

Abstract
Although coordinated molecular signaling through excitatory and modulatory neurotransmissions is critical for the induction of immediate early genes (IEGs), which lead to effective changes in synaptic plasticity, the intracellular mechanisms responsible remain obscure. Here we measured the expression of IEGs and used bioluminescence imaging to visualize the expression of Bdnf when GPCRs, major neuromodulator receptors, were stimulated. Stimulation of pituitary adenylate cyclase-activating polypeptide (PACAP)-specific receptor (PAC1), a Gαs/q-protein-coupled GPCR, with PACAP selectively activated the calcineurin (CN) pathway that is controlled by calcium signals evoked via NMDAR. This signaling pathway then induced the expression of Bdnf and CN-dependent IEGs through the nuclear translocation of CREB-regulated transcriptional coactivator 1 (CRTC1). Intracerebroventricular injection of PACAP and intraperitoneal administration of MK801 in mice demonstrated that functional interactions between PAC1 and NMDAR induced the expression of Bdnf in the brain. Coactivation of NMDAR and PAC1 synergistically induced the expression of Bdnf attributable to selective activation of the CN pathway. This CN pathway-controlled expression of Bdnf was also induced by stimulating other Gαs- or Gαq-coupled GPCRs, such as dopamine D1, adrenaline β, CRF, and neurotensin receptors, either with their cognate agonists or by direct stimulation of the protein kinase A (PKA)/PKC pathway with chemical activators. Thus, the GPCR-induced expression of IEGs in coordination with NMDAR might occur via the selective activation of the CN/CRTC1/CREB pathway under simultaneous excitatory and modulatory synaptic transmissions in neurons if either the Gαs/adenylate cyclase/PKA or Gαq/PLC/PKC-mediated pathway is activated.
Introduction
An increasing number of studies have demonstrated that the coincident integration of dopamine- and glutamate-coded signals at the cellular level in many regions of the cortex, limbic system, and basal ganglia in the brain play a pivotal role in the effective formation of motivation and memory (Berke and Hyman, 2000; Kelley, 2004). This has been demonstrated mainly by pharmacological experiments using animals, in which either the blockade of NMDAR or dopamine D1R impaired the acquisition of reward learning or drug addiction (Wolf and Khansa, 1991; Crawford et al., 1997; Baldwin et al., 2002; Kelley, 2004). Moreover, coordinated molecular signaling evoked via NMDAR and D1R pathways has been identified as a critical cellular event in the induction of immediate early genes (IEGs; Young et al., 1991; Kiba and Jayaraman, 1994; Liu et al., 1994; Granado et al., 2008). Over the past two decades, many intracellular signaling factors, such as Ca2+ and cAMP signals, multiple signaling pathways, including protein kinase A (PKA), PKC, CaMKs, and ERK/MAPK, and the transcription factor CREB have been implicated in the control of IEG expression (Kelley, 2004). Evidence indicating that the activity of NMDAR may be potentiated by stimulating GPCRs or the PKA pathway has been reported previously (Konradi et al., 1996; Raman et al., 1996; Skeberdis et al., 2006), which suggests that an enhancement in the excitatory activity of NMDAR is mediated through the modulatory effects of GPCRs.
Pituitary adenylate cyclase-activating polypeptide (PACAP) is a member of the VIP/secretin/glucagon family (Miyata et al., 1989) and acts on three types of GPCRs: PACAP-specific receptor (PAC1), VIP/PACAP receptor subtype 1 (VPAC1), and VPAC2 (Vaudry et al., 2009). PACAP is involved in the control of many neurological functions, such as synaptic plasticity and emotional behavior (Tanaka et al., 2006; Vaudry et al., 2009). Electrophysiological methods demonstrated that the stimulation of PAC1 with PACAP enhanced the response of NMDAR through signaling pathways, including Gαs/adenylate cyclase (AC)/PKA (Yaka et al., 2003a) and Gαq/PLC/PKC (Macdonald et al., 2005), both of which facilitate synaptic function in the CA1 region of the hippocampus. In addition, the involvement of NMDAR in the PACAP-induced expression of c-fos and Bdnf (Martin et al., 1995; Pellegri et al., 1998) has been suggested already. However, it remains unclear how intracellular signals evoked via NMDAR and PAC1 converge to induce the expression of IEGs.
In the present study, we used PACAP as a ligand to stimulate GPCRs and evaluated the mRNA expression of IEGs, particularly focusing on the Bdnf gene, in cultured rat cortical cells. We also examined the expression of Bdnf in mouse brain after intracerebroventricular injection of PACAP. Furthermore, we developed a bioluminescence imaging system with cultured cells prepared from a novel transgenic (Tg) mouse strain, in which a firefly luciferase (Luc) gene was introduced into the coding region of Bdnf, and examined transcription mechanisms of PACAP-induced Bdnf and IEG expression. Here, we demonstrated the novel GPCR-mediated induction of IEGs, which might be mediated by a common intracellular signaling mechanism underlying coordinated excitatory and modulatory synaptic transmissions in neurons.
Materials and Methods
Reagents.
PACAP38, PACAP27, CRF, and neurotensin were purchased from the Peptide Institute. dl-APV, CNQX, MK801, NMDA, forskolin, 12-o-tetradecanoylphorbol-13-acetate (TPA), FK506 [(3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy-3-[(1E)-2-[(1R,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propen-1-yl)-15,19-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)tetrone], and (R)-(+)-SKF38393 (2,3,4,5-tetrahydro-7,8-dihydroxy-1-phenyl-1H-3-benazepine HCl) were purchased from Sigma. Guanosine-5′-O-(2-thiodiphosphate) (GDPβS) and H89 (N-[2-(p-bromo-cinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl) were purchased from Biomol. Bisindolylmaleimide I (BisI), U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene], isoproterenol, STO609 [7H-benzimidazo(2,1-a)benz(de)isoquinoline-7-one-3-carboxylic acid], and KN93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine) were from Calbiochem.
Reporter plasmids.
pGL4.12–Bdnf-PIV was generated by inserting rat Bdnf promoter IV (Bdnf-PIV; from −629 to +281; Tabuchi et al., 2002) into the HindIII/XhoI sites of pGL4.12–luc2CP (Promega). The pGL4.12–Bdnf-PI and pGL4.12—c-fos promoter was constructed by inserting rat Bdnf-PI (from −528 to +138; Tabuchi et al., 2002) and the human c-fos promoter (from −404 to +41; Tabuchi et al., 1999) into the HindIII site of pGL4.12–luc2CP, respectively. Ca2+ signal-responsive element 1,2,3 (CaRE1,2,3) and CaRE3-mutated Bdnf-PIV (CaRE1-mutated, 5′-acATTTCGcG-3′; CaRE2-mutated, 5′-ATagTAgaAC-3′; CaRE3-mutated, 5′-TacCGTac-3′; lowercase characters denote mutated nucleotides) were generated with the QuikChange Site-Directed Mutagenesis kit (Stratagene). pGL4.11–Arc7000 was donated by Dr. H. Bito (University of Tokyo, Bunkyo-ku, Japan) (Kawashima et al., 2009).
Antibodies and antiserum.
An anti-MAP2 antibody (1:1000) was purchased from Sigma-Aldrich, anti-PAC1 antibody (1:50) from Santa Cruz Biotechnology, and anti-NMDAR1 antibody (1:300) from BD Biosceinces Pharmingen. Anti-CREB-regulated transcriptional coactivator 1 (CRTC1) antiserum (1:1000) was donated by Dr. H. Takemori (National Institute of Biomedical Innovation, Ibaraki, Japan).
Primary cultures of rat cortical cells.
A primary culture of rat cortical neuronal cells was prepared from the cerebral cortices of 17-d-old Sprague Dawley rat embryos (Japan SLC) as described previously (Tabuchi et al., 2002; Fukuchi et al., 2015). The dissociated cells were seeded at 1.8 × 106 cells in 35 mm culture dishes (for the quantitative RT-PCR analysis; Asahi Techno Glass) or 5 × 106 cells in 60 mm culture dishes [for promoter analysis, GeneChip, and chromatin immunoprecipitation (ChIP) assay; Asahi Techno Glass]. DNA transfection was performed at 3 d in culture, and GeneChip, quantitative RT-PCR, immunostaining, and ChIP assays were performed at 5 d in culture.
The dissociated cells in Figure 3, D and E, were suspended in Neurobasal medium containing 1× B27 supplement (Invitrogen), 2 μg/ml gentamicin, and 2 mm glutamine and seeded at 1.8 × 106 cells in 35 mm culture dishes (Asahi Techno Glass) coated with poly-l-lysine. Half of the conditioned medium was exchanged for fresh medium every 3 d. The experiment was performed on 5 or 14 DIV.
All animal care and experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of the University of Toyama (Authorizations S-2008 PHA-5, S-2009 PHA-24, S-2010 PHA-3, A2011PHA-1, A2012PHA-2, and A2013PHA-3).
Quantitative RT-PCR analysis.
