

Abstract
What has become standard textbook knowledge over the last decade was a hotly debated matter a decade earlier: the proposition that new neurons are generated in the adult mammalian CNS. The early discovery by Altman and colleagues in the 1960s was vulnerable to criticism due to the lack of technical strategies for unequivocal demonstration, quantification, and physiological analysis of newly generated neurons in adult brain tissue. After several technological advancements had been made in the field, we published a paper in 1996 describing the generation of new neurons in the adult rat brain and the decline of hippocampal neurogenesis during aging. The paper coincided with the publication of several other studies that together established neurogenesis as a cellular mechanism in the adult mammalian brain. In this Progressions article, which is by no means a comprehensive review, we recount our personal view of the initial setting that led to our study and we discuss some of its implications and developments that followed. We also address questions that remain regarding the regulation and function of neurogenesis in the adult mammalian brain, in particular the existence of neurogenesis in the adult human brain.
Introduction
Over the last two decades, numerous studies have demonstrated that a large majority of mammalian species retain the capacity for neurogenesis in the hippocampus into adult life. Our paper published in March 1996 (Kuhn et al., 1996), together with other concurrent studies, indicated the establishment of adult neurogenesis as a new research area, even though these studies were rediscoveries of a phenomenon described >30 years earlier by Altman, Bayer, Kaplan, and others (Altman and Das, 1965; Kaplan and Hinds, 1977; Bayer, 1983). Significant skepticism about the observation made in the early studies that new neurons are generated in the adult brain prevailed for decades. The main conceptual argument against the finding was that established neuronal networks would require stable neuronal elements and the addition of new elements would disturb network stability and thus cognition, a criticism that was later defused by computational network modeling (for review, see Deng et al., 2010) and behavioral studies (Dupret et al., 2008; Imayoshi et al., 2008; Deng et al., 2009; Arruda-Carvalho et al., 2011). Methodologically, the earliest studies were hampered by the limitations of 3H-thymidine labeling and a scarcity of specific neuronal markers because immunohistochemistry was still under development. Conceptual and technological advances were thus key factors that ultimately established adult mammalian neurogenesis as a biological concept and generated several thousand publications in the years that followed.
In the early 1990s, we and others discovered that neural stem/progenitor cells (NSPCs) could be isolated from embryonic brain tissue and propagated in vitro using defined cell culture media containing FGF-2 (Ray et al., 1993; Ray and Gage, 1994) or EGF (Reynolds et al., 1992). This was soon followed by in vitro propagation of neural stem cells from adult brain tissue (Reynolds and Weiss, 1992; Palmer et al., 1995) and provided a tangible foundation for the idea that new neurons are continually generated in the adult brain from endogenous NSPCs. High-efficiency isolation of neural stem cells from brain regions such as the dentate gyrus (DG) (Palmer et al., 1997) and the subventricular zone (Morshead et al., 1994) indicated to us that NSPCs had to exist in the adult rodent brain. Our group was also able to demonstrate that most brain areas seem to harbor progenitor cells that are capable of generating neurons and glial cells in vitro (Palmer et al., 1995, 1999), suggesting that brain regions with ongoing neurogenesis might retain not only NSPCs but also the proper molecular environment for neurogenesis, also referred to as the stem cell niche (Lim et al., 2000; Palmer et al., 2000).
The early in vitro studies were paralleled by investigations that not only replicated the observations by Altman and colleagues, but were first indications that adult neurogenesis was regulated in vivo by molecular cues. In a series of studies using 3H-thymidine labeling, Gould and Cameron studied the role of glucocorticoids and excitatory input to the dentate gyrus and gave first indications of the importance of stress and the hypothalamic-pituitary-adrenal-axis for hippocampal neurogenesis (Gould et al., 1992, 1994; Cameron et al., 1993, 1995). The study of adult neurogenesis intensified even more when novel histological labeling techniques, such as immunofluorescence against halogenated thymidine analogs in combination with cell-type specific markers, confocal microscopy and stereology, became available (for review, see Kuhn et al., 2016). Bromodeoxyuridine (BrdU) integrates into the DNA during the S-phase of the cell cycle, thereby permanently labeling cells that have undergone cell division during BrdU administration. The immunofluorescence detection of BrdU in conjunction with neuron-specific markers, such as NeuN, allowed high-resolution colocalization within individual cells with confocal microscopy and quickly became the gold standard for birthdating newly generated neurons in adult brain tissue (Kuhn and Cooper-Kuhn, 2007). Critique of this prevailing technique, which allows labeling of large cohorts of cells by systemic injection of BrdU, came from studies focusing on the possibility of false-positive labeling of cells undergoing DNA repair or aberrant cell division attempts under high stress load (Herrup and Yang, 2007). However, although it is conceivable that BrdU incorporation could lead to false-positive labeling, by taking appropriate precautions during BrdU labeling (Kuhn et al., 2016) and by applying additional detection methods, such as retroviral labeling, the generation of new neurons in the adult brain was unequivocally established.
