dorsal RGC axons was significantly higher in SPIG1-KD embryos (p < 0.01 for control = 1.48 and SPIG1-KD = 1.87 per axon; Student's t test).

Effect of SPIG1-knockdown on developmental retinotectal projections. A, Projection pattern of dorsal RGC axons by DiI labeling in control (Con) and SPIG1-KD embryos at E12. Higher magnification of the boxed area is shown in the lower part of each panel. A, Anterior; P, posterior; D, dorsal; V, ventral. Scale bars, 1 mm. B, Schematic drawings of the RGC axons in A. The tectum was subdivided into 10 areas along the AP (blue) or DV (green) axis. Tg, Tegmentum. C, Quantification of axonal branch distributions at E12. The number of branches was counted in each subdivided tectal area as shown in B. Data are mean ± SE (control, n = 8; SPIG1-KD, n = 10). D, Typical projection pattern of dorsal RGC axons in control and SPIG1-KD embryos at E14 with higher magnification. Scale bars, 1 mm. E, Schematic drawings of RGC axons in D. The tectum was subdivided into 10 areas along the AP (blue) or DV (green) axis. F, Quantification of axonal branch distributions at E14. Data are mean ± SE (control, n = 4; SPIG1-KD, n = 5). G, Typical projection patterns of dorsal RGC axons in control and SPIG1-KD embryos at E18 with higher magnification. Scale bars, 1 mm. H, Schematic drawings of G. I, Quantification of TZ at E18 (control, n = 5; SPIG1-KD, n = 5). After spatial normalization, tectal images were two-dimensionally divided into 50 × 50 pixel blocks along the AP and DV axes. We defined the terminal area of DiI-labeled RGC axons by being over a threshold (top to half-maximal intensity of fluorescence) in each reconstructed image.
Dorsal RGC axons in SPIG1-KD embryos failed to eliminate ectopic branches at E14 (8 of 10 embryos) (Fig. 2D–F), arborized ectopically, and formed diffuse TZ at E18 (11 of 14 embryos) (Fig. 2G–I). Ventral RGC axons showed normal branching at E18, and the trajectory of RGC axons within the retina appeared normal in all cases (data not shown). The overexpression of SPIG1 did not affect the projection pattern or branching of RGC axons (data not shown).
Synapse formation of RGC axons in the tectum of SPIG1-knockdown embryos
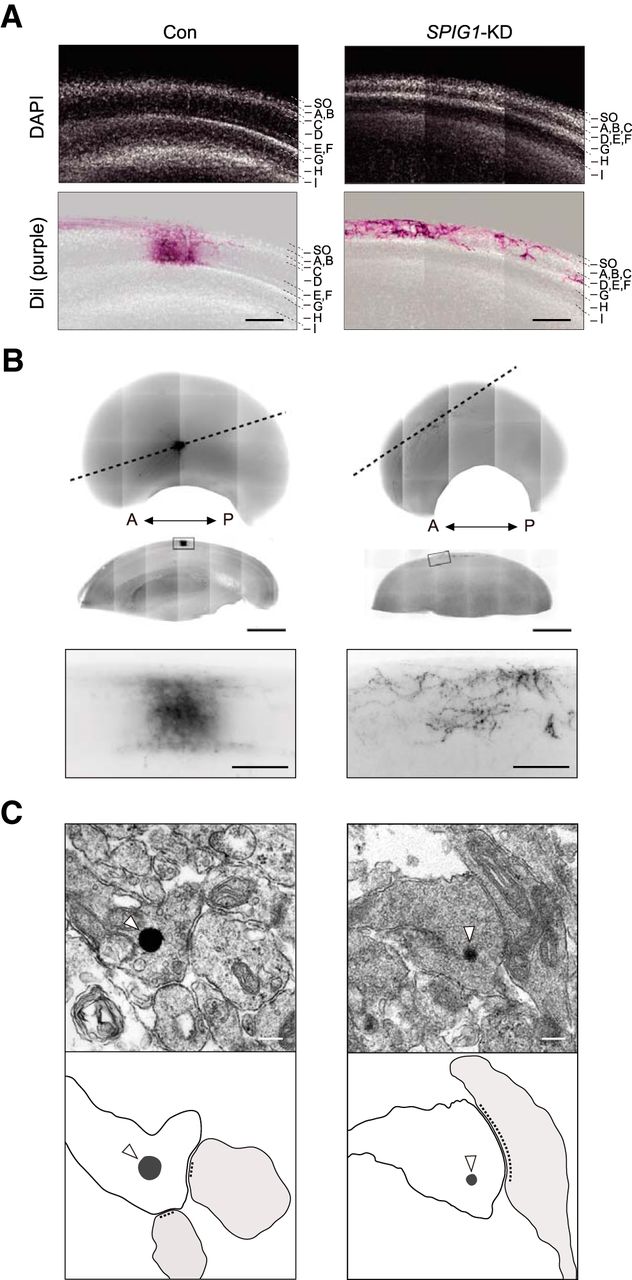
RGC axons normally elaborate arbors near their target sites and form synapses with the dendrites of tectal neurons in laminae B to F of the stratum griseum et fibrosum superficiale in the tectum (LaVail and Cowan, 1971; Yamagata and Sanes, 1995). A transverse section of the E18 tectum in SPIG1-KD embryos revealed thinner superficial laminae (A-F), and the positional distribution of the axonal arbors of DiI-labeled RGCs were highly defective (Fig. 3A,B). However, these were deserved within the A-F layers in SPIG1-KD embryos, which were essentially similar to those in control embryos. No significant differences were observed in the structure of synaptic contacts between knockdown and control chicks on electron micrographic observations of photoconverted DiI-labeled sections (Fig. 3C). These results indicated that dorsal RGC axons in the SPIG1-KD embryos arborized and formed synapses, although ectopically placed.