Total cellular RNA was extracted by the acid guanidine phenol-chloroform method using TRIsure (Bioline), and 1 μg of RNA was reverse transcribed into cDNA using SuperScript II reverse transcriptase (Invitrogen), as described previously (Fukuchi et al., 2004, 2014, 2015). Quantitative PCR amplification was performed using the Stratagene Mx3000p Real-Time PCR system and Brilliant SYBR Green QPCR Master Mix (Stratagene), as described previously (Fukuchi et al., 2004, 2014, 2015). The thermal profile for PCR included an initial denaturation at 95°C for 10 min, followed by 45 cycles of denaturation at 95°C for 45 s, annealing at 57°C for 45 s, and extension at 72°C for 1 min. Standard curves were generated for each gene using a plasmid dilution series containing the target sequences. The threshold cycle for each sample was taken from the linear range and converted to the starting amount by interpolation from the standard curve. The expression of each mRNA was normalized respective to the level of Gapdh mRNA. The primer sequences were as follows: Gapdh, 5′-ATCGTGGAAGGGCTCATGAC-3′ and 5′-TAGCCCAGGATGCCCTTTAGT-3′; activity-regulated cytoskeleton-associated protein (Arc), 5′-CGCTGGAAGAAGTCCATCAA-3′ and 5′-GGGCTAACAGTGTAGTCGTA-3′; exon IV-containing Bdnf mRNA (Bdnf-eIV), 5′-TCGGCCACCAAAGACTCG-3′ and 5′-GCCCATTCACGCTCTCCA-3′; Bdnf-eI, 5′-GACACATTACCTTCCAGCATC-3′ and 5′-GCCCATTCACGCTCTCCA-3′; c-fos, 5′-GTTTCAACGCGGACTACGAG-3′ and 5′-AGCGTATCTGTCAGCTCCCT-3′; early growth response 2 (Egr2), 5′-ATCCCAGTAACTCTCAGTGG-3′ and 5′-TGATGATGCCTTCTGGGTAG-3′; and dual specificity phosphatase 5 (Dusp5), 5′-AGGATCCCATGGAAGCTGTTG-3′ and 5′-CAGGGTAGGGAGGGAAACATT-3′.
RT-PCR analysis of mouse cerebral cortex.
Mice (C57BL/6N, male, 7 weeks old; Japan SLC) were administered an intracerebroventricular injection of PACAP27 dissolved in saline under anesthesia. Both PACAP27 and PACAP38 bind to PACAP/VIP receptors (Vaudry et al., 2009). MK801 (1 mg/kg body weight) was administered intraperitoneally to mice 30 min before the injection of PACAP27. Three hours after the injection, the brain was rapidly removed after decapitation under deep anesthesia with pentobarbital (100 mg/kg, i.p.), and the whole cerebral cortex was dissected out and frozen. Animal care and experimental protocols for these experiments were approved by the Animal Experiment Committee of the University of Toyama (Authorizations A2011PHA-18 and A2014PHA-1) and were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the University of Toyama. Total RNA was isolated using TRIsure (Bioline). Overall, 1 μg of RNA was reverse transcribed into cDNA using SuperScript II reverse transcriptase (Invitrogen). Semiquantitative real-time PCR was performed using the Stratagene Mx3000p Real-Time PCR system with SYBR Select Master Mix (Life Technologies). The thermal profile for PCR included UDG activation at 50°C for 2 min and Taq activation at 95°C for 2 min, followed by 45 cycles of denaturation at 95°C for 15 s, annealing at 57°C for 15 s, and extension at 72°C for 1 min. The primer sequences were as follows: mouse Gapdh, 5′-GCACAGTCAAGGCCGAGAA-3′ and 5′-CTTCTCCATGGTGGTGAAGAC-3′; mouse Bdnf, 5′-AAGGACGCGGACTTGTACAC-3′ and 5′-CGCTAATACTGTCACACACGC-3′; and mouse Dusp5, 5′-CTGTTGGTGGAAGGAGAAGC-3′ and 5′-GGAGGGAAACATTGTCACAATGG-3′.
Microarray analysis.
Total cellular RNA was extracted from cultured rat cortical cells using the RNeasy Total RNA Extraction kit (Qiagen). Gene expression was analyzed using a GeneChip system with a Rat Genome 230 2.0 Array (Affymetrix) and GeneChip 3′IVT Express kit (Affymetrix). We used three arrays for each sample tested in this study. Total RNA (500 ng) was used to synthesize double-stranded cDNA, and biotin-labeled cRNA was then synthesized. After fragmentation, biotinylated cRNA was hybridized to arrays at 45°C for 16 h. The arrays were washed, stained with streptavidin–phycoerythrin, and scanned with a probe array scanner. The scanned chip was analyzed using the GeneChip Analysis Suite (Affymetrix). Data were further analyzed using GeneSpring software (Silicon Genetics).
Generation of Bdnf–Luc Tg mice.
Animal care and experimental protocols for Tg mouse generation were approved by the Animal Experiment Committee of the University of Toyama (Authorizations S-2010 MED-51 and A2011PHA-18) and were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the University of Toyama.
A bacterial artificial chromosome (BAC) genomic clone (RP24-310A6) originating from the DNA of C57BL/6 mice and containing Bdnf was obtained from the BACPAC Resource Center CHORI. The counter selection BAC modification kit (Gene Bridges) was used to construct a Tg vector. The nucleotide sequence of the mouse genome was obtained from the National Center for Biotechnology Information (NCBI Map Viewer, Mus musculus Build 36.1), the translation start site in Bdnf (the A of ATG) located in exon IX referred to position +1, and the preceding residues were indicated by negative numbers in this study.
We used PCR to increase the stability of the Tg Luc gene and introduced a small-t intron sequence derived from SV40 between amino acids 414 and 415 of the firefly Luc coding sequence from pGL4.13 (Promega) to yield the plasmid piLuc. The Luc DNA fragment containing the intron (iLuc) was ligated with a self-excision-type Cre- and Neo-resistant gene expression vector flanked with loxP sequences derived from pACN (Bunting et al., 1999) to yield the plasmid piLucACN. The iLucACN DNA fragment was introduced into position +1 in Bdnf of PR24-310A6 with BAC homologous recombination to yield pTgBAC–Bdnf–iLuc.
After the purification of BAC DNA using a DNA Large-Construct kit (Qiagen), pTgBAC–Bdnf–iLuc was linearized by NotI digestion, extracted with phenol and chloroform, and precipitated with ethanol. Purified BAC DNA (10 μg) was electroporated into the ES cell line RENKA, derived from C57BL/6N mice (Fukaya et al., 2006) as described previously (Miya et al., 2008). After selection with G418, recombinant ES clones were identified by PCR with the primers BeIX-U (5′-CCACATGCTGTCCCCGAGAA-3′), BeIX-L (5′-CGCCTTCATGCAACCGAAGT-3′), and LucIX-L (5′-CTCGAAGTACTCGGCGTAGGT-3′). The resulting PCR amplified the DNA fragments of 142 and 261 bp derived from wild-type and Tg loci, respectively. Tg ES clones were injected into the eight-cell stage embryos of mouse strain CD-1. These embryos were cultured to blastocysts and transferred to pseudopregnant CD-1 mouse uteri. The resulting male chimeric mice were crossed with female C57BL/6 mice. We established two independent Tg mouse lines (lines 2 and 9).
Bioluminescence signal imaging.
Primary cultures of Bdnf–Luc mouse cortical cells were prepared from the cerebral cortices of 16.5-d-old Bdnf–Luc Tg mouse embryos. Dissociated cells were suspended in Neurobasal medium containing 1× B27 supplement (Invitrogen), 2 μg/ml gentamicin, and 2 mm glutamine and were then seeded at 1.8 × 106 cells in 35 mm glass-base dishes (Asahi Techno Glass) coated with poly-l-lysine. Half of the conditioned medium was exchanged for fresh medium every 3 d. Luciferin was added to the cells before bioluminescence signal imaging. The cells were treated with PACAP or other reagents and immediately set on the stage of an LV200 microscopic image analyzer (Olympus). Changes in the bioluminescence signal were analyzed using MetaMorph software (Molecular Devices).
DNA transfection and Luc assay.
DNA transfection was performed using calcium phosphate/DNA precipitation as described previously (Tabuchi et al., 2002; Fukuchi et al., 2004, 2015). Briefly, calcium phosphate/DNA precipitates were prepared by mixing 100 μl of plasmid DNA (6 μg, firefly Luc/Renilla Luc at 10:1) in a 250 mm CaCl2 solution with an equal volume of 2× HEPES-buffered saline (50 mm HEPES-NaOH, pH 7.05, 280 mm NaCl, and 1.5 mm Na2HPO4) and then added to each culture dish. In the experiment using dominant-negative CREB, 5.5 μg of the reporter plasmid (F-luc/R-luc at 10:1) and 0.5 μg of the expression vector [empty vector, wild-type CREB (W-CREB), dominant-negative CREB [acidic CREB (A-CREB)], or mutated CREB (M-CREB)] were cotransfected. In the experiment using shCRTC1, 3 μg of the reporter plasmid and 3 μg of the expression vector (Scramble or shCRTC1) were cotransfected. Each dish was washed twice with PBS, and fresh medium was added. Each experiment was performed 40 h after DNA transfection, except for those using shCRTC1 that were performed 72 h after DNA transfection. The knockdown of CRTC1 affected Renilla Luc expression because of an unexpected effect of the knockdown. However, a significant change was not observed in firefly Luc activity in each sample under the same conditions, which indicated that the efficacy of DNA transfection did not vary greatly; therefore, the normalization of efficacy was not required. We examined promoter activity by measuring firefly Luc activity without normalization (see Fig. 6E). Each experiment was performed after 40 or 72 h as indicated.
Knockdown of CRTC1.
