

Abstract
Prevention of nerve injury-induced tactile, but not thermal, hypersensitivity is achieved by ipsilateral lesions of the dorsal columns or lidocaine microinjection into the nucleus gracilis (n. gracilis). These and other data support the possibility that tactile hyperresponsiveness after nerve injury may be selectively mediated by a low-threshold myelinated fiber pathway to the n. gracilis. Here we identify a transmitter that might selectively mediate such injury-induced tactile hypersensitivity. Neuropeptide Y (NPY), normally not detected in the dorsal root ganglion (DRG) or in the n. gracilis of rats, became markedly upregulated at both sites and in the spinal cord after spinal nerve injury. Injury-induced NPY-IR occurred predominately in large-diameter DRG cells, and the NPY-IR in the n. gracilis was blocked by dorsal rhizotomy or dorsal column lesion. NPY microinjection into the n. gracilis of uninjured rats elicited reversible tactile, but not thermal, hypersensitivity only in the ipsilateral hindpaw. Administration of anti-NPY antiserum, but not control serum or preabsorbed serum, into the n. gracilis ipsilateral to nerve injury reversed tactile, but not thermal, hypersensitivity. Similarly, microinjection of the NPY antagonists NPY18–36 and (R)-N-[[4-(aminocarbonylaminomethyl)-phenyl]methyl]-N2-(diphenylacetyl)-argininamide trifluoroacetate, into the n. gracilis ipsilateral to the injury reversed tactile, but not thermal, hypersensitivity. Antagonist administration into the contralateral n. gracilis had no effect on injury-induced hypersensitivity. These data suggest the selective mediation of nerve injury-induced tactile hypersensitivity by upregulated NPY via large fiber input to n. gracilis. Selective reversal of injury-induced tactile allodynia by NPY receptor antagonists would have significant implications for human neuropathic conditions.
Human neuropathic pain is characterized in part by persistent pain, hyperalgesia (i.e., enhanced perception of nociceptive stimuli), and allodynia (i.e., normally innocuous stimuli perceived as nociceptive; Payne, 1986; Merskey and Bogduk, 1994). Allodynia remains poorly controlled by available therapies and has a profound negative impact on quality of life. Substantial data support the possibility that tactile and thermal hypersensitivity might be differentially mediated. In humans with nerve injury, differential nerve block showed that touch-induced pain was mediated through Aβ primary fibers, whereas thermal pain was mediated through unmyelinated fibers (Campbell et al., 1988; Koltzenburg et al., 1992, 1994). In experimental neuropathic models, intrathecal morphine or systemic resiniferatoxin, a C-fiber neurotoxin, reversed thermal but not tactile hypersensitivity (Bian et al., 1995; Lee et al., 1995;Ossipov et al., 1995; 1999), and systemic morphine or capsaicin did not block “dynamic allodynia” (Field et al., 1999). Spinal hemisection or dorsal column lesion performed ipsilateral but not contralateral to nerve injury blocked tactile hypersensitivity (Sun et al., 2001). Microinjection of lidocaine into the nucleus gracilis (n. gracilis) ipsilateral to nerve injury reversibly blocked tactile but not thermal hypersensitivity (Sun et al., 2001). Enhanced baseline and evoked activities of thalamic neurons were seen in nerve-injured rats, and these effects were abolished by disruption of the dorsal columns or the n. gracilis (Miki et al., 2000). These observations are consistent with the possibility that nerve injury-induced thermal hypersensitivity is mediated through nociceptive C-fibers, which express opioid receptors (Besse et al., 1990; Mansour et al., 1994), whereas tactile hypersensitivity may be mediated primarily by large-diameter Aβ fibers. These fibers are normally responsive to low-threshold stimuli, project directly to supraspinal dorsal column nuclei, send collaterals into laminas III and IV of the spinal dorsal horn (Bennett et al., 1983; Hendry et al., 1999), and are unlikely to express opioid receptors (Zhang et al., 1998).