Effect of SPIG1-KD on the lamina-specific projections of dorsal RGC axons. A, Retinorecipient lamina of control and SPIG1-KD embryos. Top, Sections of the E18 tectum stained with DAPI to visualize cellular nuclei. Bottom, DiI-labeled retinal terminal (purple) merged with the DAPI-stained image. SO, Stratum opticum. Scale bars, 200 μm. B, Synaptic connections of RGC axons on other neurons in the tectum. Top, Ventral halves of the tectum in control and SPIG1-KD embryos at E18. DiI in RGC axons was photoconverted to electron-dense DAB reaction products as described in Materials and Methods. A, Anterior; P, posterior. Middle, Sagittal sections along the dashed lines in the top panels. Boxed area in middle panels, Higher magnification at the bottom. Scale bars: middle panels, 1 mm; bottom panels, 100 μm. C, Electron micrographic observation of the photoconverted sections. Open arrowheads indicate electron-dense DAB reaction products. Scale bars, 200 nm. Bottom, Schematic drawings of the upper images: gray represents DAB reaction products; light gray represents postsynaptic regions.
Functional relevance between SPIG1 and BDNF
We further examined the effects of SPIG1-KD in RGCs using a primary culture system. The shRNA construct for SPIG1 was electroporated to the optic vesicle at HH stage 9 as well as the in vivo experiment, and cells were dissociated from the dorsal one-third of the E8 retina and cultured on a laminin/merosin-coated dish. Cultured cells were immunostained at 4 DIV (corresponding to E12) using an antibody to the GAG protein of the RCAS virus to identify infected cells. The KD of SPIG1 caused a significant increase in the number of branches of RGC axons but had no effect on axonal length (Fig. 4A, lower left), which was consistent with the in vivo results shown in Figure 2A.

Effect of exogenous BDNF on the branching and elongation of RGC axons. A, Primary culture of RGCs. After the electroporation of SPIG1-shRNA or control retroviral construct at HH stage 9–10, retinal cells were dissociated from the dorsal one-third of the retina at E8 and plated on laminin/merosin-coated dishes. The culture medium was replaced 1 d after plating with a medium containing the indicated concentrations of mature BDNF (mBDNF) and was cultured for an additional 3 d. Scale bars, 10 μm. B, Quantification of the branch number of RGC axons. C, Quantification of the axon length of RGCs. Data are mean ± SE of five independent experiments. *p < 0.01 (ANOVA with Scheffé's post hoc tests).
We added mature BDNF (5 or 15 ng/ml) to the culture because the number of RGC axon branches has been shown to be upregulated by mature BDNF in vitro (Marler et al., 2008). A fourfold increase in the branch number of control RGC axons was observed at both concentrations of mature BDNF (Fig. 4A, top, B), which suggested that 5 ng/ml of mature BDNF was sufficient to induce axon branching. No further increase in the branch number was observed in SPIG1-KD RGC axons upon the addition of mature BDNF (Fig. 4A, bottom, B). In contrast, mature BDNF exerted no effect on axon length in all cases (Fig. 4C).
We then determined whether endogenous BDNF levels were relevant to the increased axon branching observed in SPIG1-KD RGCs by using a neutralizing antibody to BDNF. The enhanced level of axon branching by SPIG1-KD was dose-dependently decreased by the addition of a neutralizing antibody to BDNF in the culture medium (Fig. 5A, bottom, B): The branch number reached the level of control RGCs at 5 μg/ml. The control treatment with the anti-alkaline phosphatase antibody had no effect on the branch number (Fig. 5A–C). In contrast, control RGCs were insensitive to the anti-BDNF antibody (Fig. 5A, top, B). The length of the axons was again not affected by the anti-BDNF or anti-alkaline phosphatase antibodies (Fig. 5C).