The pSUPER RNAi System (OligoEngine Platform) was used to knockdown the endogenous expression of CRTC1 according to the instructions of the manufacturer. Oligonucleotides were annealed and subcloned into the HindIII/BglII sites of the vector pSUPER, as described previously (Fukuchi et al., 2014). We evaluated the knockdown efficiency using NIH3T3 cells (overexpressing CRTC1) and rat cortical cells (expressing endogenous CRTC1) as described previously (Fukuchi et al., 2014).
Immunostaining.
Cells were seeded on poly-l-lysine-coated coverslips. At 5 d in culture, cortical cells were fixed in PBS containing 4% formaldehyde and 4% sucrose for 15 min at room temperature and then treated with blocking PBS containing 3% bovine serum albumin and 3% normal goat serum for 1 h at room temperature. After the blocking procedure, the cells were incubated with a primary antibody against PAC1, NMDAR1, MAP2, or anti-CRTC1 antiserum. After being washed in PBS, these cells were incubated with a CF488- or CF594-conjugated secondary antibody against rabbit or mouse IgG (Biotium). Nuclei were counterstained with 300 nm DAPI (Invitrogen). After another wash, coverslips were mounted on slides with Fluoromount (Diagnostic BioSystems). Confocal fluorescent images were taken with an LSM 700 confocal microscope (Carl Zeiss). The signal intensities of CRTC1 in the cytoplasm and nucleus were measured using ZEN 2009 software (Carl Zeiss) with reference to those of MAP2 (cytoplasm) and DAPI (nucleus), and the immunofluorescence intensity ratio of the nucleus to the cytoplasm was estimated. Overall, 30 neurons from six independent experiments were analyzed.
ChIP assay.
ChIP assays were performed using the Magna ChIP A kit (Millipore), according to the instructions of the manufacturer, as described previously (Fukuchi et al., 2015). The crosslinked DNA was diluted and mixed with protein A magnetic beads and anti-CRTC1 antiserum overnight at 4°C while undergoing rotation. Purified DNA was amplified using KOD FX DNA polymerase (Toyobo). The primer sequences were as follows: Bdnf promoter IV CRE, 5′-CTATTTCGAGGCAGAGGAGG-3′ and 5′-GTGGGAGTCCACGAGAG-3′; Bdnf promoter I CRE, 5′-GGGGCACGAACTTTTCTAAGAAG-3′ and 5′-GCAGCCTCTCTGAGCCA-3′; Arc promoter CRE, 5′-AGAGCCTTCCTGCGTGG-3′ and 5′-CGGCACCATAAAAGGAGAGAG-3′; c-fos promoter CRE, 5′-GGGAGCGCTTTCCCC-3′ and 5′-GCCGCTTTATAGAAGCGCTTG-3′; and Dusp5 promoter CRE (Breitwieser et al., 2007), 5′-CCATATTTGGCCTTATATGGGCAC-3′ and 5′-AAAGCTGGGGATTCCGCC-3′. The PCR product was separated on a 2% agarose gel. The quantities of genomic DNA were also measured by real-time PCR (Stratagene) with the SYBR Select Master Mix (Life Technologies) according to the instructions of the manufacturer.
Statistical analyses.
All values represent the mean ± SEM for separate experiments performed in duplicate. Statistical analyses were performed using a one-way ANOVA with Sheffé's F test. Statistical analysis was performed in Figure 2B using Pearson's correlation coefficient test.
Results
PACAP-induced expression of IEGs differs by NMDAR dependency
We compared the relative levels of NMDAR dependency for PACAP-induced mRNA expression of IEGs, such as Bdnf, Arc, c-fos, Egr2, and Dusp5. The stimulation of cortical cells with PACAP at 5 DIV induced the mRNA expression of all IEGs examined (Fig. 1A). Although increased mRNA expression levels were reduced by APV, a specific and competitive antagonist of NMDAR, the extent of the reduction varied among transcripts [mRNA reduction almost 100% for Bdnf-eIV, nearly 80% for c-fos and Arc, and ∼40% for Egr2; Fig. 1A]. In contrast, APV did not affect the PACAP-induced mRNA expression of Dusp5 (Fig. 1A), which was chosen as a representative NMDAR-independent gene for microarray analysis (Fig. 2B). The basal expression of Bdnf-eIV mRNA was reduced by APV (Fig. 1A; see Figs. 7B, 8A,C), suggesting that NMDAR might be activated in response to endogenously secreted glutamate and contribute to the basal expression of Bdnf-eIV mRNA.

Involvement of NMDAR in PACAP-induced expression of IEGs. A, Cortical cells were treated with 100 nm PACAP38 for 1 h at 5 DIV. APV (200 μm) was added 10 min before PACAP treatment. Total RNA was extracted, and changes in IEG expression were investigated by quantitative RT-PCR. Data represent the mean ± SE (n = 3–4). **p < 0.01 versus control (samples without PACAP); ##p < 0.01 versus the same sample without APV. B, Effects of CNQX on PACAP-induced expression of Bdnf-eIV mRNA. CNQX (10 μm) was added 10 min before cells were treated with PACAP. Data represent the mean ± SE (n = 3). **p < 0.01 versus control. C, Effects of GDPβS, H89, BisI, or H89 plus BisI on PACAP-induced Bdnf-eIV mRNA expression. GDPβS (100 μm), H89 (10 μm), BisI (5 μm), or H89 plus BisI was added 10 min before PACAP treatment. Data represent the mean ± SE (n = 3–8). **p < 0.01 versus control; ##p < 0.01 versus the same sample without inhibitors. D, Cells were fixed at 5 DIV and immunostained with anti-PAC1 (green) and anti-GluN1 (magenta) antibodies. Nuclei were counterstained with DAPI (blue). Images were captured by confocal microscopy (LSM 700). Scale bars, 20 μm. E, PACAP27 (50, 250, and 500 pmol) or saline (0 pmol) was injected intracerebroventricularly into mice, and, 3 h after injection, total RNA was prepared from mouse cerebral cortices. Changes in the expression of Bdnf and Dusp5 were investigated by semiquantitative RT-PCR. Data represent the mean ± SE (n = 3–4). *p < 0.05 and **p < 0.01 versus controls. F, Effects of MK801 on PACAP-induced mRNA expression. MK801 (1 mg/kg body weight) was administered intraperitoneally to mice 30 min before intracerebroventricular injection of PACAP. Three hours after PACAP injection, total RNA was prepared from mouse cerebral cortices, and changes in the expression of Bdnf and Dusp5 were investigated by semiquantitative RT-PCR. Data represent the mean ± SE (n = 4–5). *p < 0.05 and **p < 0.01 versus controls; ##p < 0.01 versus the same sample without MK801. NS, Not significant.

Involvement of the CN pathway in PACAP-induced expression of IEGs. A, Effects of FK506 on PACAP-induced mRNA expression. FK506 (5 μm) was added 10 min before PACAP treatment. Data represent the mean ± SE (n = 8–14). **p < 0.01 versus control; #p < 0.05 and ##p < 0.01 versus the same sample without FK506. B, GeneChip analysis of PACAP-induced transcripts affected by APV or FK506. Cortical cells were treated with PACAP for 1 h at 5 DIV. APV or FK506 was added 10 min before PACAP treatment. Total RNA was extracted, and GeneChip analysis was performed. The expression of 273 transcripts was upregulated significantly by >1.5-fold. The expression level of each transcript in the presence of APV or FK506 was calculated (left) and plotted to examine the relationship between NMDAR and CN dependencies using a correlation coefficient test (right). Statistical analysis was performed using Pearson's correlation coefficient test. C–F, Cortical cells were treated with 100 μm NMDA for 3 h (C, F) or PACAP for 1 h (D, E) at 5 DIV. APV, FK506, U0126 (20 μm), KN93 (10 μm), STO609 (5 μm), or H89 plus BisI was added 10 min before the addition of NMDA or PACAP. Total RNA was extracted, and changes in mRNA expression of Bdnf-eIV and Dusp5 were examined by quantitative RT-PCR. Data represent the mean ± SE (n = 3–4). **p < 0.01 versus control; #p < 0.05 and ##p < 0.01 versus the same sample without inhibitor.
Rat Bdnf consists of eight 5′-untranslated exons and one 3′ exon encoding the protein preproBDNF, whose gene transcription is regulated by alternative promoters located upstream of each exon (Aid et al., 2007). We here focused on the mRNA expression of Bdnf-eIV, which is known to be abundantly expressed in the nervous systems and controlled in an activity-dependent manner (Tao et al., 1998, 2002; Aid et al., 2007). APV had a marked inhibitory effect on the PACAP-induced expression of Bdnf-eIV mRNA. The induction was completely inhibited by the addition of 200 μm APV (Fig. 1A), as well as lower concentrations (12.5, 25, 50, and 100 μm; our unpublished observations). CNQX, a specific antagonist of AMPAR, did not significantly reduce Bdnf-eIV mRNA induction (Fig. 1B). By focusing on this complete dependency of Bdnf-eIV mRNA expression on activation of the NMDAR, we examined whether the Gαs/AC/PKA and Gαq/PLC/PKC pathways between PAC1 and NMDAR were involved, as demonstrated previously by electrophysiological analyses (Yaka et al., 2003a; Macdonald et al., 2005). GDPβS, a competitive inhibitor for the GTP-binding site on the α subunit of G-proteins, significantly reduced the PACAP-induced increase in Bdnf-eIV mRNA expression (Fig. 1C). Although Bdnf-eIV mRNA expression was reduced partially by either H89 or BisI (Fig. 1C), potent inhibitors of PKA and PKC, respectively, the combined addition of these inhibitors almost totally reduced this increase (Fig. 1C). This suggested the contribution of both the Gαs/AC/PKA and Gαq/PLC/PKC pathways from PAC1 for the induction of Bdnf-eIV mRNA via NMDAR. Furthermore, EGTA, an extracellular Ca2+ chelator, completely prevented PACAP-induced Bdnf-eIV mRNA expression, indicating that Ca2+ influx into neurons via NMDAR was critical for the induced expression of Bdnf-eIV mRNA (our unpublished observations).