Although nerve injury has been shown to increase NPY expression in the dorsal root ganglia, spinal cord, and dorsal column nuclei (Li et al., 1999), the role of this peptide in the modulation of nociception has remained inconclusive. Pharmacological neuropeptide Y (NPY) has been reported as both antinociceptive and pronociceptive (Hua et al., 1991;Jolicoeur et al., 1991; Heilig et al., 1992; Broqua et al., 1996;Mellado et al., 1996; Taiwo and Taylor, 2002), often with biphasic dose–effect curves (Xu et al., 1994, 1999). Excitatory effects of NPY have been observed in dorsal root ganglion (DRG) cells taken from rats with nerve injury (Abdulla and Smith, 1999). Fewer studies, however, have explored the role of endogenous NPY in the pathological state. This distinction is likely to be important, because nerve injury elicits (1) NPY upregulation in fibers that either did not express or expressed the peptide at very low levels and (2) both upregulation and downregulation of NPY receptors in different regions of the nervous system (Noguchi et al., 1993; Wakisaka et al., 1993; Nahin et al., 1994; Zhang et al., 1996; Hokfelt et al., 1998; Li et al., 1999;Marchand et al., 1999). Here, NPY antagonists were used to explore the potential role of upregulated endogenous NPY in nerve injury-induced pathology.
MATERIALS AND METHODS
Animals. Male Sprague Dawley rats (Harlan, Indianapolis, IN), 200–300 gm at time of testing, were maintained in a climate-controlled room on a 12-hr light/dark cycle (lights on at 6 A.M.), and food and water were available ad libitum. All testing was performed in accordance with the policies and recommendations of the International Association for the Study of Pain and National Institutes of Health guidelines for the handling and use of laboratory animals and received approval from the Institutional Animal Care and Use Committee of the University of Arizona.
Surgical procedures. Spinal nerve ligation (SNL) injury was induced using the procedure of Kim and Chung (1992). Anesthesia was induced with 2% halothane in O2 at 2 l/min and maintained with 0.5% halothane in O2. The dorsal vertebral column from L4 to S2 was exposed, and the L5 and L6 spinal nerves were identified and carefully isolated. The L5 and L6 spinal nerves were tightly ligated distal to the dorsal root ganglion with a 4-0 silk suture. The incision was closed, and the animals were allowed to recover for 5 d. Rats that exhibited motor deficiency (<10%) or failure to exhibit subsequent tactile hypersensitivity were excluded from further testing. Sham control rats underwent the same operation and handling as the experimental animals but without SNL. Behavioral evaluation of tactile and thermal hypersensitivity occurred at numerous time points after SNL surgery as indicated. Dorsal rhizotomy was performed in the same manner as SNL in anesthetized rats, except that the spinal dorsal roots between the DRG and the spinal cord at L5 were ligated and cut. Lesions of the dorsal columns or of the dorsolateral funiculus (DLF) were performed in rats anesthetized with halothane. A laminectomy was performed at T8 with the aid of a dissecting microscope to expose the spinal cord. Dorsal column lesions were preformed by careful incision of the dorsal aspect of the spinal cord with the point of a number 11 scalpel blade. Lesions of the DLF were performed by gently rotating the spinal cord to expose the ventrolateral aspect and crushing the area with fine forceps. Hemostasis was confirmed, and the wound over the exposed spinal cord was packed with Gelfoam and closed. Sham spinal surgery was performed by exposing the vertebras and performing the laminectomy but without cutting any neuronal tissue. All lesions were verified histologically at the termination of the experiments, and only animals with correctly placed lesions were included in the analysis. The rhizotomies and lesions of the dorsal column or DLF were performed 7 d before SNL. Proper placement of the spinal lesions was verified histologically by staining cord sections with Luxol fast blue to identify myelinated fiber tracts. All studies were performed after the fourth week after SNL, with the exception of time course studies for NPY upregulation.