Effect of a neutralizing antibody for BDNF on the elongation and branching of RGC axons. A, Primary culture of RGCs. One day after plating of the retinal cells prepared as above, the culture medium was replaced with a medium containing the indicated concentrations of the anti-BDNF antibody or anti-alkaline phosphatase antibody (anti-AP, as a control). The culture was continued for an additional 3 d. Scale bars, 10 μm. B, Quantification of the branch number of RGC axons. C, Quantification of the axon length of RGCs. Data are mean ± SE of four independent experiments. *p < 0.01 (ANOVA with Scheffé's post hoc tests). **p < 0.001 (ANOVA with Scheffé's post hoc tests).
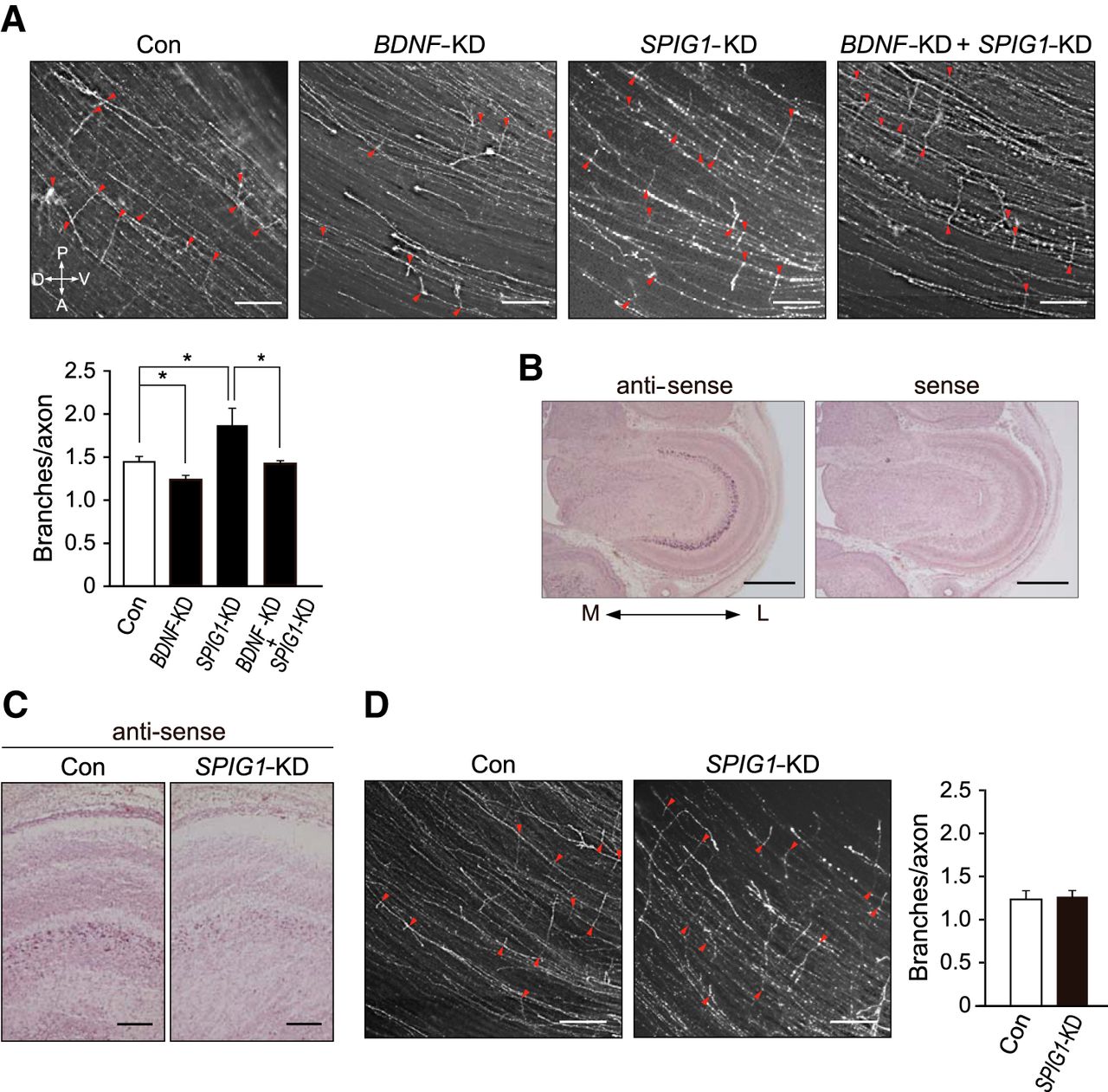
We performed a single KD of BDNF and double KD of BDNF and SPIG1 in the dorsal retina to further examine the functional relevance between SPIG1 and BDNF. In contrast to SPIG1-KD axons, the number of axonal branches was lower in BDNF-KD than in the control (Fig. 6A). Importantly, the phenotypes were cancelled when both genes were manipulated simultaneously (Fig. 6A). Because SPIG1 was also found to be expressed in the tectum (Fig. 6B), we further examined the effect of the knockdown of tectal SPIG1 expression. The expression of SPIG1 mRNA was reduced in the tectum due to electroporation of the SPIG1 shRNA construct (Fig. 6C). This knockdown had no significant effects on RGC axonal branching (Fig. 6D), which suggested that SPIG1 produced by the target tissue was not involved in the branching of RGC axons.