Most anti-PAC1 antibody-positive cells were also positively stained by the anti-GluN1 antibody (Fig. 1D), suggesting the coexpression of PAC1 and NMDAR in the same neurons. VIP did not induce a significant increase in Bdnf-eIV mRNA expression even at 100 nm (our unpublished observations). This indicated that PAC1, but not VPAC1/2, was one of the main receptors responsible for PACAP-induced Bdnf-eIV mRNA expression because PAC1 was shown previously to bind to PACAP with an affinity ∼1000-fold higher than that of VIP and VPAC1/2 bound to PACAP and VIP with an equal affinity (Vaudry et al., 2009).
Functional interactions between PAC1 and NMDAR induce the expression of Bdnf in the brain
To further examine whether PACAP-induced Bdnf mRNA expression through NMDAR could be detected in the brain, we injected PACAP27 intracerebroventricularly into the brain of mice and then dissected out the whole cerebral cortex to examine changes in the expression of Bdnf mRNA. As shown in Figure 1E, we found that the expression of Bdnf mRNA in the cerebral cortex of mice was significantly induced by intracerebroventricular injection of PACAP and was dose dependent. The expression of Dusp5 mRNA in the cerebral cortex of mice was also increased by PACAP (Fig. 1E); however, its basal expression level was markedly lower than that of Bdnf mRNA (our unpublished observations). Corresponding to changes in the expression of Bdnf and Dusp5 mRNA in cultured cortical cells (Fig. 1A), the PACAP-induced mRNA expression of Bdnf, but not Dusp5, was inhibited significantly by the intraperitoneal administration of MK801 (Fig. 1F), a specific antagonist for NMDAR. This indicated the involvement of NMDAR in the expression of Bdnf mRNA induced by PACAP in the brain. We confirmed that the effect of PACAP38 on the expression of Bdnf mRNA was similar to that of PACAP27 both in vitro and in vivo (our unpublished observation). This is consistent with previous reports that PACAP38 and PACAP27 are both potent agonists for PACAP/VIP receptors (Vaudry et al., 2009).
Relationship between NMDAR and calcineurin dependency on PACAP-induced expression of IEGs
We further investigated the intracellular mechanisms underlying the PACAP-induced expression of IEGs using cultured cortical cells. The CN pathway was shown previously by reporter assay to be responsible for the activation of CRE-dependent transcription induced by PACAP (Baxter et al., 2011). Therefore, we focused on the CN pathway and compared the levels of dependency on the PACAP-induced expression of IEGs. FK506, a potent inhibitor of CN, abolished the PACAP-induced expression of Bdnf-eIV mRNA (Fig. 2A). In contrast, the PACAP-induced increase in Arc or c-fos mRNA expression was only partially reduced by FK506, whereas that of Egr2 or Dusp5 was slightly enhanced (Fig. 2A). The same inhibitory effect was observed when cells were treated with cyclosporin A, another potent inhibitor of CN (our unpublished observations).
The inhibitory effect of APV on the PACAP-induced expression of IEGs correlated with that of FK506 (Figs. 1A, 2A). To confirm this correlation, we performed a microarray analysis. PACAP significantly increased the expression of ∼270 gene transcripts, whereas the effects of APV or FK506 varied among these transcripts (Fig. 2B). The results of the microarray analysis indicated that the transcripts, the induction of which was mostly prevented by APV, were also mostly prevented by FK506, whereas APV-insensitive transcripts were not (Fig. 2B). The correlation coefficient test revealed a strong correlation (r = 0.785, p < 0.001) between the effects of APV and FK506 on the PACAP-induced expression of IEGs (Fig. 2B).
In contrast to the PACAP-induced expression of Bdnf-eIV mRNA, which was abolished completely by FK506, NMDA-induced expression was reduced partially by FK506, U0126 (an MEK1/2 inhibitor), STO609 (a CaMK kinase inhibitor; Tokumitsu et al., 2002), and KN93 (an inhibitor of the CaMK pathway at high concentrations; Redondo et al., 2010; Fig. 2C). However, the PACAP-induced expression of Bdnf-eIV mRNA was not reduced by U0126, KN93, or STO609 (Fig. 2D), indicating the different usage of Ca2+ signaling pathways between NMDA- and PACAP-induced expression of Bdnf-eIV mRNA, namely, the selective usage of the CN pathway for the PACAP-induced expression of Bdnf mRNA.
To investigate differences in intracellular mechanisms between the expression of NMDAR/CN pathway-dependent and -independent genes, we focused on the expression of Dusp5 mRNA. The PACAP-induced expression of Dusp5 mRNA, which was independent of the NMDAR and CN pathways (Figs. 1A, 2A), was suppressed by combined addition of H89 and BisI and partially by U0126 (Fig. 2E). Thus, the induction of Dusp5 was controlled directly by the PKA and PKC pathways evoked via Gαs/AC and Gαq/PLC and possibly by the ERK/MAPK pathway via a cAMP-dependent pathway (Obara et al., 2007; Emery et al., 2013). Notably, the expression of Dusp5 mRNA was induced significantly by the direct activation of NMDAR with NMDA (Fig. 2F), and its induction was inhibited significantly by APV, U0126, STO609, or KN93 (Fig. 2F). However, FK506 did not inhibit the NMDA-induced Dusp5 expression (Fig. 2F), similar to that for PACAP-induced Dusp5 expression (Fig. 1A). This indicated that NMDAR-derived Ca2+ signaling pathways except CN are responsible for controlling NMDA-induced Dusp5 mRNA expression.
Bioluminescence imaging of PACAP-induced Bdnf expression
To further examine the PACAP-induced expression of Bdnf, we established a bioluminescence imaging system to visualize the specific PACAP-induced expression of Bdnf in living cultured cells by generating a novel Tg mouse strain, referred to as Bdnf–Luc Tg mice. Using a BAC clone containing the entire mouse Bdnf, we constructed Tg mice containing the firefly Luc gene introduced into the translation initiation site of Bdnf (Fig. 3A). We prepared a primary culture of cortical cells from Tg mice and determined whether bioluminescence signals in individual cells changed with PAC1 stimulation using a microscope-based bioluminescence signal imaging system (LV200; Olympus). Although we detected an increase in the bioluminescence signal with PAC1 stimulation at 5 DIV, a small number of cultured cells with strong signals were detected (Fig. 3B,C). However, the number of cells with strong bioluminescence signals increased greatly when PACAP was added at 14 DIV (Fig. 3B,C). Consistent with this result, the level of Bdnf-eIV mRNA expression induced by PACAP at 14 DIV was greater than that at 5 DIV (Fig. 3D). The increase in bioluminescence signals was inhibited completely by either APV or FK506 at DIV 5 and completely by APV or almost completely by FK506 at DIV14 (Fig. 3C), which corresponded to changes in Bdnf-eIV mRNA expression [Figs. 1A, 2A (at 5 DIV) and 3E (at 14 DIV)]. Although alternative promoters located upstream of all exons of Bdnf (Aid et al., 2007; Fig. 3A) were included in the BAC clone used here, the increase in bioluminescence signals was always detected depending on the NMDAR and CN pathways. The correlation of changes in Bdnf-eIV mRNA expression and bioluminescence signals indicated that Bdnf-eIV transcripts likely contribute to the generation of bioluminescence signals in neurons prepared from Bdnf–Luc Tg mice.

Bioluminescence imaging of PACAP-induced Bdnf expression. A, The generation of Bdnf–Luc Tg mice. Using the BAC clone, the Luc gene was inserted at the translation start site of mouse Bdnf. B, C, Representative images of bioluminescence signals after the addition of PACAP at 5 or 14 DIV (B, left) and sequential changes in the signal after its addition [B, right (changes in the signal in each cell), C (the mean signal value), n = 50 cells]. Cultured cells were prepared from Bdnf–Luc Tg mouse embryos and treated with PACAP at 5 or 14 DIV. Changes in bioluminescence signals were examined by time-lapse signal imaging (LV200; Olympus). Luciferin was added to the culture at a final concentration of 0.5 mm before measurements. Bioluminescence signals were measured every 10 min (exposure time, 5 min) for 11 h. APV or FK506 was added 10 min before PACAP treatment. Scale bars, 200 μm. D, At 5 or 14 DIV, cortical cells were treated with PACAP for 1 h. Changes in Bdnf-eIV mRNA expression were investigated by quantitative RT-PCR. Data represent the mean ± SE (n = 3). *p < 0.05 and **p < 0.01 versus control. E, Effect of APV or FK506 on PACAP-induced expression of Bdnf-eIV mRNA in cultured rat cortical cells at 14 DIV. APV or FK506 was added 10 min before PACAP treatment. Total RNA was extracted, and changes in Bdnf-eIV mRNA expression were investigated by quantitative RT-PCR. Data represent the mean ± SE (n = 3). **p < 0.01 versus control; ##p < 0.01 versus the same sample without APV or FK506.