Cannula placement. All rats were prepared for drug microinjection into the n. gracilis by placing anesthetized (ketamine and xylazine, 100 mg/kg, i.p.) animals in a stereotaxic head holder. The skull was exposed, and a 26-gauge guide cannula (Plastics One Inc., Roanoke, VA) was directed toward the n. gracilis (anteroposterior, −5.5 mm from interaural line; mediolateral ±0.5 mm from midline and 2 mm above the interaural line); these coordinates were obtained from the atlas of Paxinos and Watson (1986). The guide cannula was cemented in place and secured to the skull by small stainless steel machine screws. The animals were allowed to recover 5 d after surgery before any pharmacological manipulations were made. Drugs were slowly microinjected in a volume of 0.5 μl through a 33 gauge injection cannula inserted through the guide cannula and protruding an additional 2 mm into fresh brain tissue to prevent backflow of drug into the guide cannula. Progress of the injection was confirmed by monitoring the movement of an air bubble through a calibrated length of tubing. A volume of 1 μl was used in situations in which additional doses were required but higher concentrations were not possible because of solubility limits. At the termination of all experiments, the animals were killed; fast green dye was microinjected into the region; and cannula placement was verified histologically. Only those animals with correct cannula placement were included in behavioral analysis.
Drugs and solutions. The nonpeptidic, highly selective Y1 antagonist (R)-N-[[4-(aminocarbonylaminomethyl)-phenyl]methyl]-N2-(diphenylacetyl)-argininamide trifluoroacetate (BIBO 3304) (Wieland et al., 1998) and the highly selective Y2 antagonist (S)-(N(2)-[[1-[2-[4-[(R,S)-5,11-dihydro-6(6h)-oxodibenz[b,e]azepin-11-yl]-1-piperazinyl]-2-oxoethyl]cyclopentyl]acetyl]-N-[2-[1,2-dihydro-3,5(4H)-dioxo-1,2-diphenyl-3H-1,2,4-triazol-4-yl]ethyl]-argininamide (BIIE 0246) (Doods et al., 1999) were dissolved in 50% DMSO in distilled water. NPY and NPY(18–36) (American Peptide Co., Sunnyvale, CA) and anti-NPY antiserum (Peninsula Laboratories, Inc., San Carlos, CA) were dissolved in distilled water. Preadsorbed serum was prepared from the anti-NPY antiserum as a control. The preadsorbed serum was prepared by incubating 0.2 ml of the antiserum (40 mg of protein/ml) with 0.8 ml of agarose-bound protein A (Vector Laboratories, Burlingame, CA) for 24 hr at 4°C. The suspension was pelleted by low-speed centrifugation (5000 ×g for 5 min). The supernatant, which is devoid of anti-NPY IgG, was used to define the IgG-independent effects of the antiserum administration.
Evaluation of tactile hypersensitivity. Tactile hypersensitivity of the hindpaws was determined by measuring the paw withdrawal threshold in response to probing with a series of calibrated (0.41, 0.70, 1.20, 2.00, 3.63, 5.50, 8.5, and 15.1 gm) von Frey filaments (Chaplan et al., 1994). The rats were acclimated for 60 min in suspended cages with wire mesh floors to allow probing from below. The von Frey filament was applied perpendicularly to the plantar aspect of the affected paw of the rat until it buckled slightly. A positive response was indicated by a sharp withdrawal of the paw. The 50% paw withdrawal threshold was determined by the nonparametric method ofDixon (1980). An initial probe equivalent to 2.00 gm was applied, and the stimulus was incrementally increased until a positive response or the maximal stimulus was obtained and then decreased until a negative result or the minimal stimulus was observed. This “up–down” method was repeated until three changes in behavior were determined. The pattern of positive and negative responses was tabulated. The 50% paw withdrawal threshold was determined as (10[Xf + k∂])/10,000, where Xf is the value of the last von Frey filament used;k is the Dixon value for the positive–negative pattern; and ∂ is the mean (log) difference among stimuli. Significant differences among paw withdrawal thresholds at several time points after drug injection were determined by ANOVA followed by the post hocleast significant differences test. Pair-wise comparisons were performed by Student's t test. Significance was set atp = 0.05.
