

Abstract
Protein kinase M ζ (PKMζ), an atypical isoform of protein kinase C, has been suggested to be necessary and sufficient for the maintenance of long-term potentiation (LTP) and long-term memory (LTM). This evidence is heavily based on the use of ζ inhibitory peptide (ZIP), a supposed specific inhibitor of PKMζ that interferes with both LTP and LTM. Problematically, both LTP and LTM are unaffected in both constitutive and conditional PKMζ knock-out mice, yet both are still impaired by ZIP application, suggesting a nonspecific mechanism of action. Because translational interference can disrupt neural activity, we assessed network activity after a unilateral intrahippocampal infusion of ZIP in anesthetized rats. ZIP profoundly reduced spontaneous hippocampal local field potentials, comparable in magnitude to infusions of lidocaine, but with a slower onset and longer duration. Our results highlight a serious confound in interpreting the behavioral effects of ZIP. We suggest that future molecular approaches in neuroscience consider the intervening level of cellular and systems neurophysiology before claiming influences on behavior.
SIGNIFICANCE STATEMENT Long-term memory in the brain is thought to arise from a sustained molecular process that can maintain changes in synaptic plasticity. A so-called candidate for the title of “the memory molecule” is protein kinase M ζ (PKMζ), mainly because its inhibition by ζ inhibitory peptide (ZIP) interferes with previously established synaptic plasticity and memory. We show that brain applications of ZIP that can impair memory actually profoundly suppress spontaneous brain activity directly or can cause abnormal seizure activity. We suggest that normal brain activity occurring after learning may be a more primary element of memory permanence.
Introduction
The enduring nature of memory is a captivating issue in neuroscience. Changes in synaptic strength are thought to underlie learning and memory, placing the focus of neurobiological mechanisms on molecular modulation of synaptic transmission (Kandel, 2004; Morgado-Bernal, 2011). One protein product, protein kinase M ζ (PKMζ), has emerged as an attractive molecular candidate for memory maintenance based on its association with synaptic and behavioral plasticity. The activity of PKMζ is correlated with the expression of long-term potentiation (LTP) and long-term memory (LTM) (Sacktor et al., 1993; Ling et al., 2002), its overexpression results in increased memory maintenance (Shema et al., 2011), and its inhibition by ζ inhibitory peptide (ZIP) eliminates synaptic facilitation and impairs memory (for reviews, see Sacktor, 2012; Kwapis and Helmstetter, 2014).
Recent evidence has disputed PKMζ's essential role in memory. Mice with conditional and constitutive PKMζ genetic knock-outs (KOs) demonstrate normal LTP and memory. Problematically, these processes are still impaired using ZIP, a supposed specific inhibitor (Lee et al., 2013; Volk et al., 2013), suggesting that its effects are independent of PKMζ. Although some have speculated on ZIP's influence on other similar molecular targets, such as PKMζ isoform protein kinase C lambda (PKCλ) (Ren et al., 2013; Jalil et al., 2015), it may be that these disruptive influences are more general, involving other neurobiological processes.
Both general and specific protein knock-down techniques have been shown to have more widespread and catastrophic consequences for basic neurobiological function (Neumann et al., 1995; Garcia-Osta et al., 2006; Qi and Gold, 2009; Sharma et al., 2012). Our hypothesis was that ZIP might also impair ongoing neural activity. Here, we show that intrahippocampally applied ZIP (but not a scrambled version of the peptide or vehicle) dramatically inhibits neural activity that is similar in magnitude to, but longer-lasting than, that produced by the sodium channel blocker lidocaine. We feel that this illustrates the potential confound of testing the behavioral influence of so-called targeted molecular techniques without considering the intervening level of neural activity.
Materials and Methods
Subjects.
Experiments were performed on 33 adult male Sprague Dawley rats weighing 323 ± 14 g. All experimental protocols conformed to the guidelines of the Canadian Council on Animal Care and were approved by the Biosciences Animal Care and Use Committee at the University of Alberta.