Synergistic Bdnf expression was induced by the CN pathway
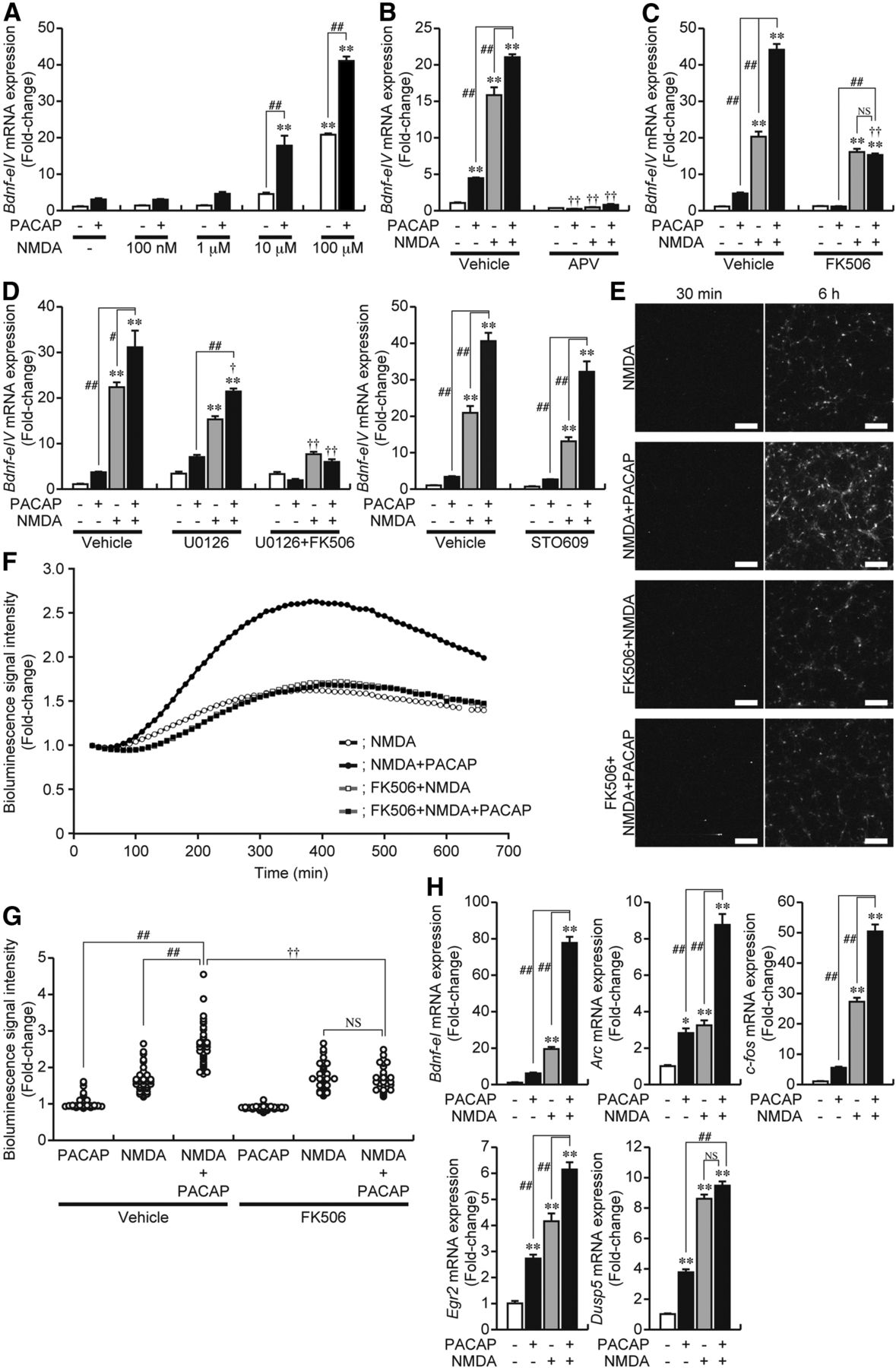
We next examined the effects of increasing the activity of NMDAR with exogenously added NMDA on the PACAP-induced expression of Bdnf using cell culture at 5 DIV. Bdnf-eIV mRNA expression was enhanced by increasing the concentration of NMDA, and the simultaneous addition of PACAP with NMDA synergistically increased the expression of Bdnf-eIV mRNA at concentrations above 1 μm NMDA (Fig. 4A), which was reduced to basal levels by APV (Fig. 4B). FK506 reduced Bdnf-eIV mRNA expression induced by the simultaneous addition of NMDA and PACAP to the same level observed for NMDA in the presence of FK506 (Fig. 4C). In contrast, although U0126 or STO609 partially reduced the NMDA-induced expression of Bdnf-eIV mRNA (Figs. 2C, 4D), a synergistic increase was still observed when PACAP was added with NMDA in the presence of U0126 or STO609 (Fig. 4D). However, further addition of FK506 with U0126 completely abolished the synergistic increase and decreased expression to the same level observed for NMDA in the presence of U0126 and FK506 (Fig. 4D).

Synergistic Bdnf expression is controlled by CN. A, PACAP was administered with NMDA to cells at the indicated concentrations at 5 DIV, and total RNA was prepared 3 h after treatment. Changes in Bdnf-eIV mRNA were measured by quantitative RT-PCR. Data represent the mean ± SE (n = 3). **p < 0.01 versus control; ##p < 0.01 versus samples without PACAP. B–D, Effects of APV (B), FK506 (C), U0126 (D), or STO609 (D) on the induction of Bdnf-eIV mRNA expression by NMDA (100 μm) and/or PACAP. An inhibitor was added 10 min before treatment. Data represent the mean ± SE (n = 3–6). **p < 0.01 versus control; #p < 0.05 and ##p < 0.01 versus samples with PACAP or NMDA alone; †p < 0.05 and ††p < 0.01 versus the same sample without inhibitors. NS, Not significant. E, F, Representative images of the bioluminescence signal 30 min and 6 h after the addition of NMDA with or without PACAP (E) and sequential changes in the signal after its addition (F, the mean signal values, n = 50 cells). Changes in the bioluminescence signal at 5 DIV were examined by time-lapse signal imaging (LV200; Olympus). Measurements were performed in the same conditions as described in Figure 3, B and C. FK506 was added 10 min before addition. Scale bars, 200 μm. G, The bioluminescence signal intensities in individual cells during treatment of cells with PACAP and/or NMDA for 6 h in the presence or absence of FK506 were plotted. n = 45–50 cells. ##p < 0.01 versus samples with PACAP or NMDA alone; ††p < 0.01 versus the same sample without an inhibitor. NS, Not significant. H, PACAP and NMDA were added simultaneously at 5 DIV, and total RNA was prepared 3 h after treatment. The expression of Bdnf-eI, Arc, c-fos, Egr2, and Dusp5 mRNA was investigated by quantitative RT-PCR. Data represent the mean ± SE (n = 4). *p < 0.05 and **p < 0.01 versus control; ##p < 0.01 versus samples with PACAP or NMDA alone. NS, Not significant.
Bioluminescence imaging was performed to further examine the synergistic expression of Bdnf. As shown in Figure 3, B and C, PACAP increased the bioluminescence signal in a limited number of cultured cells, which was completely inhibited by FK506. The number of cells responding to the direct activation of NMDAR with NMDA was markedly higher than those with bioluminescence signals induced by PACAP only (Figs. 3B,C, 4E,F), and the NMDA-induced signals were partially reduced by FK506 (Fig. 4E,F). However, these signals were synergistically enhanced in cells treated with NMDA in the presence of PACAP (Fig. 4E,F), and the number of cells with strong bioluminescence signals was increased (Fig. 4E,G). FK506 reduced the synergistic increase to the same level induced by NMDA in the presence of FK506 (Fig. 4E–G). This was consistent with the changes observed for Bdnf-eIV mRNA expression (Fig. 4C). These results indicated that the enhancing effect of PACAP on NMDA-induced Bdnf expression was completely inhibited by FK506.
The simultaneous addition of PACAP and NMDA also caused a synergistic increase in the mRNA expression of Bdnf-eI, Arc, and c-fos (Fig. 4H), all of which are CN-dependent genes (Fig. 2A). In contrast, an additive, but not synergistic, increase in mRNA expression was detected for Egr2, whereas neither a synergistic nor additive increase was observed for Dusp5 (Fig. 4H), both of which are CN-independent genes (Fig. 2A).
PACAP-induced activation of Bdnf-promoter IV by CREB and nuclear translocation of CRTC1
The Ca2+ signal-dependent activation of Bdnf-PIV is controlled by CaRE1, CaRE2, and CaRE3 (corresponding to CRE), which are located in the proximal region of Bdnf-PIV (Fig. 5A; Tao et al., 2002). Using promoter analyses, we found that the activation of Bdnf-PIV was induced by PACAP (Fig. 5), and changes in the expression of Bdnf-eIV mRNA (Figs. 1A, 2A, 4B,C) corresponded well to those in Bdnf-PIV activities (Fig. 5B,C,G,H). The disruption of CaRE3 only was sufficient to abolish the PACAP-induced activation of Bdnf-PIV (Fig. 5D). Using a dominant-negative CREB, A-CREB, that can interfere with the binding of wild-type CREB to CRE (Ahn et al., 1998), we confirmed the involvement of CREB in the activation of Bdnf-PIV (Fig. 5E). However, overexpression of M-CREB, a mutated CREB with a conserved serine at position 133 substituted for an alanine residue (Gonzalez and Montminy, 1989), did not affect PACAP-induced activation (Fig. 5F), whereas the depolarization-induced activation of Bdnf-PIV was prevented by M-CREB (our unpublished observations; Tao et al., 1998).

