

Abstract
According to the current model of neurovascular coupling, blood flow is controlled regionally through phasic changes in the activity of neurons and astrocytes that signal to alter arteriole diameter. Absent in this model, however, is how brain blood flow is tonically regulated independent of regional changes in activity. This is important because a large fraction of brain blood flow is required to maintain basal metabolic needs. Using two-photon fluorescence imaging combined with patch-clamp in acute rat brain slices of sensory-motor cortex, we demonstrate that reducing resting Ca2+ in astrocytes with intracellular BAPTA causes vasoconstriction in adjacent arterioles. BAPTA-induced vasoconstriction was eliminated by a general COX blocker and the effect is mimicked by a COX-1, but not COX-2, antagonist, suggesting that astrocytes provide tonic, steady-state vasodilation by releasing prostaglandin messengers. Tonic vasodilation was insensitive to TTX, as well as a variety of synaptic and extrasynaptic receptor antagonists, indicating that the phenomenon operates largely independent of neural activity. Using in vivo two-photon fluorescence imaging of the barrel cortex in fully awake mice, we reveal that acute COX-1 inhibition reduces resting arteriole diameter but fails to affect vasodilation in response to vibrissae stimulation. Our findings demonstrate that astrocytes provide tonic regulation of arterioles using resting intracellular Ca2+ in a manner that is independent of phasic, neuronal-evoked vasodilation.
SIGNIFICANCE STATEMENT The brain requires both phasic and tonic regulation of its blood supply to service energy needs over various temporal windows. While many mechanisms have been described for phasic blood flow regulation, how the brain accomplishes tonic control is largely unknown. Here we describe a way in which astrocytes contribute to the management of basal brain blood flow by providing steady-state vasodilation to arterioles via resting astrocyte Ca2+ and the continuous release of prostaglandin messengers. This phenomenon may be important for understanding the declines in basal brain blood flow that occur in aging and dementia, as well as for the interpretation of fMRI data.
Introduction
Neurovascular coupling is the process by which local neuronal and astrocytic elements signal to nearby vascular contractile cells to change arteriole diameter, thereby altering blood flow to meet the energy demands of resident cells (Attwell et al., 2010). Brain cells have distinctly different requirements for blood flow during enhanced activity compared with basal activity states (Howarth et al., 2012). At one extent, this requires rapid and phasic changes to arteriole diameter, whereas at the other, constant or tonic diameter control is required. Although basal metabolic demand accounts for ∼50% of total energy needs (Howarth et al., 2012), our current understanding of neurovascular coupling is largely limited to phasic, activity-dependent changes, whereby transient and regional elevations in synaptic transmission activate receptors on neurons and astrocytes. This is a feedforward process that initiates Ca2+-dependent pathways and the release of diffusible messengers that ultimately control the diameter of brain arterioles (Zonta et al., 2003; Filosa et al., 2004; Mulligan and MacVicar, 2004; Metea and Newman, 2006; Rancillac et al., 2006). For resting blood flow control, local, feedforward control mechanisms remain mostly unexplored.
In vivo experiments point to the possible contribution from local tonic blood flow control pathways. Pharmacological antagonists directed at vasoactive enzymes not only reduce evoked blood flow increases but also resting blood flow (Alkayed et al., 1996; Peng et al., 2004). In particular, cyclooxygenase-1 (COX-1) knock-outs show reduced baseline brain blood flow without an effect on functional hyperemia (Niwa et al., 2001). However, the role of a particular brain cell type that affects the diameter of parenchymal arterioles to help set resting brain blood flow locally has not been clearly described, although glial Muller cells have recently been hypothesized to play such a role in the retina (Kur and Newman, 2014).
Astrocytes influence arteriole diameter by their endfeet (Mulligan and MacVicar, 2004; Straub et al., 2006; Takano et al., 2006; Gordon et al., 2008), which are specialized compartments that directly appose and surround microvasculature (Simard et al., 2003; Mathiisen et al., 2010). Astrocytes may be well suited for tonic blood flow control due to their high resting and spontaneous Ca2+ activity (Shigetomi et al., 2012; Haustein et al., 2014), their lack of temporally precise responses to neural input (Schummers et al., 2008; Schulz et al., 2012; Nizar et al., 2013; Paukert et al., 2014; but see Lind et al., 2013), their ability to sense local metabolic changes (Gordon et al., 2008), as well as their ability to influence basal synaptic transmission in a continuous fashion (Pascual et al., 2005; Panatier et al., 2011). In particular, astrocytes have been shown to express COX-1 (Takano et al., 2006; Cahoy et al., 2008; Gordon et al., 2008; but see Lecrux et al., 2011) and the high resting Ca2+ activity in these cells may provide a means to drive Ca2+-dependent cellular pathways for arteriole communication.
Here we test the hypothesis that resting Ca2+ in astrocytes provides tonic control over arteriole diameter using a pathway that is independent of phasic neurovascular coupling. We find that intracellular delivery of the Ca2+ chelator BAPTA into astrocytes results in vasoconstriction (loss of tonic vasodilation) of nearby arterioles when BAPTA reaches endfeet. COX inhibition prevents the effect of astrocyte BAPTA, and blockade of COX-1 enzymes, but not COX-2, mimics the effect. In fully awake mice in vivo, we demonstrate that inhibiting COX-1 decreases resting arteriole diameter while leaving functional hyperemia intact. Our data reveal a type of brain blood flow regulation in which resting astrocyte Ca2+ causes the continuous release of prostaglandin messengers to provide steady-state vasodilation. This finding provides an expanded framework to aid our understanding of how the energy demands of basal brain metabolism may be met. This fundamental process may be important for understanding disease and/or age-related declines in basal brain blood flow (Iadecola, 2013).
Materials and Methods
All procedures were performed in accordance with the guidelines set forth by the Animal Care and Use committee of the University of Calgary (protocols M11002 and M11032) and thereby abided by Canadian standards for animal research.
Two-photon imaging.