Drugs.
Urethane and atropine were dissolved in distilled water (0.67 g/ml and 0.05 mg/ml, respectively) and lidocaine was dissolved in PBS at a concentration of 4% (weight by volume; Sigma-Aldrich). ZIP (Myr-SIYRRGARRWRKL-OH) and scrambled ZIP (sZIP; Myr-RLYRKRIWRSAGR-OH) were obtained from Tocris Bioscience in multiple batches and were dissolved in a minimal volume of acetic acid (maximum 7 μl) and diluted to 50 μl with PBS (10 mm; 10 nmol/μl).
Surgery.
As described previously (Wolansky et al., 2006), rats were implanted with a jugular catheter while anesthetized with isoflurane and were subsequently transferred to urethane anesthesia by slowly (0.03–0.08 ml/min) infusing intravenously to a final anesthetic dose of 1.25 ± 0.07 g/kg. Subcutaneous atropine methyl nitrate (0.05 mg/kg; Sigma-Aldrich) was administered to prevent respiratory secretions. Stereotaxic methods were used to implant bipolar Teflon-coated stainless steel (125 μm diameter) wire bilaterally in the dorsal hippocampus to straddle the CA1 pyramidal layer (−3.3 mm AP, ±2.2 mm ML from bregma). In the hemisphere ipsilateral to infusion, the electrode was fastened along the shank of a 30 gauge stainless steel cannula and inserted similarly.
Recording and infusion procedures.
Local field potential (LFP) recordings were amplified at a gain of 1000 and band-pass filtered between 0.1 Hz and 20 kHz using a 4-channel differential AC amplifier (model 1700; A-M Systems). Signals were digitized using an A-D board (Digidata 1322a; Molecular Devices), anti-alias filtered at 500 Hz, sampled at 1 kHz, and recorded on a PC computer running AxoScope (Molecular Devices).
After baseline spontaneous recordings of at least 20 min (30.51 ± 2.6), intrahippocampal microinfusions were delivered at 0.5 μl/min over 2 min (1 μl total) using a microinfusion system connected to the ipsilateral cannula (Sharma et al., 2012). These parameters replicated those used previously for behavioral assessment of dorsal hippocampal infusions of ZIP. Recordings were made continuously for at least 1 h after infusions (83.41 ± 6.7 min). After recordings, the positions of electrodes and cannulae were visualized using standard brain histological techniques that we have described previously (Sharma et al., 2012). Two individuals blinded to the experimental conditions scored successful cannula placements as those beneath the pyramidal cell layer of CA1.
Data analysis.
Data were analyzed in Clampfit (Molecular Devices) and using both built in and custom-written code in MATLAB (The MathWorks). The root mean square (RMS), the average amplitude fluctuations of LFP traces, of 10 min segments corresponding to baseline (B), ∼15 min (P15), and ∼1 h (P60) after infusion were computed. RMS was also evaluated in siding windows of 30 s that were moved in increments of 10 s. Spectral analysis was initially performed using Clampfit using a fast Fourier transform on data segments (1.50 ± 0.04 min for θ; 1.10 ± 0.04 min for slow oscillation, SO) with a Hamming window of 4096 samples length. Supplemental analyses were conducted in MATLAB (Wolansky et al., 2006), which involved a modified Welch's periodogram method (6 s, Hamming-windowed segments with 2 s overlap) on continuous data segments of at least 30 s. In all cases, power was reported and averaged across the θ (3.2–5.6 Hz) and the SO (0.25–1.7 Hz) frequency bands and were consistent with what is normally observed under urethane. Confidence limits (95%) of the spectral estimates were also computed. Spectrograms were constructed by computing spectra as above for a series of 30 s windows slid across data segments in steps of 10 s.