PACAP-induced activation of Bdnf-PIV through CRE/CREB. A, The structure of Bdnf-PIV. UBE and CRE stand for USF- and CREB-binding elements, respectively. Boxes with an oblique line indicate a mutation in the elements. B, C, Effects of APV (B) or FK506 (C) on PACAP-induced activation of Bdnf-PIV. Transfected cells were treated with PACAP for 6 h, and cell lysates were prepared for dual-Luc assay. APV or FK506 was added 10 min before treatment. Data represent the mean ± SE (n = 3). **p < 0.01 versus control; ##p < 0.01 versus the same sample without APV or FK506. D–F, Effect of mutations to CaRE1, CaRE2, and CaRE3 (D) or the coexpression of W-CREB (E), A-CREB (E), or M-CREB (F) on PACAP-induced activation of Bdnf-PIV. Data represent the mean ± SE (n = 3). *p < 0.05 and **p < 0.01 versus control; ##p < 0.01 versus Bdnf-PIV wild-type (D) or A-CREB (E). G, H, Effect of APV (G) or FK506 (H) on the activation of Bdnf-PIV by NMDA and/or PACAP. APV or FK506 was added 10 min before treatment. Data represent the mean ± SE (n = 3–6). **p < 0.01 versus control; ##p < 0.01 versus samples with PACAP or NMDA alone; ††p < 0.01 versus the same sample without APV or FK506. NS, Not significant.
CRTC1, also known as a transducer of regulated CREB 1, was shown previously to be dephosphorylated by CN and subsequently transported from the cytoplasm to the nucleus to activate CRE-dependent transcription (Conkright et al., 2003; Bittinger et al., 2004; Ch'ng et al., 2012). Although a previous study demonstrated that PACAP-induced CRTC1/CREB-dependent transcription was regulated by PKA-induced action potential firing (Baxter et al., 2011), they did not identify the target genes upregulated by the PACAP/CRTC1 pathway. In addition, the release of the receptor for activated C-kinase 1 (RACK1) from NMDAR and its translocation to the nucleus by PAC1 stimulation was reported to be responsible for the increase in Bdnf mRNA expression in hippocampal slices (Yaka et al., 2003a). However, we did not observe the involvement of RACK1 in our experimental system using rat cortical neurons (our unpublished observations). This may be attributable to an interaction between NMDAR and RACK1 in the hippocampus but not cerebral cortex (Yaka et al., 2003b). Thus, the intracellular mechanisms related to the PACAP-induced expression of IEGs are still obscure, and there is no direct evidence showing that PAC1 stimulation triggers activation of the NMDAR/CN pathway leading to the induction of Bdnf and other CN-dependent IEGs. To approach this, we investigated the subcellular localization of endogenous CRTC1 with PAC1 stimulation and found that PACAP-stimulated cortical cells facilitated the translocation of CRTC1 from the cytoplasm to the nucleus, whereas its translocation was not induced in the presence of APV or FK506 (Fig. 6A,B). As shown in Figure 6C, the translocation of CRTC1 to the nucleus peaked at 15 min after PAC1 stimulation but decreased thereafter, whereas the translocation also peaked at 15 min after NMDAR stimulation but was more likely to be maintained at the same level. The simultaneous addition of PACAP with NMDA further enhanced the level of translocation to the nucleus compared with NMDA alone, which peaked 30 min after the stimulation and was maintained for at least 60 min (Fig. 6C).

PACAP-induced activation of Bdnf-PIV through CRTC1. A, B, Subcellular localization of CRTC1 in cultured cortical cells (A). Cells were treated with PACAP for 15 min, and immunostaining was subsequently performed. APV or FK506 was added 10 min before PACAP treatment. Scale bars, 20 μm. The immunofluorescence intensity ratio of the nucleus to the cytoplasm was analyzed using commercially available software (ZEN 2009; Carl Zeiss) (B). **p < 0.01 versus control; ##p < 0.01 versus the same sample without APV or FK506. C, Cells were treated with PACAP or NMDA alone, or PACAP and NMDA simultaneously and then fixed 15, 30, or 60 min after treatment. Based on the immunofluorescence intensity of CRTC1 using ZEN 2009, the number of cells with CRTC1 in the nucleus was counted. **p < 0.01 versus CT (control); ##p < 0.01 versus samples with PACAP alone. D, Effects of shCRTC1 on the expression of overexpressed CRTC1 in NIH3T3 cells (left) and endogenous CRTC1 in rat cortical cells (right). The expression of CRTC1 was analyzed by immunoblotting (left) and immunostaining (right), respectively, at 48 h (NIH3T3 cells) or 72 h (rat cortical cells) after transfection, as described previously (Fukuchi et al., 2014). E, Effects of shCRTC1 on PACAP-induced transcriptional activation. Reporter plasmids and the expression vector for shCRTC1 or Scramble were cotransfected into cells. Seventy-two hours after transfection, cells were treated with PACAP for 6 h, and firefly Luc activity was measured. Data represent the mean ± SE (n = 5–6). *p < 0.05 and **p < 0.01 versus control; #p < 0.05 and ##p < 0.01 versus Scramble. F, A ChIP assay was performed using CRTC1 antiserum. After immunoprecipitation, purified DNA was amplified by PCR and separated on a 2% agarose gel (top). A ChIP assay was performed 15 min after PACAP treatment or not, and the quantity of purified DNA was measured by real-time PCR (bottom). Data represent the mean ± SE (n = 4).
We then examined the effects of CRTC1 shRNA (shCRTC1) on the PACAP-induced activation of Bdnf-PIV. We demonstrated that the expression of shCRTC1 decreased both overexpressed CRTC1 in NIH3T3 cells and endogenous CRTC1 in rat cortical cells, whereas scrambled CRTC1 shRNA (Scramble) had no effect on CRTC1 expression (Fig. 6D; Fukuchi et al., 2014). The PACAP-induced activation of Bdnf-PIV was reduced significantly by the expression of shCRTC1 but not by Scramble (Fig. 6E). The PACAP-induced activation of Bdnf-PI, the Arc promoter, and the c-fos promoter was also inhibited by the expression of shCRTC1 (Fig. 6E). The knockdown of CRTC1 slightly decreased the activities of Bdnf-PIV and Bdnf-PI in the absence of PACAP (Fig. 6E), suggesting the involvement of CRTC1 in controlling the basal activity of Bdnf promoters, which might be related to a decrease in the basal expression of Bdnf mRNA during the addition of APV alone (Figs. 1A, 7B, 8A,C). By ChIP assay, the precipitation of chromatin with anti-CRTC1 antiserum revealed CRTC1 binding on Bdnf-PIV, whereas a negligible level of binding was detected on the Dusp5 promoter (Fig. 6F). CRTC1 binding on the CRE-containing promoters of Bdnf-PIV, Bdnf-PI, Arc, and c-fos, but not Dusp5, was increased slightly after PACAP treatment (Fig. 6F).

Direct activation of the PKA or PKC pathway induced Bdnf-eIV mRNA expression through the Ca2+/CN pathway. A, At 5 DIV, cells were treated with forskolin (10 μm) or TPA (100 ng/ml) for 1 h, and total RNA was extracted. H89 or BisI was added 10 min before treatment. Data represent the mean ± SE (n = 4). *p < 0.05 and **p < 0.01 versus control; ##p < 0.01 versus the same sample without H89 or BisI. B, C, Effects of APV (B) or FK506 (C) on forskolin- or TPA-induced Bdnf-eIV mRNA expression. Cells were treated with forskolin or TPA for 1 h, and total RNA was extracted. APV or FK506 was added 10 min before treatment. Data represent the mean ± SE (n = 3). **p < 0.01 versus control; ##p < 0.01 versus the same sample without APV or FK506. D, E, Localization of CRTC1 in cortical cells treated with forskolin or TPA. APV or FK506 was added 10 min before treatment. Fifteen minutes after treatment with forskolin or TPA, cells were fixed and immunostained with anti-CRTC1 antiserum (green) and an anti-MAP2 antibody (magenta). Nuclei were counterstained with DAPI (blue). Images were taken by confocal microscopy (LSM 700) (D). Scale bars, 20 μm. The immunofluorescence intensity ratio of the nucleus to the cytoplasm was analyzed using ZEN 2009 software (Carl Zeiss) (E). **p < 0.01 versus control; ##p < 0.01 versus the same sample without APV or FK506. F, Cells were treated with NMDA plus forskolin or TPA for 3 h, and changes in Bdnf-eIV mRNA expression were measured by quantitative RT-PCR. Data represent the mean ± SE (n = 3). **p < 0.01 versus control; ##p < 0.01 versus the sample with forskolin, TPA, or NMDA alone.