Neurogenesis, whether observed in the embryonic or adult brain, comprises a cascade of cellular events leading to the generation of mature neurons. Lineage tracing to follow and dissect the individual steps has therefore been a crucial component of adult neurogenesis research. In vivo retroviral labeling is particularly informative because it requires nuclear membrane breakdown for stable integration of a reporter gene, which only occurs during cell division (Morshead et al., 1998; van Praag et al., 2002). It revealed cellular details, such as dendritic arborization and synaptic elements, that were previously largely missing from conventional immunohistochemical labeling of new neurons. But even more importantly, retrovirally labeled cells could be visualized in live brain slices and studied using electrophysiological methods (van Praag et al., 2002). Furthermore, the use of constitutive transgenic mouse lines with genetically encoded markers, such as Nestin promoter-based GFP-expressing mice, accurately represented NSPCs in the developing and adult nervous system and labeled a large majority of such cells (Yamaguchi et al., 2000; Kempermann et al., 2003; Encinas and Enikolopov, 2008). Finally, the development of inducible cre-lox systems permitted lineage tracing as well as lineage manipulations of developing cells in the adult brain (for review, see Enikolopov et al., 2015). Together, these tools have been extremely helpful in establishing the presence of neurogenesis in the adult brain of the numerous mammalian species studied so far, even though its existence had been heavily debated at the time (Gage, 1994, 2002).
Neurogenesis in the aging brain
Our initial paper from 1996 focused on the regulation of adult neurogenesis by age and found neurogenesis to be fully present in the 6-month-old rat (Kuhn et al., 1996). A drastic decrease in neurogenesis from adult stages toward later time points was observed; however, hippocampal neurogenesis was still detectable in the older rats (up to 27 months of age). The duration of adult neurogenesis was, at the time, acutely debated. Adult neurogenesis was seen by some as a remnant of embryonic development, declining to undetectable levels once development ended; others proposed it might provide a mechanism by which new neurons are continually added to the DG, regardless of age, to facilitate learning and memory processes. From this perspective, the hippocampus can also be seen as a brain region that never completes development. Although many changes occur in the local microenvironment of the brain during aging, newborn neurons appear to retain the potential to become fully mature and functional granule cells.
We observed a progressive decline of precursor cell proliferation during aging, with a decrease of >80% occurring between 6 and 12 months of age and stabilizing at a low level thereafter. This finding raised competing hypotheses: (1) the hippocampal stem cell pool exhaust with age; (2) the aging microenvironment does not provide the molecular cues for further proliferation; or (3) the aging stem/progenitor cells become unresponsive to environmental cues. The first hypothesis would imply that the hippocampal stem cells are depleted with time, a model that was put forward by Encinas et al. (2011), who showed that increasing numbers of astrocytes are generated from activation of quiescent neural stem cells with age. However, Song and colleagues showed that individual neural stem cells are able to undergo activation, return to quiescence and reactivation with limited depletion via astrocytic transformation (Bonaguidi et al., 2011). An increasing number of studies has focused on the second hypothesis and revealed changes in the local microenvironment as well as the systemic milieu with age involving increasing levels of inhibitory molecules or decreasing levels of neurogenesis-promoting factors (Smith et al., 2018; for recent reviews, see Mosher and Schaffer, 2018). But importantly, even at late stages of aging, hippocampal neurogenesis can be stimulated by exposing animals to both physically and mentally stimulating environments (Kempermann et al., 1998, 2002; van Praag et al., 2005; Kronenberg et al., 2006). Last, the intrinsic responsiveness of neural stem cells may also be altered due to epigenetic changes. Epigenetic mechanisms are crucial components of adult neurogenesis (Jobe et al., 2012) and changes have been observed with age (Kuzumaki et al., 2010a,b; Horvath et al., 2012). All together, what began with the observation of neurogenesis decline has led to intensive and still ongoing research of the molecular mechanisms leading to the age-related changes in hippocampal neurogenesis; and while different hypotheses are on the table, it appears highly likely that several signaling pathways are involved.