Under urethane anesthesia, spontaneous alterations occur between activated (θ) and deactivated (SO) states similar to REM and slow-wave states occurring during natural sleep (Clement et al., 2008). Because of this, the closest epoch of each state to the desired time point was chosen. After ipsilateral infusions, the power of the unaffected contralateral signals was monitored for conformance with baseline conditions to ensure equivalent comparisons. Averages values are reported together with the SEM (mean ± SEM). Data were analyzed using a repeated-measures within-subjects two-way ANOVA (VassarStats: Web Site for Statistical Computation, http://vassarstats.net) for condition. Post hoc Tukey HSD tests using only ipsilateral data confirmed any significant changes (α = 0.05) between the specified time point and baseline (VassarStats).
Results
Based on exclusion criteria for cannula position and signal strength, 12 ZIP, five sZIP, four lidocaine, and four PBS experiments were included (eight animals were removed due to misplacement of cannula position). In seven of 12 ZIP experiments and one of five sZIP experiments, the infusion induced seizures or spreading depression, which were not observed after lidocaine or PBS and had lasting suppressive effects; therefore, we excluded these from further analysis. We evaluated the regions around cannula tips and found no obvious lesions other than the cannula track for any group. No changes were observed in contralateral signals in any group (see Fig. 3B,D). These contralateral analyses are not reported because these data were analyzed in the omnibus ANOVAS below for each group.
ZIP suppresses activity
ZIP caused a marked reduction in raw ipsilateral hippocampal LFP amplitude, plateauing at an average latency of 11.95 ± 1.8 min (n = 5; Fig. 1A). Two-way ANOVAs showed a significant reduction across hemisphere (RMS: F(1,4) = 9.74, p = 0.036; θ: F(1,4) = 10.51, p = 0.032), time (RMS: F(1,3) = 44.67, p < 0.0001; θ: F(2,8) = 13.57, p = 0.0027), as well as their interaction (RMS: F(1,3) = 9.48, p = 0.0077; θ: F(2,8) = 9.93, p = 0.0068). Tukey HSD tests confirmed a significant reduction in the ipsilateral signal for both RMS and θ at P15 (RMS: 52.46 ± 9.0%, p < 0.01; θ: 37.81 ± 11.0%, p < 0.01) and P60 (RMS: 63.88 ± 6.2%, p < 0.01; θ: 50.38 ± 9.9%, p < 0.01) that outlasted the duration of every experiment (107.64 ± 13.3 min; Figs. 1B,C, 2A–C).

ZIP causes a profound reduction in neural activity. A, Intrahippocampal infusions of ZIP reduce local neural activity. Simultaneous LFP recordings from the contralateral and ipsilateral hippocampus are shown before (left) and ∼15 min after (right) ZIP infusion (expansions at top). Suppression was observed in the ipsilateral but not contralateral traces after infusion. B, Sliding RMS values over time during the same period shown in A by averaging RMS across 30 s segments of data and sliding by 10 s. A substantial decrease in RMS is observed for the ipsilateral but not contralateral postinfusion signals. C, Spectrograms demonstrating a prominent power reduction specific for the ipsilateral (bottom) signal during the same time period.

ZIP reduces RMS and signal power in the ipsilateral hippocampus. A, Amplitude distribution (left) for signals during baseline (B, black), P15 (red), and P60 (gray). Right, Net RMS is reduced in the ipsilateral (red circles), but not contralateral (black squares) hemisphere at P15 and P60. B, C, Power spectra (95% confidence intervals; shading) highlight the reduction in power of the ipsilateral signal occurring across the entire bandwidth after ZIP administration (middle) during both the activated (B) and deactivated (C) states. Black traces correspond to baseline conditions and red traces correspond to P15. As shown, no significant changes were observed for contralateral signals (left). Dotted vertical line shows the θ (activated, B) and SO (deactivated, C) frequency peaks. Average θ and SO power is shown for this experiment before (baseline: marked “B”) and after ZIP infusion (15 and 60 min; right). There was a large decrease in both θ and SO power in the ipsilateral but not the contralateral signal.