Selective stimulation of GPCR induced Bdnf-eIV mRNA expression through the Ca2+/CN pathway. A–D, Effects of APV (A, C) or FK506 (B, D) on SKF38393-, isoproterenol-, CRF-, or neurotensin-induced Bdnf-eIV mRNA expression. SKF38393 (A, B, 1 μm), isoproterenol (A, B, 10 μm), CRF (C, D, 10 nm), or neurotensin (C, D, 1 nm) was added to cortical cells at 5 d in culture. Total RNA was extracted 1 h later for quantitative RT-PCR. Data represent the mean ± SE (n = 3–5). **p < 0.01 versus control; ##p < 0.01 versus the same sample without inhibitors. We investigated the expression levels of GPCRs in primary cultures of rat cortical cells and found that D1R (Gαs), β1AR (Gαs), CRF receptor 1 (Gαs), and neurotensin receptor 1 (Gαq) were expressed functionally (our unpublished observations). E, The subcellular localization of CRTC1 in cultured cortical cells. Cells were treated with SKF38393, isoproterenol, CRF, or neurotensin for 15 min, and immunostaining was subsequently performed. APV or FK506 was added 10 min before the treatment. Scale bars, 20 μm.
Direct activation of the PKA or PKC pathways activated the NMDAR/Ca2+/CN pathway, leading to Bdnf-eIV mRNA expression
We determined whether direct activation of the AC/cAMP/PKA or PKC pathway with forskolin and TPA, respectively, induced Bdnf-eIV mRNA expression through the NMDAR/Ca2+/CN pathway. As shown in Figure 7A, these chemical activators induced the expression of Bdnf-eIV mRNA, and their effects were completely prevented by H89 and BisI, respectively, indicating the major contribution of PKA and PKC pathways to Bdnf-eIV mRNA expression. Forskolin- or TPA-induced expression of Bdnf-eIV mRNA was also inhibited completely by APV or FK506 (Fig. 7B,C). Forskolin and TPA induced the translocation of CRTC1 from the cytoplasm to the nucleus, and this was also inhibited by APV or FK506 (Fig. 7D,E). Furthermore, the simultaneous addition of NMDA with either forskolin or TPA caused a synergistic increase in the expression of Bdnf-eIV mRNA (Fig. 7F). These results indicated that direct activation of the downstream pathways of Gαs/AC- and Gαq/PLC-coupled GPCRs induced the expression of Bdnf-eIV mRNA through selective activation of the NMDAR/CN pathway.
Selective stimulation of Gαs- or Gαq-coupled GPCRs induced Bdnf-eIV mRNA expression through the NMDAR/Ca2+/CN pathway
We determined whether Bdnf-eIV mRNA expression was induced by the selective stimulation of GPCRs other than PAC1. We focused on D1R and β-adrenergic receptor (βAR), GPCRs known to potentiate NMDAR activity through the Gαs/AC/PKA pathway (Konradi et al., 1996; Raman et al., 1996). D1R agonist (SKF38393) or βAR agonist (isoproterenol) increased the expression of Bdnf-eIV mRNA, and these increases were inhibited entirely by APV or FK506 (Fig. 8A,B). The specific activation of D1R or βAR was confirmed as SKF38393- or isoproterenol-induced expression of Bdnf-eIV mRNA was reduced by specific receptor antagonists, SKF83566 (8-bromo-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3-benzazepin-7-ol) or propranolol, respectively (our unpublished observations).
To selectively stimulate other GPCRs, we used CRF and neurotensin, agonists of CRF receptor (a Gαs-coupled GPCR; Arzt and Holsboer, 2006) and neurotensine receptor (a Gαq-coupled GPCR; Kitabgi, 2006), respectively. CRF or neurotensin also increased the expression of Bdnf-eIV mRNA, and this was almost completely inhibited by APV or FK506 (Fig. 8C,D). The specific stimulation of their receptors was confirmed by the addition of an antagonist to either the CRF receptor (astressin) or neurotensin receptor [SR142948 (2-[[[5-(2,6-dimethoxyphenyl)-1-[4-[[[3-(dimethylamino)propyl]methylamino]carbonyl]-2-(1-methylethyl)phenyl]-1H-pyrazol-3-yl]carbonyl]amino]-tricyclo[3.3.1.13,7]decane-2-carboxylic acid)] (our unpublished observations). The GPCR ligands used in the present study also induced the translocation of CRTC1 from the cytoplasm to the nucleus, and this was inhibited by FK506 or APV (Fig. 8E).
Bioluminescence imaging revealed that these ligands also increased the bioluminescence signal, particularly at 12–15 DIV (Fig. 9A). The number of cells with bioluminescence varied depending on the agonists administered. To examine the synergistic effect of these agonists on Bdnf-eIV mRNA expression, we focused on CRF, because the number of cells responding were relatively low among these agonists (Fig. 9A). Although the bioluminescence signals induced by CRF only were very low or undetectable at 5 DIV (Fig. 9B,C), the increase in bioluminescence signals during NMDA treatment was enhanced in the presence of CRF (Fig. 9B,C); that is, the number of cells with strong bioluminescence signal intensity were detected more frequently during the simultaneous addition of NMDA and CRF (Fig. 9D). The low synergistic effect of CRF may be attributable to a small population of cells responding to CRF in the cortical cell culture compared with those that responded to NMDA. In addition, the synergistic effect of CRF on Bdnf-eIV mRNA expression was more clearly observed after addition of NMDA at 10 μm than at 100 μm (Fig. 9E). This synergistic induction by the simultaneous addition of CRF and 10 μm NMDA was completely inhibited by APV (Fig. 9F). Furthermore, FK506 reduced the mRNA expression induced by NMDA and CRF to the same level as that of NMDA alone (Fig. 9F), as observed for PACAP and NMDA (Fig. 4C). The level of NMDA-induced Bdnf-eIV mRNA expression was enhanced in the presence of FK506 when 10 μm NMDA, but not 100 μm NMDA, was used (Fig. 9F). This might be attributable to the inhibitory effect of FK506 on the dephosphorylation of other transcription factors, which could enhance the phosphorylation of these factors (Bito et al., 1996) and increase the levels of activity-dependent mRNA expression in neurons.

Bioluminescence imaging of Bdnf expression after the selective stimulation of GPCR. A, Representative images of the bioluminescence signal 30 min and 6 h after the addition of SKF38393, isoproterenol, CRF, or neurotensin. Changes in bioluminescence signal were examined at 12–15 DIV by time-lapse signal imaging (LV200; Olympus). Luciferin was added into the medium at a final concentration of 1.0 mm before measurements. The bioluminescence signal was measured every 10 min (exposure time, 5 min) for 11 h. Scale bars, 200 μm. B, C, Representative images of the bioluminescence signal 30 min and 6 h after the addition of NMDA with or without CRF (B) and sequential changes in the signal after the addition (C, the mean signal value, n = 50 cells). Changes in the bioluminescence signal were examined at 5 DIV by time-lapse signal imaging (LV200; Olympus). Luciferin was added to the medium at a final concentration of 0.5 mm before measurements. The bioluminescence signal was measured every 10 min (exposure time, 3 min) for 11 h. Scale bars, 200 μm. D, Bioluminescence signal intensities in individual cells during treatment with CRF and/or NMDA for 6 h were plotted (n = 49–50 cells). ##p < 0.01 versus samples with CRF or NMDA alone. E, CRF was added with NMDA to cells at the indicated concentrations at 5 DIV. Changes in Bdnf-eIV mRNA were measured by quantitative RT-PCR. Data represent the mean ± SE (n = 3–4). **p < 0.01 versus control; #p < 0.05 versus samples without CRF. NS, Not significant. F, Effects of APV or FK506 on the induction of Bdnf-eIV mRNA expression by NMDA (10 μm) and/or CRF. An inhibitor was added 10 min before treatment. Data represent the mean ± SE (n = 3–4). **p < 0.01 versus control; ##p < 0.01 versus samples with CRF or NMDA alone; ††p < 0.01 versus the same sample without an inhibitor. NS, Not significant.
Discussion
This study analyzed the PACAP-induced expression of IEGs with a focus on Bdnf and identified novel aspects of the induction of IEGs triggered by GPCR stimulation (Fig. 10). In addition to quantitative RT-PCR analyses, we developed a bioluminescence imaging system to monitor Bdnf expression in living primary cultured cells prepared from the brains of Bdnf–Luc Tg mice. This led to a more general understanding of the mechanisms underlying the GPCR-mediated expression of IEGs.

A schematic model of GPCR-mediated gene expression in neurons. The expression of Bdnf and other CN-dependent genes under the activation of GPCR was induced through the Ca2+/CN/CRTC1/CREB pathway. The direct activation of NMDAR by an excitatory input activated the expression of Bdnf via multiple Ca2+ signaling pathways, including CN, CaMK, and MAPK. Once the activation of GPCR was induced simultaneously with excitatory neurotransmission, the expression of Bdnf and other CN-dependent genes in particular were upregulated synergistically by the CN pathway. Therefore, coincident NMDAR activation by a glutamatergic input and GPCR activation by neuromodulators into neurons efficiently induced CN-dependent gene expression.
Although the role of NMDAR in the PACAP-induced expression of IEGs, such as c-fos and Bdnf, was demonstrated already (Martin et al., 1995; Pellegri et al., 1998), our comprehensive analyses of PACAP-induced IEG expression revealed different levels of NMDAR dependency for IEG induction. Furthermore, we observed a strong correlation between the dependencies on NMDAR and CN pathways for PACAP-induced IEG expression, in which an enhanced dependency on NMDAR is associated with a greater dependency on CN and vice versa. Among the IEGs tested, the PACAP-induced expression of Bdnf was characterized particularly by its absolute dependency on the NMDAR/CN pathway.