Functional interactions between SPIG1 and BDNF in branching retinal axons in the tectum. A, Effect of the SPIG1 and BNDF double knockdown. The projection pattern of dorsal RGC axons on the contralateral tectum at E12 was analyzed after the electroporation of control EGFP-shRNA (Con), BDNF-shRNA (BDNF-KD), SPIG1-shRNA#3 (SPIG1-KD), or both the BDNF- and SPIG1-shRNA#3 (BDNF-KD + SPIG1-KD) into the optic vesicle at HH stage 9–10. Arrowheads (red) indicate branch points. Scale bars, 200 μm. Bottom, Quantification of axonal branch distributions. Data are mean ± SE (control, n = 8; BDNF-KD, n = 5; SPIG1-KD, n = 10; BDNF-KD + SPIG1-KD, n = 5). *p < 0.05 (Tukey-Kramer test). B, In situ hybridization for SPIG1 expression in the tectum at E12. M, Medial; L, lateral. Scale bars, 1 mm. C, In situ hybridization for SPIG1 expression in the SPIG1-KD tectum at E12. Scale bars, 200 μm. D, No significant effect was observed on the branching of retinal axons by the knockdown of the tectal SPIG1 expression. The projection of dorsal RGC axons on the tectum at E12 was analyzed as above after the electroporation of SPIG1-shRNA#3 (SPIG1-KD) or control EGFP-shRNA (Con) into the mesencephalon at HH stage 10. Arrowheads (red) indicate branch points. Scale bars, 200 μm. The quantification of axonal branch distributions is shown as mean ± SE (control, n = 4; SPIG1-KD, n = 4).
SPIG1 binds to proBDNF but binds very weakly to mature BDNF
We then exogenously expressed SPIG1 (FLAG-tagged at the C terminus) and BDNF (nontagged) in RGCs using a primary culture to analyze subcellular localization. Marked colocalization of SPIG1 and proBDNF was observed (70% overlap) in a punctuate distribution not only in the growth cone and axon (Fig. 7A), but also in the cell soma and dendrites (Fig. 7B), suggesting an interaction between SPIG1 and BDNF in secretory granules. Based on these results, we speculated that SPIG1 negatively controls axonal branch formation by modulating the generation of mature BDNF.

Subcellular colocalization of exogenous SPIG1 and proBDNF in the chick RGC. A, Colocalization of SPIG1 and proBDNF in axon terminals. After the electroporation of SPIG1 (FLAG-tagged at the C terminus) and BDNF constructs at HH stage 9, retinal cells were dissociated from the dorsal one-third of the retina at E8 and cultured for 4 DIV. Exogenously expressed FLAG-tagged SPIG1 and proBDNF were detected with anti-FLAG (green) and anti-proBDNF (red) antibodies, respectively. Bottom, The higher magnification of the boxed areas (a, b). Scale bars, 20 μm. B, Colocalization of SPIG1 and proBDNF in the cell soma and dendrites. Bottom, The higher magnification of the boxed areas (a, b). Scale bars, 20 μm.
When SPIG1-Fc (or control Fc) was expressed in HEK293T cells together with BDNF, both proteins were detected in the cells (Fig. 8A, left) and in culture medium, expectedly (Fig. 8A, right). SPIG1-Fc or control Fc proteins in the samples were then pulled down with Protein-G beads. The results clearly suggested that SPIG1-Fc selectively bound to the proform of BDNF (proBDNF) in cells, but not to mature BDNF in the medium (Fig. 8B). Direct binding assays with purified proteins also revealed the significantly higher binding activity of purified SPIG1 proteins to proBDNF than to mature BDNF (Fig. 8C).