New technologies to address the aging of adult hippocampal neurogenesis
As mentioned earlier, transgenic approaches have been among the most powerful tools to visualize the process of adult neurogenesis and to dissect out the genetic and environmental factors that influence adult hippocampal neurogenesis in rodents. A number of transgenic mouse lines with fluorescent proteins or a Cre recombinase enzyme under the control of cell-type-specific promoters, such as the Nestin, Hes5, hGFAP, and Sox2 promoters, have been used to selectively label NSPCs and their progeny or to delete target genes in those populations in vivo (Yamaguchi et al., 2000; Lagace et al., 2007; Suh et al., 2007; Imayoshi et al., 2008). Many studies have provided significant insights into the maintenance and aging of adult hippocampal neurogenesis (Suh et al., 2007; Lugert et al., 2010; Bonaguidi et al., 2011, 2012; Encinas et al., 2011; Kempermann, 2015; Toda et al., 2018). However, observations using fixed tissues constrain our view of the dynamic processes of adult neurogenesis to fragmented time-series sampling. To overcome this technical hurdle, recently, we and others developed a novel methodology to image the DG in awake, behaving mice using multiphoton microscopy (Danielson et al., 2016, 2017; Gonçalves et al., 2016b; Pilz et al., 2016; Kirschen et al., 2017). These live-imaging systems enable us to continuously visualize the dynamics of adult neurogenesis, neuronal maturation, and neural activity in the DG with less fragmentation. Most recently, Jessberger's group successfully traced the process of adult neurogenesis from a subpopulation of adult NSPCs to neurons over 2 months in the live adult mouse hippocampus using multiphoton imaging (Pilz et al., 2018). Live-imaging of adult hippocampal neurogenesis uncovered a variable neurogenic competency, survival rate, and fate commitment among cell clones, which have been difficult to estimate with fixed tissue. Similarly, live-imaging developing dendrites of adult-born dentate granule cells revealed an unexpected homeostatic dendritic pruning process in which facilitation of dendritic branching by an exposure to enriched environmental is counteracted by earlier and more intensive pruning (Gonçalves et al., 2016b). Future experiments using long-term live imaging will reveal more precise dynamics of the aging process in adult neurogenesis, including when the development of DG stops and when the aging of adult neurogenesis starts, as well as the heterogeneous nature of adult NSPCS, the effects of environment and genetic factors.
In parallel with live imaging, recent progress in single-cell RNA sequencing with optimized next generation sequencing technology provides a higher-resolution view of cellular heterogeneity and better insight into the function of an individual cell (Shin et al., 2015; Habib et al., 2016; Lacar et al., 2016; Artegiani et al., 2017; Yuzwa et al., 2017; Hochgerner et al., 2018; Jaeger et al., 2018). This technology allows us not only to resolve the heterogeneous nature of the transcriptome but also to capture developmental dynamics, the effects of environmental changes on a specific population, and the differences in cellular state within the same population. The evolution of single-cell technology is now pushing forward our understanding of the complex nature of adult neurogenesis.
In addition to live-imaging and sequencing technology, molecular tools to manipulate the process of adult neurogenesis have been evolving. Viral tools, including retroviral, lentiviral and adeno-associated viral tools, have been widely used to label adult-born cells and manipulate genes of interest in these populations (Lie et al., 2005; Tashiro et al., 2006; Zhao et al., 2006; Kirschen et al., 2017). These viral tools can also express optogenetic (e.g., channelrhodopsins, halorhodopsins) (Gu et al., 2012; Danielson et al., 2016; Zhuo et al., 2016) and chemogenetic proteins (e.g., the synthetic receptor hMd3 and synthetic ligand clozapine-N-oxide) (Alvarez et al., 2016; Anacker et al., 2018) to selectively manipulate neural activity in a specific population at a specific time. In combination with a retrograde rabies-viral tracing methodology (Wickersham et al., 2007), this research has revealed the dynamic reorganization of circuitry of adult-born neurons during maturation (Vivar et al., 2012; Deshpande et al., 2013; Bergami et al., 2015; Alvarez et al., 2016; McAvoy et al., 2016; Sah et al., 2017). For example, local and long-distance afferents from local interneurons and cortical neurons onto newborn DG neurons were significantly increased with environmental enrichment (Bergami et al., 2015), and optogenetic and chemogenetic tools helped to reveal that disynaptic circuits via local interneurons mediated the effect of environmental enrichment (Temprana et al., 2015; Alvarez et al., 2016). In addition to manipulating neural activity, the evolution of optogenetic tools has enabled us to manipulate several biological processes, including protein localization, protein degradation, organelle transport, signaling pathways, and gene regulation (Imayoshi et al., 2013; Rost et al., 2017), any of which could be manipulated for the direct regulation of adult neurogenesis.