The degree, latency, and duration of suppression from ZIP were compared with infusions of sZIP, PBS (both negative controls), and lidocaine (a positive control). As with ZIP, lidocaine yielded a significant reduction across hemisphere (RMS: F(1,4) = 33.63, p = 0.010; θ: F(1,3) = 10.16, p = 0.0498), time (RMS: F(1,3) = 26.96, p = 0.001; θ: F(2,6) = 18.03. p = 0.0029), and their interaction (RMS: F(1,3) = 22.19, p = 0.00169; θ: F(2,6) = 45.78, p = 0.00023). This ipsilateral reduction was only significant at P15 (RMS: 51.00 ± 1.4%, p < 0.01; θ: 27.48 ± 5.1%, p < 0.01), but values returned to baseline at P60 (p > 0.05; Fig. 3A,C,E). The lidocaine effect occurred rapidly, plateauing at 8.5 ± 1.4 min and lasting 34.75 ± 1.5 min. There were no significant reductions after sZIP across hemisphere (RMS: F(1,4) = 0.10, p = 0.768; θ: F(1,3) = 0.067, p = 0.81), time (RMS: F(1,3) = 1.66, p = 0.266; θ: F(2,6) = 3.66, p = 0.092), and their interaction (RMS: F(1,3) = 1.25, p = 0.35; θ: F(2,6) = 2.32, p = 0.18), although all probabilities for θ power were close to significance. No reductions were observed for PBS infusions across hemisphere (RMS: F(1,4) = 5.12, p = 0.109; θ: F(1,3) = 0.17, p = 0.71), time (RMS: F(1,3) = 4.56, p = 0.062; θ: F(2,6) = 1.03, p = 0.41), and their interaction (RMS: F(1,3) = 3.912, p = 0.0818; θ: F(2,6) = 0.83, p = 0.48). These data are shown in Figure 3, B and D. A similar pattern of results was observed for the SO state (data not shown).

ZIP causes a specific and profound inhibition of neural activity comparable in magnitude but longer lasting than that of lidocaine. A, Average and individual normalized RMS values for ipsilateral hippocampal signals are shown for baseline, P15, and P60. Intrahippocampal infusions of ZIP (n = 5; i), sZIP (n = 4; ii), PBS (n = 4; iii), and lidocaine (n = 4; iv). Significant reductions (*p < 0.05) are observed for ZIP at both time points and for lidocaine at P15. B, Same data as in A but for the contralateral hemisphere. There are no significant reductions in any group. C, Average and individual normalized power values in the θ (3.5–4.6) range during the activated state for ipsilateral signals are shown for the same conditions as in A. Significant reductions are observed for ZIP at P15 and P60 and for lidocaine at P15. D, Same data as in C but for the contralateral hemisphere. There are no significant reductions in any group. E, Subset of experiments (n = 3) in which recordings were made for at least 2 h after infusion. The significant reduction at P15 and P60 was maintained at P120.
A subset of ZIP experiments (n = 3) were extended for at least 2 h (130.51 ± 7.5 min; P120; Fig. 3E). There was a significant reduction across hemisphere for θ power but not RMS (RMS: F(1,2) = 2.08, p = 0.286; θ: F(1,2) = 13.34, p = 0.067), time (RMS: F(3,6) = 28.65, p = 0.00059; θ: F(3,6) = 13.38, p = 0.0046), and interaction for θ power but not RMS (RMS: F(3,6) = 1.94, p = 0.224; θ: F(3,6) = 5.08, p = 0.04). However, post hoc tests showed significant ipsilateral reductions at P15 (RMS: 67.08 ± 6.4%,p < 0.01; θ: 56.94 ± 4.8%, p < 0.01), P60 (RMS: 74.00 ± 4.4%, p < 0.05; θ: 66.02 ± 6.9%, p < 0.01), and P120 (RMS: 69.42 ± 6.6%, p < 0.01; θ: 61.50 ± 7.6%, p < 0.01).