Binding of SPIG1 to BDNF. A, Protein expression from plasmids in the cell and culture medium was verified by Western blotting with anti-human IgG (to detect SPIG1-Fc or conrol Fc protein) or anti-BDNF. The closed arrow indicates the expected size for proBDNF. Signal intensities quantified by densitometry are shown as a percentage of the control cells (Fc). Data are mean ± SE of four independent experiments. *p < 0.05 (Student's t test). B, Pull-down assay with SPIG1-Fc or control Fc protein from the cell extract and culture medium. Protein complexes pulled-down with protein G beads from the cell extracts or culture media were analyzed by Western blotting. The open arrow and closed arrows indicate the expected size for mature BDNF and proBDNF, respectively. The results obtained show that SPIG1 interacted with proBDNF in the cells. C, Binding assays with purified SPIG1-Fc. Left, Coomassie Brilliant Blue (CBB) staining of purified SPIG1-Fc and Fc proteins (100 ng) on a SDS-PAGE gel. SPIG1-Fc (or control Fc) proteins were applied to an ELISA plate coated with proBDNF or mature BDNF, and binding was detected using anti-Fc as described in Materials and Methods. Right, Specific binding of SPIG1-Fc determined by subtracting nonspecific binding to control Fc. Data are mean ± SE of three independent experiments. a.u., Arbitrary units. *p < 0.05 (Tukey-Kramer test).
SPIG1 suppresses BDNF maturation
In the expression assay using HEK293T cells, the amount of mature BDNF proteins was significantly decreased by the coexpression of SPIG1 in both cells and the culture medium (Fig. 8A). We validated these results in NGF-differentiated PC12 cells. SPIG1 and BDNF proteins were colocalized in vesicle-like structures in neuronal cells (Fig. 9A) as was also observed in chick RGCs (Fig. 7). The amount of mature BDNF in the cell was decreased by the coexpression of SPIG1 in differentiated PC12 cells (Fig. 9B, normal) as well as HEK293T cells (Fig. 8A). SPIG1 expression similarly reduced the amount of mature BDNF proteins secreted into the medium under both normal and high K+ conditions (Fig. 9C). The relative secretion of mature BDNF (defined as the ratio of secreted mature BDNF to cellular mature BDNF) remained unchanged (Fig. 9D). Thus, SPIG1 may downregulate BDNF maturation; therefore, mature BDNF secretion from neuronal cells was attenuated by its coexpression.

Effect of SPIG1 on BDNF expression in PC12 cells. A, Colocalization of SPIG1 and proBDNF in vesicle-like structures. Twelve hours after transfection with the indicated constructs, PC12 cells were differentiated with NGF as described in Materials and Methods. Exogenously expressed SPIG1 and BDNF were detected with anti-FLAG (green) and anti-proBDNF (red) antibodies, respectively. Nuclei were stained with DAPI (blue). The higher magnifications of the three boxed areas (a–c) are shown individually on the right side. Scale bars, 20 μm. B, Effect of the overexpression of SPIG1 on BDNF expression. PC12 cells were transfected with the indicated construct and then differentiated as in A. After 1 h of incubation with normal aCSF (6 mm K+) or high K+ (60 mm) aCSF, the amount of the indicated proteins in the cell extracts was analyzed by Western blotting. The applied protein amounts were verified by CBB staining (left panel). Signal intensities quantified by densitometry are shown as a percentage of control Fc-transfected cells. Data are mean ± SE of four independent experiments. *p < 0.05 in the same treatment (Student's t test). C, Effect of SPIG1 on mature BDNF secretion. BDNF secreted to the medium was concentrated as described in Materials and Methods and analyzed by Western blotting. Densitometry and data analyses were performed as in B. D, Relative secretion of mature BDNF, which was defined as the ratio between secreted mature BDNF (shown in C) and cellular mature BDNF (shown in B) under each condition. Data are mean ± SE of the experiments shown in B and C.
BDNF/TrkB signaling is increased in the hippocampus of SPIG1-KO mice
We previously generated SPIG1-KO mice by inserting the EGFP gene in frame into the SPIG1 gene (Yonehara et al., 2008). EGFP (i.e., SPIG1) expression was detected in the cornu ammonis (CA) region and dentate gyrus of the hippocampus (Yonehara et al., 2008). The marked colocalization of FLAG-tagged SPIG1 and proBDNF was again observed in vesicle-like structures when expressed exogenously in primary cultured hippocampal neurons (Fig. 10A).