Evaluation of thermal hypersensitivity. The method ofHargreaves et al. (1988) was used for the determination of thermal hyperalgesia of the hindpaws. Rats were allowed to acclimate within Plexiglas enclosures on a lightly frosted glass plate transparent to infrared radiation. A radiant heat source in the form of a calibrated infrared generator (Stoelting Co., Wood Dale, IL) was activated and focused onto the plantar aspect of a hindpaw. Paw withdrawal resulted in termination of the stimulus and a timer. A maximal cutoff latency of 40 sec was used to prevent tissue damage. Paw withdrawal latencies were determined to the nearest 0.1 sec. Significant differences among paw withdrawal latencies at several time points after drug injection were determined by ANOVA followed by the post hoc least significant differences test. Pairwise comparisons were performed by Student's t test. Significance was set at p= 0.05.
Immunofluorescent labeling of NPY. The rats were deeply anesthetized with ketamine and xylazine and perfused transcardially with 250 ml of PBS (0.1 m) pH 7.4, containing 15,000 IU/l heparin, followed by 4% paraformaldehyde in PBS for 20 min. The brain, spinal cord, and L4–L6 DRGs were removed, postfixed in 4% paraformaldehyde for 4 hr, and cryoprotected in 30% sucrose in PBS overnight at 4°C. Frontal sections (20 μm) were cut through the coronal plane of the L4–L6 spinal cord and caudal medulla at the level of the n. gracilis. Frozen sections (20 μm) were also obtained from the DRG at L4–L6. The sections were mounted on gelatin-coated glass slides and preincubated in a solution of 4% normal goat serum in PBS with 1% bovine serum albumin and 0.2% Triton X-100 for 30 min. The sections were then incubated in a polyclonal rabbit antiserum to NPY (1:10,000; Peninsula Laboratories) for 1–3 d at 4°C. They were then washed in PBS (three times for 10 min) and incubated with Alexa Fluor 594 goat anti-rabbit IgG (1:400; Molecular Probes, Eugene, OR) for 60 min at room temperature. After two 5 min washes in PBS, the sections were air-dried overnight and coverslipped with DPX. The sections were examined with a Nikon (Melville, NY) E800 fluorescence microscope equipped with standard filters for Texas Red. Images were acquired with a Hamamatsu C5810 color CCD camera and its proprietary Image Processor software (Hamamatsu Photonic Systems, Bridgewater, NJ). The acquired images were then processed with Adobe PhotoShop (Abode Systems Inc., San Jose, CA). Measurement of cross-sectional diameters of neuronal cell bodies was accomplished with the aid of the imaging and analysis software Metamorph 5.5 (Universal Imaging Corp., West Chester, PA).
RESULTS
NPY distribution in uninjured or sham-operated rats
NPY-IR was below the level of detection in either the n. gracilis or the DRG in uninjured or sham-operated rats. NPY-IR was seen in the spinal cord and was localized predominantly to nerve fibers in the outer laminas of the dorsal horn. These findings are consistent with previous studies demonstrating that NPY-IR is found in nerve terminals and varicosities throughout the dorsal horn, with the greatest concentration being in the superficial laminas (Hokfelt et al., 1981;Hunt et al., 1981b; Sasek and Elde, 1985; Doyle and Maxwell, 1994). The intensity of NPY-IR in the spinal dorsal horn was unchanged after dorsal rhizotomy (data not shown), suggesting that basal expression of spinal NPY is unlikely to originate from primary afferent fibers in the uninjured state. Lesions of the DLF also did not result in alterations in spinal NPY-IR (data not shown); thus NPY was unlikely to arise from bulbospinal projections through this region of the spinal cord. Previous studies using confocal microscopy found neuronal cell bodies in laminas III and IV that express NPY (Polgar et al., 1999). These observations and those in the present study are consistent with the suggestion that spinal NPY is primarily derived from spinal interneurons (Hunt et al., 1981a,b; Gibson et al., 1984).
NPY distribution after SNL
The n. gracilis, which is normally devoid of NPY-IR, contained a significant level of immunofluorescence for NPY after SNL (Fig.1). Such NPY-IR was restricted to the side ipsilateral to the nerve injury. Importantly, the NPY-IR appeared to be localized within nerve fibers and not cell soma. NPY-IR was clearly evident in the n. gracilis on the third day after SNL, and expression appeared maximal by the seventh day after SNL (Fig. 1). The NPY-IR in the n. gracilis remained elevated throughout the course of the experiment (4 weeks after L5 and L6 SNL) (Fig. 1). The time course of the enhanced NPY-IR correlates well with the onset and persistence of SNL-induced tactile and thermal hypersensitivity (Burgess et al., 2002).