New technology always brings us novel insights. There are more ongoing technological developments in areas, such as single-cell proteomics/genomics and computational modeling. Implementation of new technologies will surely uncover heretofore unrecognized aspects of adult hippocampal neurogenesis and reveal how aging of the whole organism affects the neurogenesis process (Fig. 1).
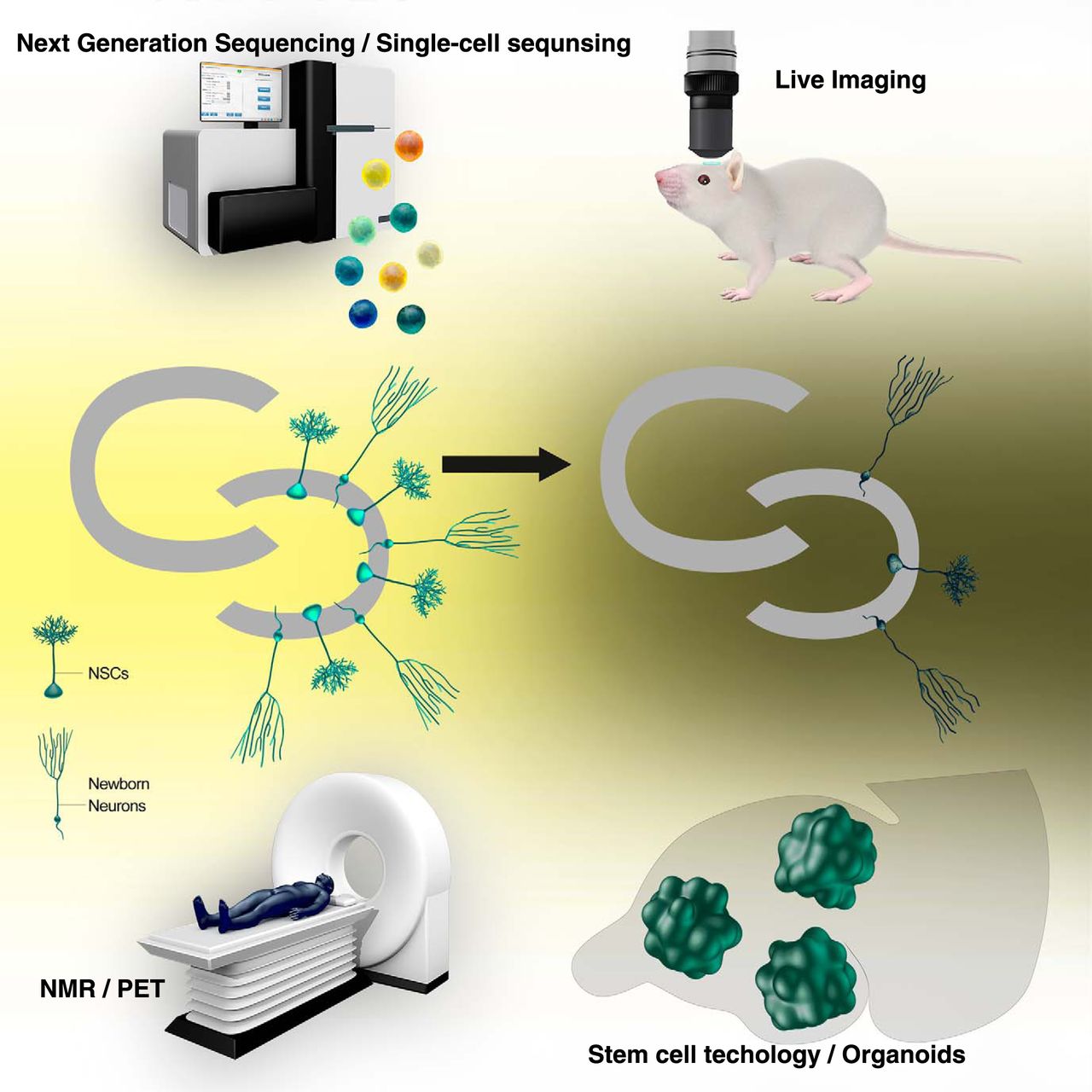
A schematic view of implementation of emerging technology to study adult hippocampal neurogenesis throughout the life span. Technology that will be implemented to study adult hippocampal neurogenesis is next generation sequencing/single-cell sequencing (top left), live imaging (top right), noninvasive imaging (e.g., NMR, nuclear magnetic resonance spectroscopy or PET, positron emission tomography) (bottom left), and stem cell technology/organoids (bottom right), but not limited to these technologies.