Functional interactions between SPIG1 and BDNF in hippocampal cells. A, Colocalization of exogenous SPIG1 and BDNF. Hippocampal cells isolated from E18.5 mouse brains were electroporated with the indicated constructs and then cultured for 5 d. The expressed SPIG1 and proBDNF was detected with anti-FLAG (green) and anti-proBDNF (red) antibodies, respectively. Nuclei were stained with DAPI (blue). The higher magnifications of the boxed areas are shown in the bottom (a–c). Hippocampal cells incubated only with the secondary antibodies without the first antibodies did not show any immunofluorescent signals (data not shown). Scale bars, 20 μm. B, Golgi staining of the hippocampal tissues from WT and SPIG1-KO mice at postnatal day 10 (P10). Representative photomicrographs of CA1 pyramidal neurons. Scale bars, 40 μm. The number of dendritic filopodia (arrowheads) is shown as mean ± SE (n = 6 for each). *p < 0.01 (Student's t test). C, Analysis of the dendritic segments of CA1 pyramidal neurons. We analyzed spine numbers in Class I neurons with a single apical dendrite. Scale bars, 10 μm. Spine density and dendritic length are shown as mean ± SE (n = 6 for each). *p < 0.01 (Student's t test). D, Comparison of BDNF expression in the hippocampus of WT and SPIG1-KO mice at P14. The applied protein amounts were verified by CBB staining. The amounts of BDNF are presented by densitometric units normalized to the value for the WT. Data are mean ± SE (n = 14 for each). *p < 0.05 (Student's t test). E, Increased tyrosine phosphorylation of TrkB in the hippocampus of SPIG1-KO mice. Overall tyrosine phosphorylation patterns of the total protein and expression of TrkB in the hippocampus at P14 were examined by Western blotting using anti-phosphotyrosine (4G10) and anti-TrkB antibodies, respectively (left). The tyrosine phosphorylation of TrkB proteins immunoprecipitated with an anti-Trk antibody from the hippocampus was analyzed using 4G10 and anti-TrkB (right). Densitometric data are presented as a percentage of the WT control (bottom). Data are mean ± SE (WT, n = 6; SPIG1-KO, n = 8). *p < 0.05 (Student's t test).
Mature BDNF has been shown to increase spine density in the apical dendrites of CA1 pyramidal neurons (Tyler and Pozzo-Miller, 2003). We then examined whether SPIG1 was involved in dendritic spine formation in the mouse hippocampus by Golgi-Cox staining. The mean filopodia number and spine density in the apical dendrites of CA1 pyramidal neurons in SPIG1-KO mice were 2.2- and 1.4-fold higher than those in wild-type (WT) mice, respectively (Fig. 10B,C), notwithstanding that the dendritic length was similar between the two genotypes (Fig. 10C).
Western blot analyses showed that the amount of mature BDNF in the hippocampal tissue of SPIG1-KO mice was 1.3-fold higher, whereas that of proBDNF was 0.7-fold lower than that of WT mice (Fig. 10D). Immunoprecipitation experiments revealed that the tyrosine phosphorylation of TrkB, a receptor of BDNF, in the hippocampus was significantly higher in SPIG1-KO mice than in WT mice (Fig. 10E). These results strongly suggest that BDNF/TrkB signaling is upregulated in SPIG1-KO mice, resulting in an increased spine density in pyramidal neurons in their hippocampi.
Discussion
Recent studies have suggested that proBDNF and mature BDNF antagonistically regulate branch formation, although the regulatory mechanisms controlling BDNF maturation have not been fully elucidated. In the present study, we showed that SPIG1 negatively regulated the maturation of BDNF and thereby modulated BDNF/TrkB signaling. SPIG1-KD in chick RGCs in vivo showed defects in the regulated branch formation of dorsal RGC axons in the tectum; the number of branches was increased and the distribution of these branches expanded toward the anterior of the tectum. Moreover, ectopic arborization and synapse formation were observed. A primary culture of SPIG1-KD in RGCs showed that this increase in the branch number was suppressed by the addition of a neutralizing antibody to BDNF. The alteration in axonal branching by SPIG1-KD was consistently canceled when SPIG1 and BDNF were simultaneously knocked down in RGCs. SPIG1 and proBDNF colocalized in the axons of RGCs, including the growth cones. SPIG1 bound with proBDNF, and the exogenous expression of SPIG1 decreased the amount of mature BDNF in both the cells and culture medium in cell-based assays. Furthermore, mature BDNF levels were markedly increased and the tyrosine phosphorylation of TrkB was enhanced in the hippocampus of SPIG1-KO mice. These results suggest that SPIG1 negatively regulates BDNF maturation by binding to proBDNF and thereby suppresses axonal branching and spine formation. Therefore, the abnormal phenotypes induced by SPIG1-KD appeared to be a consequence of an increase in the secretion of mature BDNF in RGC axons.