Fluorescence imaging was performed on two custom two-photon laser-scanning microscopes: one optimized for acute brain slices and patch-clamp electrophysiology (Rosenegger et al., 2014) and one optimized for awake in vivo experiments (Tran and Gordon, 2015). Each microscope was equipped with a Ti:Sapph laser (Ultra II, ∼4 W average power, 670–1080 nm, Coherent), objectives (Zeiss 40× NA 1.0, Nikon 16× NA 0.8), a green bandpass emission filter (525–40 nm), an orange/red bandpass emission filter (605–70 nm), and associated photomultiplier tubes (GaAsP Hamamatsu). For acute slice experiments, time-series images using bidirectional scanning (512 pixels2 at 1 Hz temporal resolution) were acquired at a single focal plane incorporating the cells of interest and the middle of an arteriole lumen. Z-stacks were used to assess the extent of Alexa-488 diffusion through the astrocyte network. To image Alexa-488 during astrocyte network filling, the Ti:Sapph laser was tuned to 780 nm. In experiments that quantified the decrease in Rhod-2 fluorescence by BAPTA, the Ti:Sapph was tuned to 850 nm. For in vivo imaging, Ca2+ signals and penetrating arterioles were imaged using bidirectional scanning (512 pixels2 at 4 Hz). When imaging GCaMP3 mice and Rhod-dextran, the laser was tuned to 940 nm.
Acute brain slice preparation.
Acute coronal slices of the neocortex from male Sprague Dawley rats (p21-p30) were prepared. Animals were anesthetized with isoflurane (5% induction, 2% maintenance). After tail vein injection (see below), animals were decapitated with a rodent guillotine, and their brains were quickly and carefully removed with surgical tools. The brains were then placed into a beaker containing ice-cold slicing solution continuously bubbled with carbogen (95% O2, 5% CO2) for 2 min. Slicing solution contained the following (in mm): 119.9 N- methyl d-glucamine, 2.5 KCl, 25 NaHCO3, 1.0 CaCl2-2H2O, 6.9 MgCl2-6H2O, 1.4 NaH2PO4-H2O, and 20 glucose. Slices were cut on a vibratome (Leica VT 1200S) and subsequently laid on mesh submerged in an ACSF recovery chamber maintained at 34°C in a water bath. Slices received continuous bubbling with carbogen and recovered for 45 min. ACSF contained the following (in mm): 126 NaCl, 2.5 KCl, 25 NaHCO3, 1.3 CaCl2, 1.2 MgCl2, 1.25 NaH2PO4, and 10 glucose. The arteriole preconstrictor U-46619 (100 nm) was added to the bath for all experiments to preconstrict arterioles by ∼25% (Metea and Newman, 2006; Blanco et al., 2008). Imaging was performed at 22°C and perfused at ∼2 ml/min using a sealed solution delivery system driven by pressure via a carbogen tank and regulator.
Patch clamping astrocytes.
Astrocytes targeted for patch clamp were within ∼30–100 μm from the arteriole under study and at a depth between ∼30–60 μm from the surface of the slice. A Giga-Ohm seal was maintained for 15–30 min to allow patch-related effects to arteriole diameter to subside (Zonta et al., 2003). Once 5 min of stable baseline was observed, whole-cell configuration was then obtained to initiate filling of astrocytes with internal solution containing Alexa-488 (100–200 μm) (to visualize the extent of diffusion), and different concentrations of BAPTA or EGTA. As a base, intracellular solutions contained the following (in mm): 8 KCl, 8 sodium gluconate, 2 MgCl2, 10 HEPES, 4 potassium ATP, and 0.3 sodium GTP. To achieve different levels of intracellular Ca2+ buffering, to the base intracellular solution, we added (in mm): 10 potassium BAPTA with 68 potassium gluconate; or 40–60 potassium BAPTA; or 0.1–1 potassium EGTA with 108 potassium gluconate. All internal solutions were corrected for osmolarity to ∼285 mosmol and corrected for pH with KOH to 7.2. The astrocyte cell type was confirmed by a low input resistance (10–20 mΩ), extensive dye transfer between coupled cells via gap junctions, and visibly loaded endfeet apposed to microvasculature. Astrocytes were voltage clamped at −80 mV.
Loading dyes in astrocytes, neurons, and arterioles.
Brain tissue was bulk loaded with Rhod-2/AM Ca2+ indicator (15 μm) (0.1% DMSO; 0.006% pluronic acid; 0.0002% Cremophore EL). Astrocytes were identified by their bright uptake of Rhod-2/AM and by their endfeet (Simard et al., 2003; Mulligan and MacVicar, 2004). Ca2+ signals were calculated as follows: ΔF/F = ((F1 − F0)/F0) × 100, where F is fluorescence, 1 is at any given time point, and 0 is an average baseline value. To image and quantify arteriole diameter changes, FITC-dextran (2000 kDa) (7.5 mg in 0.2 ml lactated Ringer's) or Rhodamine-dextran (70 kDa) (11 mg in 0.2 ml lactated Ringer's) was tail vein injected to fill the vasculature with dye before in vivo imaging or taking acute slices. We quantified arteriole diameter as a change in lumen area centered on the brightest endfoot Alexa signal and shortest tau value, and took the lowest value in all experimental groups. Either paired or unpaired Student's t tests (as appropriate) were used to compare between two groups. ANOVA was used when comparing more than two groups.
Awake in vivo two-photon imaging.
Fully awake mouse imaging experiments were performed as previously described (Tran and Gordon, 2015). Briefly, a custom head-bar was surgically installed using standard aseptic surgical procedures (ethanol and betadine), anesthesia (isoflurane 5% induction, 2% maintenance), pain relief (buprenorphine 0.05 mg/kg), and antibiotics (enrofloxacin 2.5 mg/kg) in male GLAST-Cre-ERT × GCaMP3 mice (age range p50-p90). After 2 d recovery from surgery, animals began training for head fixation on a passive, air-supported, spherical treadmill (Dombeck et al., 2007; Tran and Gordon, 2015). In Session 1, a given mouse was head-fixed for 30 min. In Session 2 on the following day, the mouse was head-fixed on the treadmill for 45 min. In each session, the mouse was acclimatized to a 5 s puff of air onto the contralateral whiskers. Two days following training Session 2, a craniectomy over the barrel cortex was performed that involved bone and dura removal and the implantation of a fully sealed cranial window (Holtmaat et al., 2009; Tran and Gordon, 2015). The animal recovered from anesthesia head-fixed on the treadmill and was ready for imaging after a 45 min hiatus. Animal behavior was constantly monitored using an infrared LED and camera while on the treadmill. Arterioles could be readily distinguished from venules due to regular pulsations and a time-locked vasodilation to whisker stimulation.
GLAST-Cre-ERT × GCaMP3 mice.
To achieve a genetically encoded Ca2+ indicator in astrocytes, we crossed GLAST-Cre-ERT (The Jackson Laboratory, 012586) with LSL-GCaMP3 (The Jackson Laboratory, 014538) mice. Male mice positive for both inserts received three tamoxifen injections (100 mg/kg i.p.) (corn oil vehicle) on consecutive days starting between p16 and p36. Animals were allowed at least 3 weeks for recombination to take place before head-bar implantation and treadmill training.