Roles of environment in the aging of adult neurogenesis
One prominent feature of adult hippocampal neurogenesis is that an animal's experiences impact the neurogenesis process. Positive experiences, such as learning, exposure to enriched environment, and physical activity, can partially reverse the age-related decline of neurogenesis (Kempermann et al., 1997; Gould et al., 1999a; van Praag et al., 1999, 2005; Kempermann, 2015). In addition, other environmental components, such as stress, diet, sleep, and life events, have significant impacts on adult hippocampal neurogenesis as reported and reviewed by others (Gould et al., 1998; Mirescu et al., 2004; Stangl and Thuret, 2009; Leuner and Gould, 2010; Snyder et al., 2011; Anacker and Hen, 2017). In this review, we focus on aging, which is an unavoidable biological process that gradually compromises brain function and plasticity, including adult neurogenesis-dependent structural/functional plasticity and mood regulation in the hippocampus. Therefore, understanding how environmental factors can potentiate brain plasticity through the activation of adult neurogenesis has been a fundamental challenge. The proliferation rate of NSPCs, the fraction of adult-born cells that differentiate into neurons, and the survival rate of adult-born neurons are all significantly decreased with age, presumably due to both cell-intrinsic and cell-extrinsic changes (Renault et al., 2009; Lugert et al., 2010; Encinas et al., 2011; Yousef et al., 2015; Leeman et al., 2018). These include changes in metabolic status, transcriptional and epigenetic programs, hormonal regulation, systemic milieu, and neurotrophic signaling (Kuhn et al., 1996; Cameron and McKay, 1999; Villeda et al., 2011, 2014; Kuipers et al., 2015; Moore et al., 2015; Yousef et al., 2015; Corenblum et al., 2016; Beckervordersandforth et al., 2017; Castellano et al., 2017). The reduction of neurogenic capability can be partially reversed by environmental enrichment and physical activity, reducing corticosteroid levels as well as systemic factors transferred from young to old animals (Falkenberg et al., 1992; Kempermann et al., 1998, 2002; Cameron and McKay, 1999; Imayoshi et al., 2008; Villeda et al., 2011, 2014; Speisman et al., 2013; Yousef et al., 2015; Castellano et al., 2017). The exact mechanisms by which environmental enrichment, physical exercise, and systemic milieu from young animals potentiate neurogenesis are not clear yet, but presumably they include neurotrophic, Wnt/FGF, neurotransmitters, and MHC signaling (Oliff et al., 1998; Imayoshi et al., 2008; Kobilo et al., 2011; Okamoto et al., 2011; Vivar et al., 2013; Kang and Hébert, 2015; Smith et al., 2015; Fan et al., 2017). A recent report demonstrated that aging also delays the maturation and integration of adult-born neurons (Trinchero et al., 2017), even though the morphological features of new neurons in the aged brain are similar to those generated in the young brain (van Praag et al., 2005). It is not clear whether this age-dependent delay is beneficial for the aging brain, but the delayed morphological maturation and synaptic integration can be reversed by enhancing neurotrophic signaling or physical activity (Trinchero et al., 2017). It is likely that other, yet unknown factors involved in aging contribute to this delay. These factors would include not only local environmental changes in the brain, such as decreased synaptic activity, reduced neurotrophic factors, reduced mitochondrial activity, and age-dependent inflammation, but also systemic changes induced by a lack of mobility and altered metabolism in aged animals themselves. Further work will uncover which age-dependent changes compromise which steps of adult neurogenesis.