Given that individual spectral estimates can be assessed statistically using confidence intervals, we examined the proportion of cases in which the reduction of power in θ (3–5 Hz) or SO (0.16–1.5 Hz) bandwidths demonstrated no overlap of the 95% confidence limits at P15 and P60 compared with baseline for both ipsilateral and contralateral recordings. By using a pseudorandomization model, we showed that, for all frequencies across experiments, a maximum of 16.7% showed significant reductions for the contralateral signal. Consistently larger proportions than this were observed at P15 for the ipsilateral signal during both θ and SO after ZIP, sZIP, and lidocaine infusions. This was also the case at P60, but only for ZIP infusions (Fig. 4).
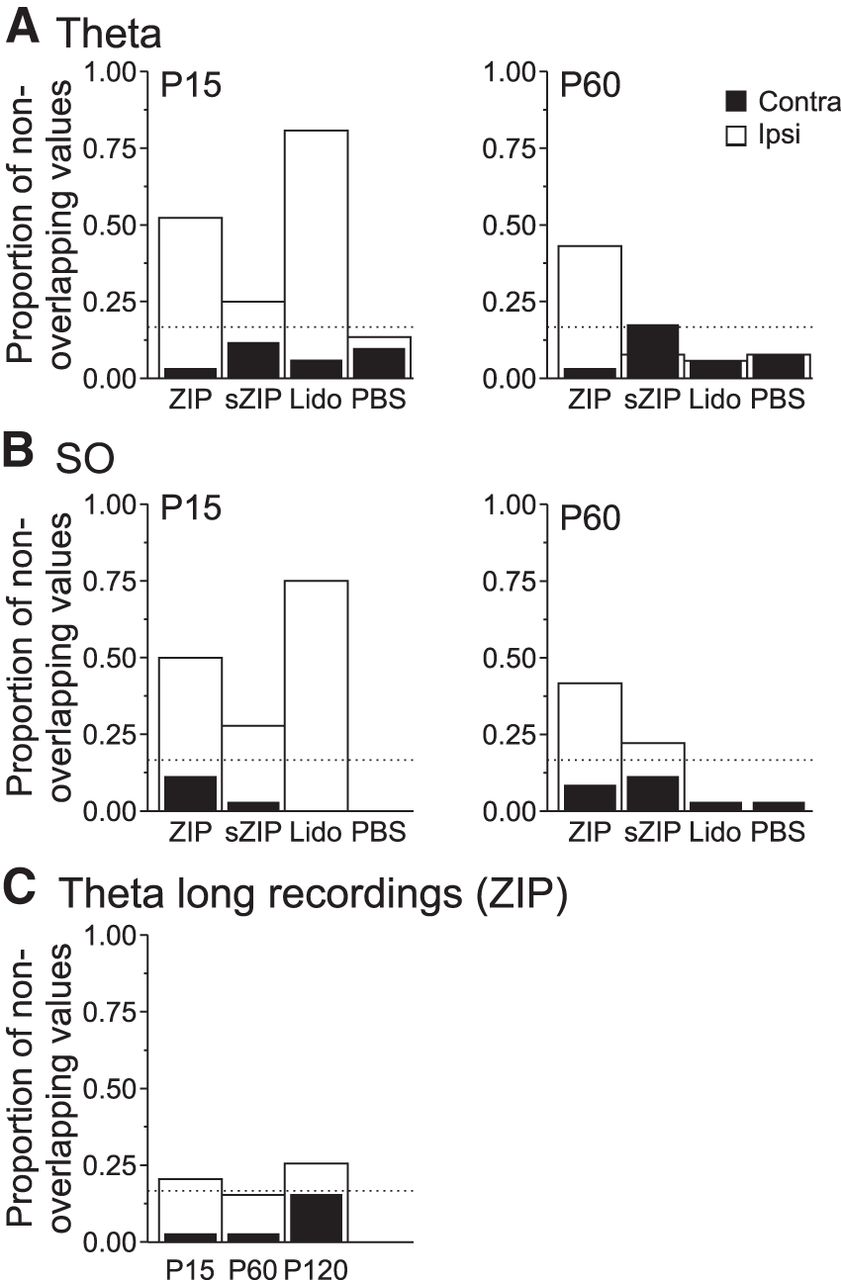
A significant proportion of spectral frequencies show significant reductions across individual experiments after infusions of ZIP, sZIP, and lidocaine. A, Significant proportion (>0.167; threshold dotted line) of spectral lines across experiments showed nonoverlapping confidence intervals in the θ band for the ipsilateral hemisphere at P15 after ZIP, sZIP, and lidocaine (but not PBS) infusions. This effect was only maintained for ZIP at P60. As shown, there are no effects for the contralateral hemisphere in any group. B, A similar pattern is observed for reductions of SO band activity. C, These reductions were also observed for the long-term (>2 h) ZIP experiments.