Functional hyperemia.
For in vivo experiments, air puff to the contralateral whisker field was applied using a picospritzer III (General Valve) using as little air pressure as possible that visibly deflected the whiskers. The air output was divided to two mounted glass capillary tubes that aimed at separate groups of vibrissa, in an attempt to stimulate as many whiskers as possible without impacting the face. An arteriole was deemed responsive and included for analysis if >50% of air puff trials resulted in a time-locked vasodilation above the previous 4 s of baseline. All trials, with clear responses or not, were averaged to obtain the measurement in a given animal.
Results
Ca2+ chelation in astrocytes decreases arteriole diameter
To test the hypothesis that resting Ca2+ in astrocytes enables tonic communication with arterioles to control blood vessel tone, we used a whole-cell patch-clamp technique to reduce intracellular free Ca2+ in astrocytes. We began by patching astrocytes that were directly connected to an arteriole, enabling us to diffuse the Ca2+ chelator BAPTA into adjoining endfeet. We coloaded with Alexa-488 (100–200 μm) to estimate the rate and extent of BAPTA diffusion through the patched astrocyte and into endfeet apposed to the arteriole. We first sealed onto an astrocyte membrane without rupturing it and waited for any patch-generated Ca2+ transients or arteriole responses caused by the patching process itself (Zonta et al., 2003) to return to baseline and stabilize (∼15–30 min). Rupturing the membrane resulted in rapid diffusion of Alexa-488 and BAPTA (10 mm) (Fig. 1A,B). Subsequent to the arrival of the Alexa-488 signal into astrocyte endfeet, we observed a prominent vasoconstriction (−13.97 ± 2.35%, n = 10, p < 0.001; Fig. 1C,D). As absolute fluorescence changes are susceptible to image depth differences between experiments, to quantify the efficacy of astrocyte filling, we measured the tau of the monophasic exponential arrival of the Alexa-488 signal in endfeet (tau = 138.2 ± 43.4 s, n = 10; Fig. 1C,D). This helped control for differences in patch access resistance and different distances from the patched astrocyte to the arteriole examined within and between experimental groups. For example, experiments in which the tau could not be calculated around the arteriole due to limited dye infiltration caused by either insufficient cell access and/or too great of a patching distance from the arteriole revealed little change to arteriole diameter (0.70 ± 0.81%, p > 0.05, n = 11; Fig. 1D). Additionally, if the patch electrode became physically disconnected during the whole-cell event, no astrocyte filling was observed and an immediate, large vasodilation occurred (data not shown), likely caused by a rapid increase in extracellular potassium from the internal solution. Next, we tested whether the observed BAPTA-induced vasoconstriction was the result of the close proximity of the patched astrocyte to the arteriole or from the intended intracellular manipulation. We did this by patching an astrocyte that was not directly connected to the arteriole. Astrocytes are coupled to their neighbors via gap junctions (Rouach et al., 2008). Whole-cell patching a single astrocyte allows access to the intracellular environment of the astrocyte network, enabling us to diffuse BAPTA into a cluster of astrocytes surrounding an arteriole. To ensure adequate BAPTA infiltration to endfeet, we raised the concentration to 40–60 mm (denoted 50 mm) when patching distant astrocytes. We found that we could still elicit a vasoconstriction (diameter: −9.34 ± 0.96%, p < 0.001, n = 11; Fig. 1D) similar in magnitude to when the astrocyte was patched at close range with 10 mm BAPTA (p > 0.05), even though the tau of endfoot filling was significantly slower (tau = 310.9 ± 52.9 s, p < 0.05; Fig. 1D).

Intracellular diffusion of BAPTA through the astrocyte network causes vasoconstriction in nearby arterioles. A, Left, Cortical penetrating arteriole (lumen filled with FITC-dextran) 1 min before obtaining a whole-cell configuration on an astrocyte (Giga-Ohm seal). Patch pipette contains Alexa-488 and 10 mm BAPTA (not seen, out of image plane). Right, Eight minutes after obtaining whole cell. Alexa and BAPTA have diffused through the astrocyte network via gap junctions, illuminating somata and endfeet wrapping the arteriole. B, Max projection Z-stack image showing the extent of the Alexa and BAPTA fill, which infiltrated numerous astrocytes and endfeet. C, Left, Close-up of the same arteriole in A, showing a decrease in arteriole diameter after the arrival of Alexa plus BAPTA to the astrocyte endfeet. Top image, No endfoot fill 1 min before whole cell. Bottom image, Endfoot fill 8 min after whole-cell and associated vasoconstriction. Right, Top traces representing the rise and eventual plateau of the Alexa signal in endfeet (green top trace) as well as the decreasing arteriole diameter (gray bottom trace). Bottom traces have an expanded time scale to show the onset of the entry of Alexa and BAPTA to the endfoot and the initiation of the vasoconstriction. The Alexa signal was quantified by obtaining the tau of the monoexponential. D, Summary data showing no astrocyte fill controls, the magnitude of vasoconstriction caused by astrocyte BAPTA (10 mm close astrocyte and 50 mm distant astrocyte, each ***p < 0.001), and the corresponding tau values of the Alexa fill (*p < 0.05). E, Top, Arteriole lumen depicted 1 min before going whole-cell on an astrocyte. Pipette solution is a BAPTA-free control (standard EGTA 0.1–1 mm). Bottom, Eight minutes after the onset of the diffusion of the BAPTA-free intracellular solution through the astrocyte network, which fails to change arteriole diameter despite good infiltration into endfeet. F, Trace data from the same experiment in A, showing the rise of the Alexa signal in an endfoot (top green trace) and the corresponding measure of arteriole diameter (bottom gray trace). G, Summary data showing no vasoconstriction during BAPTA-free network infusions, which is different compared with all BAPTA experiments (**p < 0.01). The corresponding tau values for these experiments show similar efficacy of infiltration, suggesting that the lack of diameter change by the BAPTA-free solution was not due to a difference in network filling. Data are mean ± SEM.
We then tested whether the diffusion of intracellular solution from the patch pipette, rather than BAPTA per se, could be responsible for the vasoconstriction we observed. Patching astrocytes that were either directly coupled to the arteriole or not, we found that a BAPTA-free internal solution (containing standard levels of EGTA, 0.1–1 mm) had no impact on arteriole diameter (−0.44 ± 1.66%. n = 9, p > 0.05; Fig. 1E–G), despite similar infiltration of the internal solution into endfeet (tau = 299.4 ± 55.0 s, n = 9, p > 0.05 compared with all BAPTA experiments; Fig. 1F,G). These data suggest that resting Ca2+ in astrocytic endfeet provides tonic, steady-state control of arteriole diameter that becomes disrupted by intracellular Ca2+ chelation.