Interestingly, the mechanisms underlying the maintenance of adult NSPCs and the effect of aging on that process seem to be different across neurogenic niches (Molofsky et al., 2006; Lim et al., 2009). An increased expression of p16INK4a in neural progenitors of subventricular zone significantly affected neurogenic capability but did not affect neurogenic functions in the DG (Molofsky et al., 2006). This could be due to an intrinsic difference in adult NSPC populations, distinct environmental changes, or both. It would be interesting to examine whether differences between neurogenic regions are evolutionally conserved or, even conversely, vary depending on species.
Adult hippocampal neurogenesis in the human brain
The first evidence of adult hippocampal neurogenesis in the human brain was demonstrated by using the gold-standard BrdU labeling of dividing cells with cell-type-specific markers, such as NeuN and GFAP, to identify BrdU-positive adult-born neurons by confocal microscopy (Eriksson et al., 1998). Since then, using immunohistochemical, carbon14 birth dating and tissue culture techniques, several independent laboratories have found evidence of adult hippocampal neurogenesis in the DG of the human hippocampus (Roy et al., 2000; Palmer et al., 2001; Knoth et al., 2010; Spalding et al., 2013; Dennis et al., 2016; Mathews et al., 2017; Boldrini et al., 2018), as well as in nonhuman primates (Gould et al., 1999b; Kornack and Rakic, 1999; Leuner et al., 2007). In addition, adult hippocampal neurogenesis in the DG is highly conserved across mammalian species with few exceptions (Patzke et al., 2015), implying significant roles for adult hippocampal neurogenesis in brain function. Many studies have shown an exponential reduction of hippocampal neurogenesis along with aging despite the fact that molecular signatures of continuous adult neurogenesis and proliferation have been found (Knoth et al., 2010; Spalding et al., 2013; Dennis et al., 2016; Mathews et al., 2017). Given the size of the adult human DG (500–150 mm3) and the number of newborn neurons identified per day by a carbon dating method (∼700 cells) (Spalding et al., 2013; Dillon et al., 2017), one can assume that adult neurogenesis in the human DG is sparse. The decline of adult hippocampal neurogenesis with age could attenuate forms of structural and functional plasticity, and the level of adult hippocampal neurogenesis has been linked to cognitive abilities both in rodents and nonhuman primates (Aizawa et al., 2009). Hippocampus-dependent cognitive abilities also decline with age in humans (Yassa et al., 2011), but it is not clear yet whether the levels of adult neurogenesis correlate with cognitive abilities in humans.
Recently, using an unbiased stereology method with several common markers of neurogenesis, Boldrini et al. (2018) showed that healthy human brains maintained similar levels of neurogenesis from 14 to 79 years of age, raising the possibility of higher neural plasticity in the human DG than was expected from previous studies. However, in contrast, Sorrells et al. (2018) used the same markers (DCX, PSA-NCAM) but reached a different conclusion, suggesting that neurogenesis in the human DG quickly decreased after birth and became undetectable before adulthood. Where does this contradiction come from?
One possible explanation is technical differences between the studies, including the duration of postmortem delay, fixation and sample preservation methods, and staining protocols. These factors are critical to reliably detect markers of adult-born neurons. The duration of postmortem delay in particular is crucial not only for the detection of DCX, but also the morphology of DCX signals (Boekhoorn et al., 2006). Sorrells et al. (2018) used brains with longer postmortem delays (up to 48 h) compared with other studies (Eriksson et al., 1998; Boldrini et al., 2018), which could be a critical factor in underestimating the number of adult-born neurons. Another major difference was the use of stereology (Boldrini et al., 2018), a method for unbiased quantification in 3D tissues from serial sections. The method provides accurate estimation in terms of the number of adult-born neurons compared with counting cells from a few sections of tissues, and it has been adapted to study adult neurogenesis in rodents (Kuhn et al., 1996; Kempermann et al., 1997). Usage of stereology should be encouraged to obtain an accurate picture of adult neurogenesis in the human brain.