Control mechanisms for retinotectal map formation by SPIG1
Topographical maps of neuronal connectivity occur in various brain regions. RGC axons have been shown to substantially overshoot their appropriate TZs along the AP axis of the optic tectum/SC during the development of the visual system in chicks and rodents. RGC axons from a given DV location are also broadly distributed along the DV (or mediolateral) tectal axis with a peak in axon density around the proper DV location of the TZ (Simon and O'Leary, 1992a, 1992b, 1992c; Hindges et al., 2002). Topographically appropriate connections are then established by selective branching formed along the axon shaft with a bias at the AP location of their future TZ (Simon and O'Leary, 1992a; Yates et al., 2001) and preferential extension toward the TZ along the DV axis (Nakamura and O'Leary, 1989; Hindges et al., 2002; McLaughlin et al., 2003a). Thus, the target position-specific branching of RGC axons on the tectum is essential to establish a topographic retinotectal map. However, the molecular mechanisms underlying this process have not been fully elucidated.
EphA receptor tyrosine kinases and their ligands, ephrin-As, have been proposed as modulators for the topographic branch formation of the RGC axon along the AP axis (Yates et al., 2001; Sakurai et al., 2002). Ephrin-As are glycosylphosphatidylinositol-anchored molecules that require transmembrane coreceptors to transduce their signals into cells. TrkB and neurotrophin receptor p75NTR have been proposed as coreceptors for ephrin-As (Lim et al., 2008; Marler et al., 2008). The mature BDNF ligand promotes an interaction between TrkB and ephrin-As, and this interaction increases the branching of RGC axons (Marler et al., 2008). On the other hand, the proBDNF ligand was shown to promote an interaction between p75NTR and ephrin-As, and inhibited the mature BDNF-induced branching of RGC axons (Marler et al., 2010). Consistent with these findings, aberrant anterior shifts in RGC axon terminations in the SC were observed in p75NTR-KO mice (Lim et al., 2008). These antagonistic actions of mature BDNF and proBDNF suggest that topographic branching is controlled not only by the EphAs/ephrin-As signal, but also by the balance between mature BDNF and proBDNF.
Together, the branching mechanisms of RGC axons on the tectum can be summarized as follows: EphA receptor signaling suppresses branching in the distal part of RGC axons by binding to ephrin-A ligands expressed in an anterior-low/posterior-high gradient in the tectum, whereas the signaling of ephrin-As, which are activated by EphAs expressed in an anterior-high/posterior-low gradient in the tectum, suppresses mature BDNF-mediated branching in the proximal part of RGC axons (Fig. 11, top). The phenotype in SPIG1-KD embryos, in which the distribution of the branches was shifted toward the anterior on the tectum, may have been caused by an increase in the ratio of mature BDNF to proBDNF: The signaling of mature BDNF/ephrin-A-TrkB was increased, whereas the signaling of proBDNF/ephrin-A-p75NTR in RGC axons was decreased. Therefore, RGC axons formed branches at more anterior positions in SPIG1-KD embryos (Fig. 11, bottom).

A mechanism model for the position-specific branching of RGC axons. Both ephrin-A/EphA signaling (light yellow) in the distal part and proBDNF/ephrin-A-p75NTR signaling (yellow) in the proximal part have been shown to mediate the suppression of branching for the axon (Marler et al., 2008). Branches are only formed in the region in which mBDNF/ephrin-A-TrkB signaling (blue line) predominates these two suppression signals (see Wild type). The ratio of mBDNF to proBDNF was increased in SPIG1-KD RGCs, and proBDNF/ephrin-A-p75NTR signaling was decreased. The region in which mBDNF/ephrin-A-TrkB signaling predominated proBDNF/ephrin-A-p75NTR signaling resultantly extended more in the anterior direction and slightly in the posterior direction. Therefore, axon branching occurred in a more anterior region in the tectum (see SPIG1-KD). mBDNF, Mature BDNF; A, anterior; P, posterior.
Ephrin-B/EphB and Wnt3/Ryk signaling pathways have been suggested to play a role in mapping along the DV (mediolateral) of the tectum/SC (Hindges et al., 2002; McLaughlin et al., 2003a; Schmitt et al., 2006). We demonstrated that the trajectory of dorsal RGC axons and branch distribution were shifted toward the dorsal direction in SPIG1-KD embryos (Fig. 2C,F). These phenotypes have been topographically matched with the expression pattern of SPIG1 along the DV axis in the retina, which suggests that SPIG1 is also involved in the formation of the retinotectal map along the DV (mediolateral) axis of the tectum. The trajectories of dorsal axons diverged to the dorsal direction in BDNF-KD chick embryos (data not shown). We speculate that SPIG1 may control retinotectal mapping along the DV axis by generating a BDNF gradient along the DV axis in the retina; however, the involvement of mature BDNF or proBDNF in this process has not yet been established.