A given endfoot communicates with the section of the arteriole to which it is directly apposed to and not to more lateral regions along the vessel (Mulligan and MacVicar, 2004). We reasoned that the BAPTA-induced vasoconstriction should be of larger magnitude in the section of the arteriole where the infiltration of the intracellular solution into endfeet was greatest. To detect a localized effect on the vessel, we examined a subset of experiments in which the patched astrocyte was close to the arteriole and where there was a sufficient length of arteriole lumen visible in the image plane (>150 μm). We found no significant change to arteriole diameter at distances of 50–100 μm lateral (one direction) along the length of the arteriole where the tau of the fill was significantly slower (far diameter: −3.03 ± 0.64%, p > 0.05; far tau = 707.9 ± 200.9 s) compared with clear constrictions where the Alexa signal was strongest and had the shortest tau values (near diameter: −9.62 ± 0.90%, p < 0.001; near tau = 113.9 ± 9.2 s, n = 6, p < 0.05; Fig. 2A–F). These data suggest that the BAPTA-induced vasoconstriction is spatially confined to the regions in which endfeet exhibit the greatest degree of Ca2+ buffering.

BAPTA-induced vasoconstriction is localized to the region of most Ca2+ buffering in endfeet. A, Green images represent before (left) and after (right) obtaining a whole-cell configuration on an astrocyte with Alexa-488 in the intracellular solution. Three ROIs are indicated for endfeet at different distances/locations from the patched astrocyte. B, The same arteriole and astrocytes labeled with SR-101 to better show the three endfeet ROIs in A. C, Monoexponential rises of the Alexa signal in the three different endfeet during astrocyte filling from the ROIs in A and B. Endfeet close to the patched astrocyte show brighter signal with shorter tau time values compared with more distance endfeet. D, Image of an arteriole and a patched astrocyte filled with BAPTA plus Alexa-488. Locations on the arteriole that are near (1) and far (2) from the patched astrocyte are indicated. E, Trace data from the arteriole shown in D. Near to the patched astrocyte, Alexa plus BAPTA infiltration is strong, resulting in a short tau time value and appearance of vasoconstriction. Farther from the patched astrocyte, infiltration is weak, yielding a long tau time value of the fill and no change to arteriole diameter. F, Summary data showing the magnitude of vasoconstriction at locations near to the patched astrocyte compared with far. The corresponding tau values for these different locations along the arteriole show that vasoconstriction only occurs within the region of highest BAPTA and Alexa infiltration (*p < 0.05,***p < 0.001). Data are mean ± SEM.
To confirm that the BAPTA-induced vasoconstriction was associated with a decrease in resting astrocyte Ca2+, we quantified relative changes in fluorescence of the high-affinity Ca2+ indicator Rhod-2/AM. We found that the decrease in arteriole diameter was associated with a reduction in somata and endfeet Ca2+ that corresponded with network filling (ΔF/F −14.89 ± 3.05%, p < 0.001, n = 13; Fig. 3A–C,E). BAPTA-free experiments did not display a drop in astrocyte Ca2+ (ΔF/F −0.47 ± 2.6%, n = 12; Fig. 3D,E) compared with trials with BAPTA (p < 0.01). The fact that the BAPTA-free control failed to reduce astrocyte Ca2+ also verified that the observed drop in experiments with BAPTA was not due to washout of Rhod-2 via the patch solution. Lateral along the arteriole wall where the tau of the endfoot fill was significantly slower and where there was no adjacent vasoconstriction, we failed to detect a drop in astrocyte Ca2+ (ΔF/F 3.45 ± 2.47%, p > 0.05, n = 5). These data demonstrate that vasoconstriction is associated with a decrease in resting endfoot Ca2+.

Vasoconstriction is associated with a decrease in astrocyte Ca2+. A, Image of a cortical arteriole filled with FITC-dextran and surrounding astrocytes loaded with Rhod-2. B, Image sequence from boxed region in A, showing a Rhod-2 loaded endfoot (top pseudocolored images), and the incoming BAPTA and Alexa-488 signal arising from astrocyte network filling (bottom green images). The baseline Ca2+ fluorescence decreases as the endfoot fills with dye. C, Trace data from the same experiment in A and B. Top green trace, Rate of endfoot BAPTA and Alexa filling. Bottom red trace, Decrease in Ca2+ as BAPTA infiltrates the endfoot. D, Control experiment traces show no decrease in endfoot Ca2+ using BAPTA-free intracellular solution despite good infiltration into endfeet. E, Summary data demonstrate a significant decrease in endfoot Ca2+ caused by astrocyte BAPTA compared with BAPTA-free internal solution (**p < 0.01, ***p < 0.001). Data are mean ± SEM.
Astrocytes provide tonic vasodilation via constitutive COX-1 activity
COX activity and the production of vasoactive prostaglandins are a proposed mechanism for how astrocytes regulate arteriole diameter in response to uncaging Ca2+, pharmacological agonists, and afferent activity (Zonta et al., 2003; Takano et al., 2006; Gordon et al., 2008). We pondered whether constitutive COX activity was responsible for tonic diameter control by astrocyte endfeet. One possibility is that astrocytes are tonically producing vasodilatory prostaglandins in a Ca2+-dependent manner. In this case, we would anticipate that reduction of resting Ca2+ by astrocyte BAPTA would attenuate prostaglandin production and result in the loss of steady-state vasodilation (i.e., appearance of vasoconstriction). We tested this idea by performing BAPTA delivery into the astrocyte network in the continual presence of bath indomethacin (50 μm), a broad-spectrum antagonist of COX enzymes. In this condition, astrocyte BAPTA failed to elicit vasoconstriction (−2.50 ± 0.78%, p > 0.05, n = 5; Fig. 4A–C). This was despite a high concentration of BAPTA (50 mm) and relatively fast Alexa-488 tau values in endfeet (195.3 ± 93.6 s, n = 5; Fig. 4B,C), indicating sufficient network filling. We also verified that BAPTA reduced astrocyte Ca2+ (ΔF/F −27.90 ± 3.83%, p < 0.01, n = 5; Fig. 4B,C) without causing vasoconstriction when in the presence of indomethacin. We ran a control experiment to test whether the lack of effect by BAPTA on diameter was due to an inability to observe vasoconstriction caused by the presence of indomethacin, such as a floor effect. However, bath application of the thromboxane receptor agonist U46619 (200 nm) in the presence of indomethacin was able to cause vasoconstriction (−10.72 ± 2.22%, p < 0.01, n = 5; Fig. 4D), arguing against this possibility. Collectively, these data suggest that astrocytes provide steady-state vasodilation to arterioles by constantly releasing prostaglandin messengers.