In addition, the criteria used for defining adult-born neurons in the human brain were different in the two studies. Sorrells et al. (2018) defined only DCX+PSA-NCAM+ cells as adult-born neurons; they did not count DCX−PSA-NCAM+ cells, claiming that the latter exhibited more mature morphological features based on their criteria. However, the developmental time course of adult-born neurons in the human DG has not been clearly characterized, and neurons in higher mammals take at least 6 months to fully mature (Kohler et al., 2011). Furthermore, our knowledge of the markers of adult-born neurons has been derived from studies using rodent models; therefore, we do not know the exact expression time course of neuronal markers in adult-born neurons in the human DG. In addition, adult-born DG neurons show slower kinetics of maturation/survival and different patterns of genetic programs/marker expression compared with perinatally born DG neurons in rodents (Dayer et al., 2003; Shi et al., 2004; Overstreet-Wadiche et al., 2006; Jessberger et al., 2008; Andersen et al., 2014; Urbán and Guillemot, 2014; Cahill et al., 2017). Based on these technical limitations, many researchers in the field, including us, questioned the strong conclusion from Sorrells et al. (2018; see also Kempermann et al., 2018). The discrepancies between these studies underscore that we need to clearly determine the expression time course of neurogenesis markers in the human DG. Importantly, there are still many open questions as discussed below. We believe that this debate stimulates and facilitates the field to develop advanced means as well as a technical standard to move the research of human adult hippocampal neurogenesis forward.
The future comes with more questions
Since the discovery of adult hippocampal neurogenesis, remarkable progress has been made in understanding the molecular mechanisms and functional contributions of adult neurogenesis. However, we still have fundamental questions that need to be resolved. Here we summarize and discuss some of these questions.
First, although adult neurogenesis declines with age, it is still not clear how the dynamics of adult neurogenesis are affected by aging. Such dynamics include the activation of quiescent adult NSPCs as well as the differentiation, maturation, and integration of adult-born cells. Because adult NSPCs seem to be a heterogeneous population (Jhaveri et al., 2015; Pilz et al., 2018), a distinct subpopulation may be differently affected by aging. Using multiphoton imaging, long-term live imaging of adult hippocampal neurogenesis throughout the entire life of animals (both in rodents and nonhuman primates) would reveal the nature of adult hippocampal neurogenesis in aging. Along the same line, we need to understand the heterogeneous nature of adult NSPCs and their progeny. It would be intriguing to examine whether distinct populations of adult NSPCs generate different subtypes of dentate granule cells, whether they differentially respond to environmental stimuli, how genetic and epigenetic regulations differ between subtypes, and whether the heterogeneity of adult hippocampal neurogenesis is preserved during evolution. Single-cell technologies, including single-cell RNA-seq, single-cell epigenetic methods, and single-cell proteomics, will be promising approaches to address these questions. The same approaches can be applied to pathological conditions as well to reveal the effect of pathology for each individual cell type.
Second, the developmental time course of adult-born neurons in the human brain needs to be determined. In the case of nonhuman primates, it takes at least several months to express mature neuron markers (Kohler et al., 2011), which means that it could take longer than several months for them to be fully mature. Given that humans have longer developmental time courses and lifetimes, it is reasonable to speculate that the maturation process of adult-born neurons in the human hippocampus should take longer. Characterizing the maturation process along with the expression of molecular markers is critical because most studies in the human brain rely on using the postmortem brain. However, as we discussed above, we do not know exactly which markers correspond to which developmental time points in human adult-born neurons. Furthermore, because the duration of the highly plastic maturation period in adult-born neurons could impact the entire neural network of the hippocampus through feedforward and feedback mechanisms (Toda et al., 2018), it is critical to determine the duration of the maturation period to estimate the role of adult-born neurons in the human hippocampus. Although it is difficult to conduct these experiments using human brains, combining recently developed hippocampal organoids with a transplant strategy may allow us to address this issue (Sakaguchi et al., 2015; Mansour et al., 2018). An alternative approach will be noninvasive in vivo imaging of neurogenesis using nuclear magnetic resonance spectroscopy or positron emission tomography (Manganas et al., 2007; Rueger et al., 2010; Tamura et al., 2016). These technologies are still under development, and the methodology needs to be refined to increase the spatial resolution and specificity of detection. Stem cell technology can help to identify specific markers of adult neural stem cells and adult-born neurons that could be used for noninvasive in vivo imaging. Advances in these technologies will also allow us to identify cognitive metrics relating to adult hippocampal neurogenesis in humans. In addition, this line of study has the potential to identify biomarkers for the reduction of adult neurogenesis, which could be beneficial for clinical screening.