The results of bioluminescence imaging indicated that the number of cells with strong bioluminescence signals after PACAP treatment increased as the duration of the culture was extended from 5 to 14 DIV. This suggests the maturation of glutamate-releasing synapses and an increase in the number of cells with matured synapses in this culture. In support of this, cultured cortical cells showed a synchronized oscillation in Ca2+ at 14 DIV but not 4–5 DIV (Fukuchi et al., 2014), which could be generated spontaneously at later stages of the culture with the maturation of synapses (Murphy et al., 1992; Wang and Gruenstein, 1997). However, APV reduced the basal expression of Bdnf-eIV mRNA in cultured cells at 5 DIV, indicating that endogenous glutamate could act on NMDAR to control the basal expression of Bdnf. In addition, exogenous addition of NMDA synergistically enhanced PACAP-induced Bdnf expression through the CN pathway. Together, it is likely that PACAP-induced Bdnf expression via the NMDAR/CN pathway was efficiently activated in cortical cells with matured synapses that could secrete glutamate, and, therefore, the activation of NMDAR by endogenously secreted glutamate is necessary for the GPCR-induced Bdnf expression via the NMDAR/CN pathway.
Of note, the NMDA-induced expression of Bdnf-eIV mRNA was controlled at similar levels by the CN, MAPK, and CaMK pathways, in contrast to the PACAP-induced Bdnf-eIV expression. Together with the observation that synergistic induction of Bdnf by the simultaneous addition of PACAP and NMDA was controlled exclusively by CN, it is evident that PAC1 stimulation selectively activates the NMDAR/CN pathway for the efficient induction of Bdnf and other CN-dependent IEGs. In contrast, although NMDA-induced Dusp5 expression via NMDAR was controlled by the MAPK and CaMK pathways, the induction was not dependent on the CN pathway. Furthermore, PACAP-induced Dusp5 expression was not dependent on the NMDAR/CN pathway. Thus, the dependency of Dusp5 expression on NMDAR was relieved under PAC1 stimulation, probably because of its inability to respond to the CN pathway. In addition, PACAP-induced Dusp5 expression was mainly dependent on the PKA and PKC pathways evoked by Gαs/AC and Gαq/PLC, respectively. Thus, the different regulation of Bdnf-eIV and Dusp5 expression also supports the idea of selective activation of the NMDAR/CN pathway during PAC1 stimulation.
These observations regarding the regulation of Dusp5 support the idea that the NMDAR/CN pathway is activated postsynaptically by PAC1 stimulation. If the effects of PACAP on the regulation of mRNA expression are caused by an increase in presynaptic glutamate release, PACAP-induced Dusp5 mRNA expression should be mediated by NMDAR and controlled by multiple Ca2+ signaling pathways, including CaMK and MAPK, and also PACAP-induced Bdnf-eIV mRNA expression should be, at least in part, inhibited by FK506, as well as U0126, STO609, and KN93. Evidence for the postsynaptic induction of IEGs was further supported by the observation that CN pathway-mediated synergistic expression was increasingly detected under PAC1 stimulation as the activity of NMDAR increased. Thus, the stronger the activity of NMDAR, the greater the modulatory effects of PAC1 stimulation on the NMDAR/CN pathway leading to Bdnf-eIV mRNA expression. Therefore, the modulatory effect of PAC1 stimulation on NMDAR activity is responsible for PACAP-induced Bdnf and CN-dependent IEG mRNA expression. This modulation of NMDAR might be controlled by its phosphorylation by several protein kinases (Yaka et al., 2003a; Macdonald et al., 2005) and/or mechanisms orchestrated by A-kinase anchoring protein 79/150, a multivalent scaffolding protein that coordinates the subcellular localization of PKA, PKC, and CN (Fraser and Scott, 1999; Hoshi et al., 2005; Zhang and Shapiro, 2012).
Our results suggest that selective activation of the CN pathway could be responsible for the effective nuclear translocation of CRTC1 and CREB-dependent transcription of Bdnf and CN-dependent IEGs (Fig. 10). Furthermore, the binding of CRTC1 to the promoters of CN-dependent IEGs, but not CN-independent IEGs, increased under PAC1 stimulation. Thus, GPCR-mediated selective activation of the NMDAR/CN pathway induced the nuclear localization of CRTC1 and might be involved directly in the activation of Bdnf-PIV and CN-dependent transcription of IEGs, probably independently of the phosphorylation of CREB. Although neuronal activity can affect several mechanisms involved in Bdnf mRNA processing (Tongiorgi et al., 1997; Chiaruttini et al., 2008; Fukuchi and Tsuda, 2010; Vaghi et al., 2014) and BDNF secretion (Kolarow et al., 2007; Park et al., 2014), GPCR-mediated Bdnf expression is regulated mainly at the transcriptional level because changes in Bdnf-PIV activity corresponded to those in Bdnf-eIV mRNA expression.
The direct activation of the PKA or PKC pathway with forskolin and TPA, respectively, as well as the selective stimulation of Gαs/q-coupled GPCR, such as D1R, βAR, CRF receptor, or neurotensin receptor, increased the expression of Bdnf-eIV mRNA through the NMDAR/CN pathway and translocation of CRTC1 to the nucleus. Our imaging analyses revealed that the number of cells with increased bioluminescence signal varied during treatment with specific agonists, probably because of specific cell-type responses to particular agonists. These results support that selective activation of the CN pathway is induced by any kind of stimulation of Gαs-coupled or Gαq-coupled GPCRs if the PKA and/or PKC pathway is activated in neurons with active NMDAR (Fig. 10). Blockade of D1R or NMDAR by administration of specific antagonists to animals blocked the expression of IEGs and induced behavioral changes (Wolf and Khansa, 1991; Huang and Kandel, 1995; Moratalla et al., 1996). Furthermore, the deletion of CRTC1 in mice decreased the mRNA expression levels of Bdnf and other IEGs in the hippocampus (Breuillaud et al., 2012). In this study, intracerebroventricular injection of PACAP to mice demonstrated a functional interaction between PAC1 and NMDAR in the cerebral cortex to induce Bdnf expression. These findings strongly support the GPCR-induced selective activation of the CN/CRTC1/CREB pathway to control the expression of CN-dependent IEGs in the brain. If excitatory and modulatory synaptic neurotransmissions occur in the same dendritic spines in the brain, the GPCR-mediated modulation of NMDAR might occur concurrently, causing the CN pathway to be activated efficiently in the spines. Because CRTC1 is transferred from dendritic spines to the nucleus (Ch'ng et al., 2012), the GPCR-induced expression of IEGs might be a general mechanism for the transfer of signals evoked strongly at spines that receive the coordinated integration of excitatory and modulatory neurotransmissions to the gene level in the nucleus.
The GPCR-mediated expression of IEGs might mediate the actions of antidepressants or psychoactive drugs in the brain, because the blockade of neurotransmitter uptake by these drugs reinforces the activity of GPCRs, thereby causing modulation of the NMDAR/CN pathway. In support of this, administration of antidepressants in animal models of depression increased the level of Bdnf mRNA expression and recovered the decreased size of pyramidal neurons in the CA3 of the hippocampus (Duman et al., 1997). Conversely, it was demonstrated previously that disruption of Bdnf-PIV-driven expression of Bdnf transcripts or mutations of CRE at Bdnf-PIV impaired neuronal activity-dependent BDNF expression, resulting in the abnormal development of cortical inhibition (Hong et al., 2008; Sakata et al., 2009). These reports support strongly the idea that the transcriptional activation of Bdnf could reflect the expression of BDNF protein and participate in the regulation of neuronal functions. Furthermore, it was suggested that synthesized BDNF can accumulate intracellularly (Danzer and McNamara, 2004) and be secreted in an activity-dependent manner, the disruption of which causes neuronal dysfunction (Egan et al., 2003). Thus, it seems likely that the intracellular regulation of BDNF expression is fundamentally related to neuronal plasticity (Park and Poo, 2013), in which GPCR-mediated Bdnf expression might facilitate the accumulation of BDNF in neurons receiving coordinated excitatory and modulatory neurotransmissions at spines. This then might result in the strengthening of specific neuronal networks in the brain. Overall, the GPCR-mediated expression of Bdnf and other CN-dependent IEGs might be involved widely in the control of plasticity-related phenomena in the brain, such as memory consolidation, drug addiction, and antidepressant effects, whereas its disruption may be related to the pathogenesis of neurodegenerative or psychiatric disorders.
Footnotes
This study was supported in part by Japan Society for the Promotion of Science Research Funds Grant-in-Aid for Scientific Research (B) Grant 20390023 (M.T.) and Grant-in-Aid for Young Scientists (B) Grant 25870256 (M.F.) and the Mitsubishi Foundation (M.T.). We thank Dr. M. R. Cappechi (Howard Hughes Medical Institute, University of Utah, Salt Lake City, UT) for providing pACN.
The authors declare no competing financial interest.
- Correspondence should be addressed to Masaaki Tsuda, Department of Biological Chemistry, Faculty of Pharmaceutical Sciences, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Sugitani 2630, Toyama 930-0194, Japan. tsuda{at}pha.u-toyama.ac.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}