Cyclooxygenase antagonism blocks astrocyte BAPTA-induced vasoconstriction. A, Images 1 and 2 represent an arteriole before and after astrocyte network BAPTA filling in the continual presence of the COX inhibitor indomethacin. Arteriole diameter remains unchanged despite sufficient endfoot infiltration of Alexa plus BAPTA. Rhod-2 (image 3) and its merge with the Alexa signal (images 2 + 3) are also shown. Circle ROI is the location of the astrocyte Ca2+ measurement show in B. B, Trace data from the same experiment in A. Even with good endfoot BAPTA filling (top green trace) that achieves a drop in astrocyte Ca2+ (bottom red trace), diameter does not change in the presence of indomethacin (middle gray trace). C, Summary data of diameter, astrocyte Ca2+ (**p < 0.01) and the Alexa fill in response to BAPTA filled astrocyte in the presence of indomethacin. D, Arterioles are still capable of vasoconstriction in indomethacin. Images (left) and trace data (top right) represent vasoconstriction in response to the thromboxane A2 agonist U46619 (200 nm) in the presence of indomethacin. Bottom right, Summary data. Data are mean ± SEM.
Under the framework of constitutive COX activity and the ongoing production of vasodilatory prostaglandins, we would predict the occurrence of vasoconstriction by acute application of COX antagonists. In the presence of TTX to avoid any unexpected changes in neural activity caused by COX inhibition, we found that the bath application of indomethacin (10–50 μm) indeed caused vasoconstriction (−20.70 ± 2.42%, p < 0.001, n = 7; Fig. 5A–C). The COX-1 selective antagonist SC560 (250 nm to 1 μm) in TTX also caused a robust vasoconstriction (−14.97 ± 1.26%, p < 0.001, n = 7; Fig. 5C), which was not different from indomethacin (p > 0.05). In contrast, the COX-2 selective antagonist (SC58215 1–2 μm) showed only a minor decrease in arteriole diameter (−5.34 ± 0.68%, n = 8; Fig. 5C), which was different from the degree of constriction caused by SC560 (p < 0.05). Astrocyte metabotropic glutamate receptor 5 is a potential means through which cyclooxygenase becomes activated to change arteriole diameter (Zonta et al., 2003; Takano et al., 2006). We asked whether basal mGluR5 activity provided tonic vasodilation to arterioles. However, application of the mGluR5 antagonist MTEP (5 μm) failed to affect arteriole diameter (−1.58 ± 1.12%, p > 0.05, n = 7; Fig. 5C). The detection of a drop in astrocyte Ca2+ by intracellular BAPTA in the presence of indomethacin without the appearance of vasoconstriction suggested that COX activity was downstream of astrocyte Ca2+ signaling. To further test this notion, we examined fluorescence changes in Rhod-2/AM by acute application of SC560. We found no change in astrocyte Ca2+ by SC560 (ΔF/F 0.89 ± 1.95%, p > 0.05, n = 7; Fig. 5D,E). These data suggest that COX-1 enzymes tonically provide a steady-state vasodilation to arterioles in a manner that is downstream of Ca2+ signaling and independent of mGluR5 activity.

COX-1, but not COX-2, blockade causes vasoconstriction. A, Images showing a vasoconstriction in a cortical arteriole in response to indomethacin in the presence of TTX. B, Trace data from the same experiment in A. C, Summary data showing percentage vasoconstriction caused by the broad-spectrum COX blocker indomethacin (***p < 0.001), the COX-1 selective antagonist SC560 (***p < 0.001), the COX-2 selective antagonist SC58125, and the mGluR5 antagonist MTEP. D, Traces showing that SC560 decreases arteriole diameter (bottom gray trace) without a drop in astrocyte Ca2+ (top red trace), suggesting that COX-1 is downstream of Ca2+ signaling. E, Summary data of astrocyte Ca2+ in response to bath application of SC560. Data are mean ± SEM. n.s., Not significant.
Astrocytes provide tonic vasodilation independent of spontaneous neuronal activity
Neurons in cortical brain slice preparations retain some degree of spontaneous action potential activity and neurotransmission. We asked whether astrocytes were sensing this ongoing activity as a means of regulating resting Ca2+, COX activity, and thus providing steady-state diameter control to the arteriole. Because the voltage-gated sodium channel blocker TTX would eliminate spontaneous action potentials and spike-triggered glutamate release as well as GABA release, we tested whether BAPTA-induced vasoconstriction was reduced in the presence of TTX (500 nm). Interestingly, vasoconstriction was unabated in this condition (−11.69 ± 2.66%, n = 9, p < 0.01, p > 0.05 compared with control; Fig. 6A–C), with similar Alexa-488 tau values (187.3 ± 39.3 s; Fig. 6A–C) and decreases in astrocyte Ca2+ (ΔF/F −16.27 ± 2.40%; Fig. 6A–C) to control experiments. This result suggests that astrocytes do not provide tonic diameter control by sensing spontaneous neural activity and also that astrocyte BAPTA is not changing spontaneous neural activity (Panatier et al., 2011; Poskanzer and Yuste, 2011) in a manner that influences arteriole diameter. We next tested whether miniature transmitter release events were detected by the astrocyte to enable steady-state diameter control. To do this, we used several synaptic and extrasynaptic receptor blockers targeting AMPA receptors (DNQX 5 μm), NMDA receptors (D,L-APV 50 μm), mGluR1 (LY456236 20 μm), GABAA receptors (pictotoxin 50 μm), and GABAB receptors (CGP54626). However, in the presence of TTX, bath application of this blocker cocktail did not decrease arteriole diameter (−1.50 ± 0.80%, n = 7, p > 0.05; Fig. 6D), indicating that miniature neurotransmission was also not involved.