Third, we need to identify the roles of adult hippocampal neurogenesis in humans in both physiological and pathological conditions. Accumulating evidence using animal models has uncovered significant roles for adult-born neurons in cognitive function and mood regulation (Shors et al., 2001; Kropff et al., 2015; Aimone, 2016; Anacker and Hen, 2017; Toda and Gage, 2018). In contrast, evidence linking adult hippocampal neurogenesis to cognitive function in humans is still limited and indirect (Toda et al., 2018). Although it is hard to manipulate the levels of adult neurogenesis in the human brain, the development of noninvasive in vivo functional imaging at cellular resolution would help to monitor neural activity of adult-born neurons and their contribution in cognitive function and mood regulation. Further technical development is desperately needed to advance our understanding.
Fourth, the mechanisms underlying the development and maintenance of the neurogenic niche in the subgranular zone of the DG are still unclear. Although past achievements in the field have revealed a number of essential factors in the maintenance of neurogenic capability, it is still totally unclear why specific regions of the brain, such as the subgranular zone, can possess and maintain neurogenic properties. What are the cellular and molecular components necessary for the development and maintenance of neurogenic regions in the adult brain? Since recent evidence suggests that systemic factors in serum contribute to the regulation of adult neurogenesis, the neurogenic niche may be involved not only in local cellular/tissue components within the DG, but also in other organs; even the microbiota of the gut may contribute as remote components of the neurogenic niche (Ogbonnaya et al., 2015). It would be intriguing to examine how other organs contribute to the maintenance of the neurogenic niche in the DG and how aging affects these communications.
Fifth, the cell-autonomous mechanisms underlying the long-term maintenance of multipotency/quiescence of adult neural stem cells need to be determined. The importance of cell cycle regulators as well as transcriptional/epigenetic factors has been investigated (Gonçalves et al., 2016a; Toda et al., 2018). However, most adult neural stem cells maintain a quiescent state despite the fact that a variety of stimuli in the niche can activate them; therefore, one can assume that there are very robust cell-autonomous mechanisms underlying the maintenance of the quiescent state. Intriguingly, some nuclear proteins were identified as long-lived proteins, including histones, nuclear lamins, and nucleoporins (Savas et al., 2012; Toyama et al., 2013). These proteins interact with chromatins and work as a structural foundation for cell-type-specific gene regulation (Ibarra and Hetzer, 2015; Jacinto et al., 2015; Ibarra et al., 2016; Toda et al., 2017). Because these proteins accumulate damage with age presumably due to their low turnover rates (D'Angelo et al., 2009), they may lead to age-dependent deterioration of gene regulation. These mechanisms could be fundamental in maintaining not only adult NSPCs but also the plasticity that is observed to some extent in any somatic stem cells throughout our lifetime.
In conclusion, what started in the early 1990s as an expedition to probe the possible existence of somatic stem cells within the adult brain led to the establishment of a new area of neuroscience research. For adult neurogenesis to receive significant (even though not always undisputed) recognition, the development of novel tools was essential, and they made possible the firm establishment of the phenomenon already described by Altman and colleagues in the 1960s. We were immediately aware that a central dogma of neurobiology, declared by Ramón y Cajal in 1928, “Everything may die, nothing may be regenerated … ” (Ramón y Cajal, 1991) had been repudiated. But even >20 years later, the role that adult neurogenesis plays within the context of hippocampal function, neuroplasticity, and brain repair brings up many unsolved questions. We therefore call upon the next generation of scientists to embrace the rest of Ramón y Cajal's famous declaration: “… It is for the science of the future to change, if possible, this harsh decree” (Ramón y Cajal, 1991).
Footnotes
This work was supported by National Institutes of Health R01 MH095741, National Institutes of Health U01 MH106882, G. Harold and Leila Y. Mathers Charitable Foundation, Leona M. and Harry B. Helmsley Charitable Trust Grant 2012-PG-MED00, Annette C. Merle-Smith, JPB Foundation, McKnight Foundation, Swedish Research Council Vetenskapsrådet K2015-63X-20117-I0-4, Swedish Childhood Cancer foundation Barncancerfonden MT2017-0013, and Swedish governmental support under the LUA/ALF Agreement ALFGBG-72654. T.T. was supported by Japan Society for the Promotion of Science, the Kanae Foundation, and Paul F. Glenn Center for Biology of Aging Research. We thank Mary Lynn Gage for comments on the manuscript; Veronika Mertens for the illustration; and Drs. Sara B. Linker and Sarah Parylak for discussion.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Fred. H. Gage, Laboratory of Genetics LOG-G, The Salk Institute for Biological Studies, 10010 N. Torrey Pines Road, La Jolla, CA 92037. gage{at}salk.edu





{kind=link}