Time course of nerve injury-induced upregulation of NPY-IR in the n. gracilis. The caudal brainstem was obtained from male Sprague Dawley rats at the stated time (days) after SNL. There was no detectable NPY-IR in the n. gracilis of sham-operated rats. Increased NPY-IR ipsilateral to SNL was clearly apparent by the third day (3d) after nerve injury. NPY-IR appeared maximal by the seventh day after SNL and remained elevated throughout the experiment (i.e., 28d). There was no increase in immunofluorescence for NPY in the contralateral n. gracilis at any of the time points examined. Scale bar, 200 μm. The bottom left panel shows a confocal image from day 28 demonstrating that NPY-IR in the n. gracilis is found in fibers. Scale bar, 50 μm.
The time course of expression of NPY observed in the DRG was coincident with that seen in n. gracilis (data not shown). Significant levels of NPY-IR were evident in the ipsilateral L5 and L6 but not L4 or S1 DRG on day 3 after SNL; the levels of NPY-IR persisted throughout the 4 week time course of the experiment. Immunofluorescence for NPY in the lumbar DRG contralateral to the side of the nerve injury was not different from that seen in sham controls (Fig.2). A survey of 450 DRG neurons revealed that NPY-IR was predominantly associated with medium- to large-diameter neurons. The preponderance of NPY-IR occurred among neurons with diameters of 31–35 μm (17.6%), 36–40 μm (23.3%), and 41–50 μm (36.0%). Neurons with diameters of <30 μm accounted for 6.7% of the cell count, and cells with diameters of >50 μm represented 16.4%. These findings are consistent with previous reports usingin situ hybridization, in which NPY was expressed primarily in large-diameter neurons (i.e., mean diameter of 46 μm, calculated from a reported mean cross-sectional area of 1700 μm2; Noguchi et al., 1993).

Nerve injury-induced upregulation of NPY-IR in the DRG. The L4–L6 and S1 DRG were harvested 28 d after the ligation of the L5 and L6 spinal nerves. Cell bodies immunoreactive for NPY were observed in the DRG at L5 and L6 taken ipsilateral (left panels) but not contralateral (right panels) to SNL. There was no detectable NPY-IR in the ipsilateral or contralateral L4 or S1 DRG. Scale bar, 200 μm.
In the spinal cord, increased immunofluorescence for NPY was seen in the outer laminas of the dorsal horn ipsilateral to SNL and in the region around the central canal. Moreover, the NPY-IR extended into the deeper laminas of the spinal dorsal horns, clearly indicating an increased region of distribution of NPY after peripheral nerve injury. In contrast, there was no apparent change in NPY-IR in the dorsal horn contralateral to L5 and L6 SNL. There was no evidence of an upregulation of NPY-IR in spinal sections taken outside the L5–L6 region (data not shown). The time course of SNL-induced NPY-IR in the spinal cord was consistent with that seen in n. gracilis and DRG. Spinal NPY-IR was evident by day 3 after SNL and persisted for at least 4 weeks (data not shown).
Effects of dorsal column lesion and dorsal rhizotomy on SNL-induced NPY upregulation in spinal nerves, DRG, spinal cord, and n. gracilis
Sections were obtained from the L5 spinal nerve at the region of ligation. A clear abundance of NPY-IR was seen in the region proximal to the ligation and an absence of immunofluorescence in the nerve section distal to the ligation (Fig. 3). This observation demonstrated that NPY is likely to be produced within the DRG and transported to the axon terminals. On the basis of this observation, it would be expected that manipulations that disrupt the continuity of the large-diameter afferent fibers would result in an absence of NPY-IR in the terminal field. Accordingly, it was found that dorsal rhizotomy, which did not block the SNL-induced upregulation of NPY-IR in the DRG, blocked the upregulation of NPY-IR in the ipsilateral spinal dorsal horn and in the n. gracilis after SNL. This observation is consistent with previous reports (Li et al., 1999). Furthermore, lesions of the dorsal columns made ipsilateral to SNL also blocked the upregulation of NPY-IR in the n. gracilis (Fig.4A,B) but did not prevent the upregulation of NPY-IR in either the spinal dorsal horn (Fig. 4C) or the DRG (Fig. 4D). These observations strongly suggest that increased synthesis of NPY occurs in large-diameter neurons of the DRG and that NPY is transported to the terminal fields of these neurons in the spinal dorsal horn and the n. gracilis.