Astrocyte-mediated tonic vasodilation is not dependent on neural activity. A, Arteriole before astrocyte network BAPTA filling in the continual presence of TTX. Circle ROI is the location of the astrocyte Ca2+ measurement and Alexa-488 measurement shown in B. B, Trace data from the arteriole in A. Alexa-488 and BAPTA filling (top green trace) achieves a drop in astrocyte Ca2+ (bottom red trace) and a corresponding decrease in arteriole diameter (middle gray trace) in the presence of TTX. C, Summary data of diameter (**p < 0.01), astrocyte Ca2+ (**p < 0.01), and the tau values for the Alexa endfoot fill in response to astrocyte BAPTA in the presence of TTX. D, Summary trace showing that arterioles are not responsive to a mixture of synaptic and extrasynaptic receptor antagonists in the presence of TTX, suggesting that miniature neurotransmitter release is not involved in generating tonic vasodilation. Data are mean ± SEM.
Awake mouse in vivo imaging reveals tonic COX-1 vasodilation
We explored whether COX-1-dependent tonic diameter control exists in vivo. To avoid the confounding side effects of anesthesia on resting blood flow (Masamoto et al., 2010), we performed two-photon imaging of the microcirculation in the barrel cortex of fully awake mice using an air supported spherical treadmill (Dombeck et al., 2007; Shih et al., 2012; Tran and Gordon, 2015). We quantified arteriole diameter changes across time as a measure of resting blood flow. We imaged astrocytes using GLAST-Cre-ERT × LSL-GCaMP3 mice and visualized microvasculature with Rhod-dextran. To affect endfeet relatively rapidly and to visualize the timing of our manipulation, we delivered the COX-1 antagonist SC560 through a tail artery cannula (80 μm in 0.2 ml lactated Ringer's to achieve ∼10 μm blood concentration), which was coloaded with FITC-dextran (0.2 mg) to estimate the arrival of the drug to the brain. The rationale was that SC560 is blood–brain barrier permeable and thus should access astrocyte endfeet upon its arrival. Consistent with our results in acute brain slices, we found that acute COX-1 blockade with SC560 reduced arteriole diameter that was timed with the detection of FITC-dextran in the arteriole lumen (diameter: −19.3 ± 4.7%; lumen tau: 788.2 ± 62.5 s, n = 7, p < 0.01; Fig. 7A–E). SC560 changed neither astrocyte endfoot spontaneous Ca2+ transients (Fig. 7F) nor endfoot resting Ca2+ (ΔF/F 3.2 ± 6.2%, p > 0.05, n = 5; Fig. 7F,G), consistent with the idea that COX-1 is downstream of Ca2+ for arteriole diameter control by endfeet. The delivery of FITC-dextran alone in control saline solution had no impact on arteriole diameter (3.9 ± 3.4%, p > 0.05, p < 0.01 compared with SC560, n = 5; Fig. 7D,E), and there was no difference in the exponential rise of the FITC signal in saline (lumen tau: 592.4 ± 71.7 s, n = 5; Fig. 7E) compared with SC560 (p > 0.05). Furthermore, saline infusion did not affect astrocyte endfoot Ca2+ (ΔF/F 0.6 ± 8.6%, p > 0.05, n = 4; Fig. 7G). Finally, we examined our behavioral data (acquired simultaneously during two-photon imaging) for any change in running behavior during tail infusions that might account for the change in arteriole diameter. We found no difference in the total time animals spent running comparing baseline to treatment, whether SC560 was infused (baseline: 156.4 ± 27.3 s; SC560: 139.8 ± 19.5 s, p > 0.05) or control saline (baseline: 153 ± 39.9 s; saline: 147.4 ± 35.2 s, p > 0.05, n = 5; Fig. 7H).

COX-1 antagonism with SC560 decreases arteriole diameter in awake mice in vivo. A, Image of a head-restrained mouse on the spherical treadmill under the two-photon microscope. B, Wide-field two-photon max projection z-stack image of the barrel cortex from a GLAST-Cre GCaMP3 mouse with vasculature labeled with Rhod-dextran. C, Rhod-dextran labelled penetrator arteriole (top images) at four different time points before and during the infusion of SC560 plus FITC-dextran (middle images) through a tail artery cannula. Bottom images, The merge. The arrival of the green FITC-dextran signal was met with a decrease in arteriole diameter. D, Top, Arteriole diameter measurement over time in response to intraluminal infusion of either SC560 (blue trace) or saline control (gray trace). Bottom, Traces represent the rise of FITC-dextran signal in the vasculature, indicating the arrival of SC560 or saline control in the lumen. SC560 plus FITC-dextran decreases arteriole diameter, whereas control saline FITC-dextran infusion fails to affect arteriole diameter. E, Summary data of maximum arteriole diameter decrease (left, **p < 0.01) and tau values of lumen infusion (right, p > 0.05) comparing SC560 versus saline control. F, Ca2+ measurements from endfeet via GCaMP3 from four different experiments show no change in response to luminal SC560 infusion. G, Astrocyte GCaMP3 Ca2+ signal summary data, showing no effect of SC560 or saline control on resting astrocyte Ca2+. H, Mouse running behavior was also unchanged during SC560 infusion or control saline infusion. Data are mean ± SEM. n.s., Not significant.
That astrocyte BAPTA-induced vasoconstriction was insensitive to TTX and that synaptic/extrasynaptic receptor blockade failed to mimic the effects of astrocyte BAPTA, suggested that tonic COX-1-dependent vasodilation would be independent of phasic neurovascular coupling. Thus, we tested the hypothesis that functional hyperemia would remain intact during COX-1 blockade. We quantified vasodilations evoked by vibrissae stimulation with air puff (5 s) onto the contralateral whisker field to elicit sensory-evoked hyperemia (Fig. 8A). Indeed, we found that whisker-evoked vasodilations were not different (p > 0.05, n = 8) comparing before (individual animal peak average: 24.9 ± 4.7%, Fig. 8B; all experiments averaged over time: 14.8 ± 3.3%, Fig. 8C,D) to during SC560 infusion (individual animal peak average: 20.2 ± 3.5%, Fig. 8B; all experiments averaged over time: 12.6 ± 3.0%, Fig. 8C,D). We also examined the failure rate of evoked vasodilation on a trial-by-trial basis in each animal. We found no effect of SC560 (control: 18.3 ± 12.1%; SC560: 18.4 ± 7.8%, p > 0.05; Fig. 8E), suggesting that COX-1 inhibition did not affect the fidelity of activity-dependent neurovascular coupling. Finally, we asked whether COX-1 antagonism reduced naturally occurring functional hyperemic events in the awake animal. We examined our data for the degree of fluctuation in arteriole diameter over time when providing no experimental stimulus. We reasoned that a change in the standard deviation of diameter measurements would suggest an alteration in ongoing phasic neurovascular coupling. However, we failed to detect a consistent change to arteriole diameter fluctuation during SC560 infusion (control: 8.17 ± 1.22; SC560: 8.92 ± 1.40, n = 5; Fig. 8F). These data suggest that the tonic COX-1-dependent pathway that enables steady-state vasodilation is functionally separate from neural activity evoked vasodilation.