Abnormality in SPIG1-mutant mice
In contrast to the robust defects observed in SPIG1-KD chick embryos, we could not detect any clear alterations in retinocollicular projections in SPIG1-KO mice (Yonehara et al., 2008; our unpublished data). Several reasons have been suggested, including: (1) species differences in developmental mechanisms, (2) the existence of SPIG2 (KC505335 for chicks and AF374460 for mice), (3) the acute inactivation via shRNA did not induce the compensatory mechanisms typically invoked after germline inactivation, and (4) shRNA mediated off-target effects on other genes involved in retinocollicular map formation.
Because the same defects were observed in chicks with two distinct shRNA constructs (shRNA#1 and shRNA#3) targeting different mRNA positions, the last possibility is small (data not shown). An increase in mature BDNF and a decrease in proBDNF were observed in the hippocampus of SPIG1-KO mice. Consistent with this finding, BDNF receptor TrkB phosphorylation was increased in the hippocampus of SPIG1-KO mice, which explains the increase in spine density in CA1 pyramidal neurons (Fig. 10C) (Tyler and Pozzo-Miller, 2003). These results indicate that SPIG1 negatively regulates BDNF/TrkB signaling and thereby controls spine density in CA1 pyramidal neurons.
SPIG1 is a negative regulator for BDNF maturation
BDNF production and secretion are regulated in neurons through multiple steps, including the use of distinct promoters, mRNAs, and protein transport systems, and processing of proBDNF to mature BDNF (Barker, 2009). Numerous chaperon proteins, including sortilin, carboxypeptidase E, and translin, have been shown to regulate the subcellular localization of BDNF mRNA and protein in neurons (Chen et al., 2005; Lou et al., 2005; Chiaruttini et al., 2009).
Sortilin was previously shown to traffic proBDNF to the regulated secretory pathway (Chen et al., 2005). Reductions in sortilin levels or the expression of a mutant sortilin lacking the cytoplasmic tail reduced BDNF secretion by the regulated secretory pathway (Chen et al., 2005). A recent study demonstrated that huntingtin-associated protein-1 (HAP1) bound to the prodomain of proBDNF, and also that proBDNF formed a complex with HAP1 and sortilin (Yang et al., 2011). These two proteins function to assist in not only trafficking to axons and sorting to the secretory pathway, but also protecting proBDNF from degradation. This complex was also shown to assist in the cleavage of proBDNF by furin to produce mature BDNF (Yang et al., 2011). Thus, complex formation is required for the intracellular stabilization of proBDNF and its proper processing to mature BDNF. The absence of sortilin expression in HEK293T and PC12 cells should also be considered as it may explain why we could not detect proBDNF in the culture medium (Nykjaer et al., 2004).
ProBDNF is known to be processed either intracellularly by furin or prohormone convertases (Edwards et al., 1988; Seidah et al., 1996) or extracellularly by plasmin (Pang et al., 2004) or metalloprotease MMP7 (Lee et al., 2001) at conserved cleavage sites with dibasic amino acids to yield mature BDNF. Previous studies demonstrated that sortilin bound to the prodomain of proBDNF and protected proBDNF from cleavage by the serine protease, plasmin (Teng et al., 2005). We investigated whether SPIG1 protected proBDNF from cleavage by furin or prohormone convertase 1 (PC1) in vitro using the recombinant SPIG1-Fc protein. However, we could not detect any protection of proBDNF from furin or PC1 (our unpublished data). This suggests that SPIG1 may be a member of the complex above to control proBDNF processing together with other members.
In addition, we speculate that SPIG1 may play some other extracellular roles because we detected the binding of SPIG1 to Trks (TrkA, TrkB, and TrkC), but not p75NTR by immunoprecipitation (our unpublished data). Therefore, further studies are required to fully understand the functions of SPIG1 in BDNF/TrkB signaling in the CNS. In summary, we demonstrated that SPIG1 played an important functional role in the formation of axonal branches and dendritic spines. SPIG1 intracellularly functioned as a negative regulator of BDNF maturation to reduce the ratio of mature BDNF to proBDNF in neurons. The results of the present study have provided a new insight into the mechanisms underlying the position-specific branching of axons.
Footnotes
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan Grants-in-Aid. We thank M. Goto and N. Nakanishi for technical assistance and A. Kodama for secretarial assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Masaharu Noda, Division of Molecular Neurobiology, National Institute for Basic Biology, 5-1 Higashiyama, Myodaiji-cho, Okazaki, Aichi 444-8787, Japan. madon{at}nibb.ac.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}