Fluorescence micrograph of a sagittal section of the L5 spinal nerve at the site of ligation. NPY-IR is accumulated on the side proximal to the DRG. NPY-IR is negligible at the distal end. The distribution of the NPY-IR suggests that NPY associated with axon terminals is not likely to be produced by de novosynthesis but originates from the soma via axonal transport. Scale bar, 100 μm.
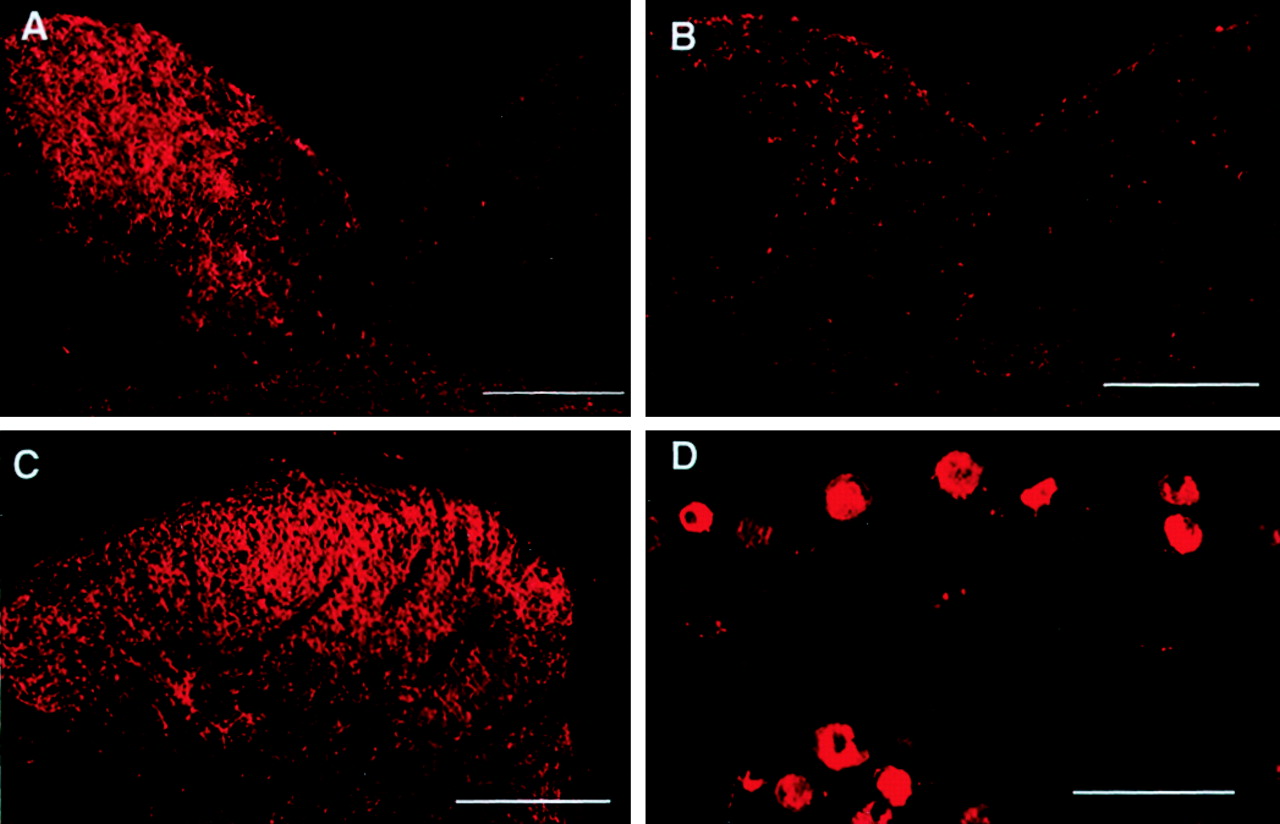
Effects of ipsilateral sham or dorsal column lesion on the level of NPY-IR in the n. gracilis (A, B), superficial laminas of the ipsilateral dorsal horn of the spinal cord at the L5 level (C), and the ipsilateral L5DRG (D). Male Sprague Dawley rats received unilateral lesions of the dorsal columns or sham surgery 7 d before L5 and L6 SNL surgery. Spinal nerve injury was performed ipsilateral to the dorsal column lesion or sham. Tissues were harvested 28 d after L5 and L6 SNL. Rats with SNL but with sham lesions of the dorsal columns showed upregulation of NPY-IR in the ipsilateral n. gracilis (A). Lesions of the dorsal columns before SNL blocked this increase in NPY-IR (B). Dorsal column lesions did not block the upregulation of NPY-IR in either the spinal dorsal horn (C) or the L5 DRG (D). Scale bar, 200 μm.