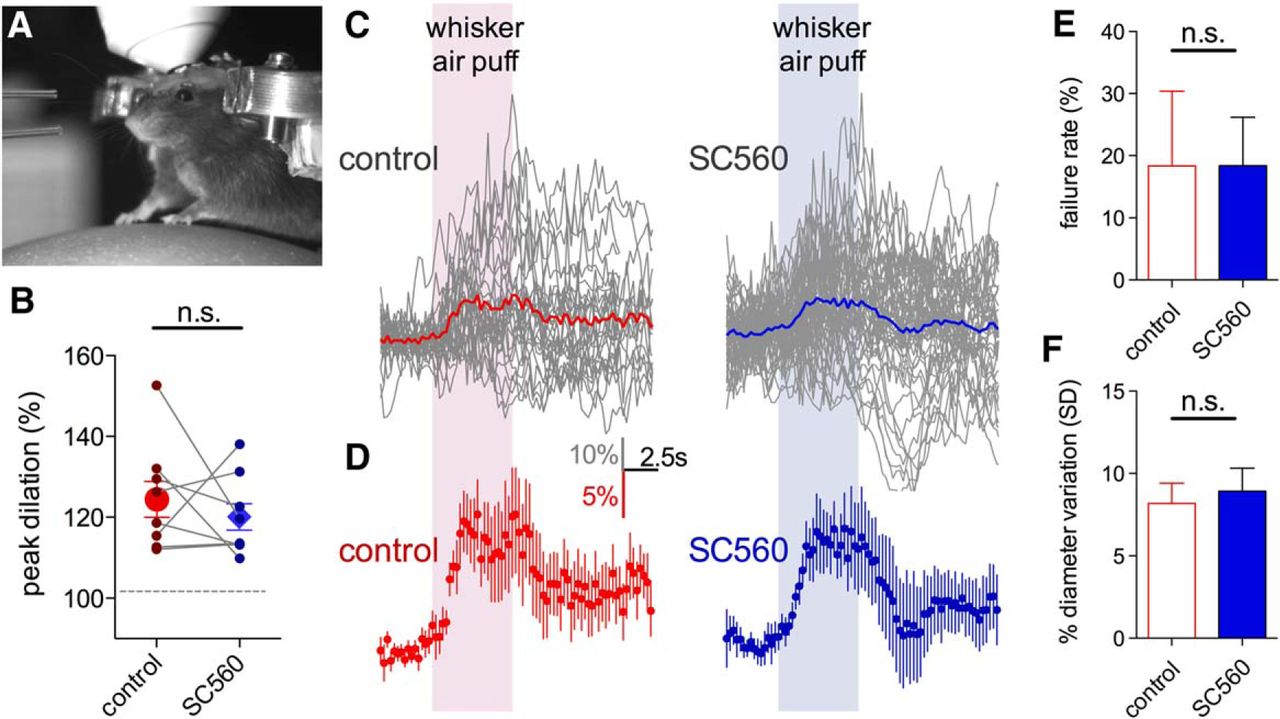
Functional hyperemia is preserved during COX-1 antagonism in awake mice in vivo. A, Picture of a mouse on the spherical treadmill with air puffers visible on the left for vibrissae stimulation. B, Peak vasodilations in response to vibrissae stimulation in each animal before and during luminal SC560 infusion. Paired observations (connecting lines) and summary (large points with SEM) show no difference on peak dilation during SC560. C, Raw arteriole diameter traces (gray) of all trials in all animals in response to vibrissae stimulation during the control period (left) or during SC560 (right). Colored traces represent the average of all trials. D, Summary data showing the overall average obtained from individual animal averages during control (left, red) and during SC560 (right, blue). SC560 fails to affect functional hyperemia at a dose that decreases baseline arteriole diameter. E, SC560 does not alter the failure rate of sensory-evoked vasodilation. F, SC560 does not change naturally occurring fluctuations in arteriole diameter, suggesting no effect on ongoing functional hyperemic events. Data are mean ± SEM. n.s., Not significant.
Discussion
Here we describe a previously unrecognized form of steady-state arteriole tone control by astrocyte endfeet, a process we termed tonic brain blood flow control. Our data suggest that astrocytes provide a constant vasodilation through the constitutive release of prostaglandins via resting Ca2+ and COX-1 activity. This particular pathway appears to operate independent of phasic neurovascular coupling, as (1) a relatively low dose of a COX-1 inhibitor decreases baseline arteriole diameter without affecting evoked vasodilation caused by brief increases in synaptic activity, (2) astrocyte BAPTA vasoconstrictions still occur in TTX, and (3) glutamate and GABA receptor blockers do not mimic the effect of astroctye BAPTA. To this point, resting blood flow control in the brain has been largely attributed to autoregulation by the vasculature itself via the myogenic response, so that flow can be kept constant across various blood pressures (Davis and Hill, 1999). Additionally, pO2 and pCO2 have also been well described to help determine resting blood flow via peripheral and central chemoreception (Marshall, 1994) and local feedback effects via pH on vascular ion channels (Willie et al., 2014). However, an additional contribution from local feedforward pathways that use cell Ca2+ and the release of diffusible messengers onto adjacent arterioles has not been assigned to resting brain blood flow control previously. Our data detail an intriguing mechanism of tonic blood flow regulation via astrocytes, independent of rapid changes in neural activity, pO2, pCO2, and pressure-mediated responses. Whereas functional hyperemia likely helps restore the energy deficits associated with rapid neural signaling from action potentials and synaptic transmission (Howarth et al., 2012), local neurons and glia have numerous other energy-consuming tasks, such as maintaining resting membrane potential, protein synthesis, cytoskeletal regulation, etc., which must be met by the basal blood supply. Thus, tonic arteriole control by astrocytes may be linked to the basal metabolic demands of the tissue.