Discussion
We evaluated the hypothesis that intrahippocampal infusions of ZIP using infusion methods shown previously to weaken memory in behaving animals impairs ongoing neural signaling. Indeed, a marked and lasting decrease in LFP activity comparable in magnitude with—although delayed in latency and longer lasting than—infusion of 4% lidocaine was observed. Given that intrahippocampal infusions of lidocaine before retention testing produce memory deficits in hippocampal-dependent tasks such as the Morris water maze (Bohbot et al., 1996; Broadbent et al., 2006), we propose that experiments in which ZIP is similarly infused just before retention testing (the most common protocol used; Kwapis and Helmstetter, 2014) may produce memory deficits due to its detrimental influence on neural activity as opposed to any specific effect on PMKζ. Indeed, neural inactivation (or even epileptic disruption, as we also observed) after ZIP administration may have detrimental effects on memory measures beyond the normal 2 h post-ZIP application retention test. At a minimum, our findings caution against conclusions of a specific molecular effect of ZIP on memory, especially when its influence on neural activity has not been documented.
Role of PKMζ in synaptic plasticity and memory
Although PKMζ has been dubbed “the memory molecule,” the veracity of this claim is presently much less clear (Frankland and Josselyn, 2013; Jones, 2013; Yong, 2013). The importance of PKMζ in synaptic plasticity and memory was based on persistent increases in expression during LTP (Osten et al., 1996), its role in upregulation of plasticity-related protein synthesis (including its own; (Westmark et al., 2010), and its presumed necessity and sufficiency for maintaining LTP and LTM. Its critical role in synaptic plasticity and memory processes has been seriously questioned after the demonstration that constitutive and conditional PKMζ KO mice show intact LTP and LTM (Lee et al., 2013; Volk et al., 2013). Interestingly, and highly relevant for our results, applications of ZIP in these mice impair both LTP and LTM in the absence of PKMζ, strongly suggesting a PKMζ-independent mechanism of action for this supposed specific peptide inhibitor.
The supposed specificity of ZIP for the inhibition of PKMζ has been commensurate with its use in behavioral paradigms (Ling et al., 2002; Pastalkova et al., 2006; Serrano et al., 2008). Although lower doses of ZIP seem to act selectively on PKMζ, higher doses affect at least one other PKC isoform, PKCλ (Ren et al., 2013). Given the 100-fold higher concentration of ZIP used in behavioral studies compared with in vitro, nonspecificity is very likely an issue that may extend to non-PKC isoforms such as CaMKII (calcium/calmodulin-dependent protein kinase II) that are also involved in synaptic plasticity (Ling et al., 2002; Lisman, 2012). Our data emphasize that arguments regarding ZIP's biochemical profile may not be as imperative as our demonstration of a fundamental disruption of general neurophysiological function.