NPY in the n. gracilis induces tactile hypersensitivity in uninjured rats
The microinjection of NPY (1 nmol) into the n. gracilis of uninjured rats resulted in reversible tactile hypersensitivity of the ipsilateral but not contralateral hindpaw to light mechanical stimuli. The paw withdrawal threshold was significantly decreased from 15 ± 0 to 6.18 ± 1.38 gm in the ipsilateral hindpaw within 20 min (Fig. 5A). The responses returned to baseline values within 60 min. Contralateral hindpaw thresholds were unchanged after microinjection into the n. gracilis; the preinjection baseline was 15 ± 0 gm, and the postinjection threshold ranged between 13.7 ± 0.82 and 15 ± 0 gm. Furthermore, the animals demonstrated an apparent increased sensitivity to normal handling with increased touch-induced vocalization during the period of tactile hypersensitivity. In contrast, the microinjection of NPY into the n. gracilis did not alter behavioral responses to noxious radiant heat (Fig. 5B). Paw withdrawal latencies did not differ significantly from the baseline value of 16.1 ± 0.83 sec over the 60 min observation period. The microinjection of the water vehicle into the n. gracilis did not produce any changes in response to tactile or thermal stimuli (Fig. 5A,B).

Acute effects of microinjection of NPY into the n. gracilis on tactile (A) and thermal (B) sensory thresholds. Male Sprague Dawley rats were prepared with guide cannulas directed at the n. gracilis. NPY (1 nmol) or vehicle (distilled water) was microinjected through an injection cannula projecting 2 mm beyond the guide cannula and into fresh brain tissue. Withdrawal thresholds of the hindpaws to probing with von Frey filaments were significantly decreased after microinjection of NPY but not vehicle into the n. gracilis (A). Microinjection of 1 nmol of NPY into the n. gracilis did not produce any changes in paw withdrawal latencies to noxious radiant heat (B). *p< 0.05.
Blockade of SNL-induced tactile hypersensitivity by microinjection of antiserum to NPY in n. gracilis
Peripheral nerve injury was induced with SNL, and tactile and thermal hypersensitivity were firmly established within 3 d and were still present at the same level at the fourth week after SNL. At postinjury day 28, the paw withdrawal threshold was significantly decreased from a pre-SNL baseline value of 15 ± 0 to 1.46 ± 0.38 gm, indicative of tactile hypersensitivity. The microinjection of 20 μg of anti-NPY antiserum into the n. gracilis produced a time-related reversal of tactile hypersensitivity (Fig.6A). The paw withdrawal thresholds were significantly increased within 15 min of microinjection of the antiserum and reached a maximum of 13.3 ± 0.81 gm before returning to baseline levels at 90 min. The microinjection of NPY antiserum did not alter paw withdrawal thresholds of sham-operated rats (Fig. 6A). Furthermore, the microinjection of preadsorbed antiserum did not produce any significant change in paw withdrawal thresholds of SNL or sham-operated rats (Fig.6A). Control (nonimmune) serum, which was obtained from the same source as the antiserum but without exposure to antigen, also did not alter sensory thresholds (data not shown).