Before our work, some evidence for a tonic feed forward pathway comes from the use of inhibitors against CYP450 and COX, which decrease resting blood flow by 20%–50% (Alkayed et al., 1996; Peng et al., 2004). In the case of COX, this effect has been appreciated for more than three decades (Dahlgren et al., 1981). Recent human studies have observed similar phenomena, in which an attenuation in baseline brain blood flow was observed with the general COX inhibitor indomethacin but not with the COX-2-selective inhibitor Celebrex, suggesting a COX-1-medated effect (Beaudin et al., 2014). A common assumption was that the effect on resting blood flow reflected a reduction in ongoing functional hyperemic events. This is supported in part, as the COX-1 inhibitor SC560 has been used in vivo to blunt functional hyperemia, although high doses were typically applied to the brain surface (500 μm) (Takano et al., 2006; Liu et al., 2012). Our experimental strategy was to use as little SC560 as possible and deliver via the blood, so that the compound would be in close apposition to the perivascular endfeet. This allowed us to demonstrate a decrease in arteriole diameter without affecting activity-dependent vasodilation in response to whisker stimulation. Our result is consistent with earlier gene knock-out work that demonstrated a decrease in resting blood flow with no effect on functional hyperemia in COX-1 null mice (Niwa et al., 2001), whereas there was a significant reduction of activity-dependent blood flow increases in COX-2 knock-outs (Niwa et al., 2000). Additionally, evoked increases in CBF have been shown to be reduced by either COX-2 or nonselective COX inhibitors, but not by a COX-1 specific inhibitor (Lecrux et al., 2011). This suggests separate roles for COX-1 and COX-2 in blood flow control. Our data indicate that the separate role for COX-1 occurs via tonic control by astrocytes. Finally, although some work has failed to detect COX-1 in astrocytes using single-cell RT-PCR (Lecrux et al., 2011), transcriptome analysis using FACS with astrocyte-specific promoters (Cahoy et al., 2008; Zhang et al., 2014) and immunohistochemistry analysis (Takano et al., 2006; Gordon et al., 2008) have detected COX-1 in astrocytes. Our occlusion experiment demonstrating a lack of BAPTA-induced constriction in the presence of indomethacin indicates at least part of a COX pathway operating in astrocytes as a key component regulating arteriole tone. However, it remains possible that only upstream Ca2+-dependent components of this pathway, such as cPLA2, which produce diffusible arachidonic acid, are expressed in astrocytes (Farooqui et al., 1997) and that COX-1 is localized elsewhere, such as the blood vessel itself.
In vitro data unequivocally demonstrate that elevations in astrocyte endfeet Ca2+ affect arteriole diameter (Mulligan and MacVicar, 2004; Straub et al., 2006; Gordon et al., 2008); yet recent in vivo data raise doubts on astrocyte involvement during transient blood flow increases in response to rapid and discrete bursts of neural activity (Nizar et al., 2013; Bonder and McCarthy, 2014). The presence (Takano et al., 2006; Winship et al., 2007; Petzold et al., 2008) versus absence (Nizar et al., 2013; Bonder and McCarthy, 2014) of correlative astrocyte Ca2+ transients during functional hyperemia in vivo may be explained by the difficulty in observing ultrafast Ca2+ signals (Lind et al., 2013), isolating fine process Ca2+ signals (Otsu et al., 2015) or by the idea that astrocytes modulate blood flow only to sustained neural activation (Schulz et al., 2012). Our description of tonic diameter control by astrocytes via a COX-1 pathway that is independent of functional hyperemia raises a question as to whether this mechanism works in tandem with previously described phasic, activity-dependent processes. More experiments are required to understand how astrocytes participate in both forms of control, such as the compartmentalization of constitutive COX-1 versus inducible COX-2 pathways. Notably, immunohistochemistry analysis targets COX-1 expression largely to endfeet apposed to arterioles over other astrocyte compartments (Takano et al., 2006). Although previous uncaging experiments provide good evidence that it is the astrocyte endfoot that communicates with the arteriole (Mulligan and MacVicar, 2004; Straub et al., 2006; Gordon et al., 2008), our measures of endfoot BAPTA/Alexa filling that correspond with vasoconstriction are, however, correlative. Nevertheless, that the BAPTA infusion must come close to the arteriole to observe the vasoconstriction suggests that either endfeet or astrocyte fine process closely apposed the arteriole are involved.
Numerous pathways are known for functional hyperemia; thus, additional pathways likely exist for tonic arteriole control. For example, a recent study conducted in retinal explants has described a tonic vasoconstriction of arterioles via constitutive ATP release. Although a role for glia was not unequivocally demonstrated with patch electrodes, the effect was sensitive to fluorocitric acid, suggesting a potential contribution from Muller cells (Kur and Newman, 2014). This pathway may work in parallel to constitutive COX effects: the former for tonic constriction and the latter for tonic dilation. What transmitters or intracellular factors modulate or select for these processes remains unclear. Metabolic factors, such as the oxygen, lactate, and adenosine levels, can dictate the polarity of the vasomotor response caused by astrocytes in response to uncaging Ca2+ (Gordon et al., 2008); thus, these variables may also play a role in influencing the control of arteriole tone. Additionally, the processes regulating resting intracellular Ca2+ in astrocytes are not known. If, for instance, specific patterns of neural activity could induce long-lasting changes to resting Ca2+, this could serve as means to change the degree of steady-state vasodilation by astrocytes to accommodate plastic changes in the brain.
Finally, understanding tonic blood flow control mechanisms may be important for the proper interpretation of fMRI data. Functional hyperemia is the basis of the BOLD fMRI signal, which is an indirect measure of neural activity that is used extensively in research and medicine. Importantly, the magnitude of the BOLD signal can be influenced by the resting condition, such as the amount of resting blood flow, resting blood volume, and basal rate of oxygen consumption (Brown et al., 2003; Sicard and Duong, 2005). Additionally, the degree of arteriole tone affects the dynamic range of the vessel (i.e., the degree to which arterioles can dilate or constrict) (Blanco et al., 2008), which would presumably change deoxygenated hemoglobin dynamics. Consistent with this idea, deleterious mutations in the COX-1 gene in humans impact the magnitude of BOLD signals (Hahn et al., 2011). By understanding the factors and/or conditions that regulate tonic arteriole diameter control by astrocytes, this may lead to better interpretation of fMRI data and perhaps new experimental standards.
Footnotes
This work was supported by the Canada Institutes of Health Research and the Heart and Stroke Foundation of Canada. G.R.G. was supported by Canada Research Chairs and the Heart and Stroke Foundation Alberta. D.G.R. was supported by Alberta Innovates Health Solutions. C.H.T.T. was supported by Alberta Innovates Health Solutions. We thank Dr. Jaideep Bains for help in editing the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Grant R. Gordon, Room 1B40A, Building HRIC, 3330 Hospital Drive NW, Calgary, Alberta T2N 4N1, Canada. gordong{at}ucalgary.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}