Impact of molecular manipulations on activity
Our study adds to the mounting evidence that biochemical manipulations can affect neuronal excitability and network functionality. For example, translational inhibitors suppress ongoing activity profoundly (Sharma et al., 2012; Shires et al., 2012). Other neurobiological dysfunctions, such as disruption of cortical motor map expression (Kleim et al., 2003) and catastrophic, long-term disruptions in neuromodulator release (Canal et al., 2007; Qi and Gold, 2009; Sadowski et al., 2011), have been observed after translational inhibition. Although perhaps compelling for those who consider online and offline neural signaling to be essential for enduring memory processes, the reaction of some researchers has been to suggest that protein-specific molecular targeting methods are immune to these potential problems (Stern and Alberini, 2013). This is a curious contention because it has also been documented that antisense inhibition of specific neuronal proteins reduces network activity (Garcia-Osta et al., 2006), as well as the excitability of single neurons, even when stimulated antidromically (Neumann et al., 1995). The downregulation of cAMP-response element-binding protein (CREB), an essential element in the long-term expression of synaptic potentiation, results in memory impairments, but may do so by simply reducing neuronal excitability (Han et al., 2006; Jancic et al., 2009). Conversely, the upregulation of CREB causes enhancement of memory that is paralleled by increased neuronal excitability (Lopez de Armentia et al., 2007; Josselyn, 2010). Here, we demonstrate that the use of ZIP, which has been suggested to interfere selectively and posttranslationally with either or both PKMζ and PKMλ (Jalil et al., 2015), also has broader neurobiological effects that confound interpretations of behavioral experiments.
Although prior studies have implied that ZIP does not impair spontaneous or evoked network activity, these studies only reported data for time windows beyond 2 h after infusion (Madroñal et al., 2010; Barry et al., 2012). Our study demonstrates significant inhibition of local network activity within that time frame. This timing is similar to the suppressive effect of anisomycin (Sharma et al., 2012), but slower than agents acting directly on ion channels (present study and van Duuren et al., 2007), which suggests that it acts via a (yet unknown) biochemical signaling mechanism. Furthermore, the presence of epileptiform activity after ZIP administration in seven of 12 (∼60%) of our ZIP experiments (despite the fact that urethane anesthesia is known to dampen seizure activity; Cain et al., 1989) presents additional interpretational problems for prior studies. For example, although ZIP was suggested to interfere with place cell stability (Barry et al., 2012), electrophysiological data were only reported after at least a 2 h period after ZIP administration. In the absence of clear neurophysiological data within this time frame, it is difficult to eliminate the possibility of an activity-modulating influence of ZIP, especially because it is known that epileptic activity can cause remapping of place cells (Zhou et al., 2007). In the future, it will be important to extend our studies to awake and behaving animals to further assess the relevance of ZIP-induced activity disruption on behavior.
Conclusion
We show that intrahippocampal microinfusions of ZIP at concentrations that have been shown previously to disrupt LTM suppressed neural activity robustly. Therefore, prior work using this peptide to test the involvement of PKMζ (or other related isoforms) in memory maintenance may be confounded by a disruption of neural signaling. More generally, we can conclude that even “targeted” molecular techniques that purportedly interfere with single proteins may have broader neurobiological consequences. We caution that the interpretation of behavioral effects induced by molecular manipulations of subcellular processes must take into account intervening neurobiological levels, including that of neural excitability.
Footnotes
This work was supported by the Natural Science and Engineering council of Canada (NSERC Grant 249861 to C.T.D.). T.L.M. was additionally supported by a Branch Out Neurological Foundation Summer Student Scholarship. We thank Jonathan Dubue and Claire Scavuzzo for contribution to data collection and Tara Whitten, Stacey Greenberg, Jesse Jackson, Kyle Mathewson, Claire Scavuzzo, and Lisa Rimstad for helpful editorial comments.
The authors declare no competing financial interests.
- Correspondence should be addressed to Clayton T Dickson, Department of Psychology, University of Alberta, Edmonton, Alberta T6G 2E9, Canada. clayton.dickson{at}ualberta.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}