Acute effects of microinjection of an anti-NPY antiserum on the tactile hypersensitivity (A) and thermal hyperalgesia (B) induced by L5 and L6 SNL. The rats were prepared with guide cannulas directed at the n. gracilis. A, The microinjection of an anti-NPY antiserum (20 μg) into the n. gracilis produced a significant reversal of tactile hypersensitivity. Microinjection of a preadsorbed antiserum had no effect. Neither NPY antiserum nor the preadsorbed antiserum produced any changes in paw withdrawal thresholds in sham-operated rats.B, Microinjection of NPY antiserum did not block thermal hyperalgesia. NPY antiserum did not produce any changes in paw withdrawal latencies in sham-operated rats. *p < 0.05.
Thermal hyperalgesia in SNL rats was indicated by a significant reduction in paw withdrawal latency to 9.28 ± 0.63 sec from a pre-SNL baseline value of 15.7 ± 0.54 sec (p < 0.05). In contrast to tactile hypersensitivity, however, the paw withdrawal latencies of either SNL or sham-operated rats were not changed by microinjection of antiserum to NPY in the n. gracilis (Fig. 6B). The microinjection of a preadsorbed antiserum into the n. gracilis also did not significantly alter the paw withdrawal latencies in either sham-operated or nerve-ligated rats (Fig. 6B). Microinjection of the antiserum into the contralateral n. gracilis had no effect on either tactile or thermal thresholds (data not shown). Control (nonimmune) serum also had no effect on sensory thresholds (data not shown).
Blockade of SNL-induced tactile hypersensitivity by microinjection of NPY receptor antagonists in the n. gracilis
Results similar to those with antiserum to NPY were obtained with the peptidic, nonselective NPY antagonist NPY(18–36). The microinjection of NPY(18–36) (0.5 nmol) into the n. gracilis blocked SNL-induced tactile hypersensitivity, as indicated by the significant increase in paw withdrawal thresholds from a baseline value of 3.02 ± 0.78 to 9.52 ± 1.3 gm (Fig.7A). In contrast, the administration of NPY(18–36) into the n. gracilis of rats with SNL did not produce any significant changes in thermal responses. The response latencies to noxious heat did not increase from the baseline value of 10.1 ± 0.38 sec (Fig.7B). Furthermore, the microinjection of NPY(18–36) into the n. gracilis of sham-operated rats did not produce any significant changes in behavioral responses to either tactile or thermal stimuli (Fig. 7A,B). The microinjection of vehicle into the n. gracilis also did not alter these behavioral end points in either the SNL or sham-operated rats (data not shown). Microinjection of the antagonist into the n. gracilis contralateral to nerve injury did not elicit any behavioral effects (data not shown).

Acute effects of microinjection of a nonselective NPY receptor antagonist, NPY(18–36), on the tactile hypersensitivity (A) and thermal hyperalgesia (B) induced by L5 and L6 SNL. The rats were prepared with guide cannulas directed at the n. gracilis.A, Microinjection of 0.5 nmol of NPY(18–36)into the n. gracilis produced a partial reversal of tactile hypersensitivity when compared with the preinjection threshold *p < 0.05. B, Microinjection of NPY(18–36) into the n. gracilis of the same rats did not block thermal hyperalgesia. Microinjection of the NPY antagonist into the n. gracilis of sham-operated rats did not produce any changes in responses to either tactile or thermal stimuli.
The selective Y1 antagonist BIBO 3304 was used to further validate the possible actions of NPY in the n. gracilis and to attempt to define the NPY receptor(s) that mediate the antagonism of SNL-induced tactile hypersensitivity. Tactile hypersensitivity was indicated by a significant reduction in the paw withdrawal threshold from pre-SNL baseline values of 15 ± 0 to 1.65 ± 0.51 and 1.58 ± 0.44 gm in separate groups of animals. The microinjection of 15 and 30 μg of BIBO 3304 (23.3 and 46.6 nmol, respectively) produced significant increases in the paw withdrawal threshold to 10.1 ± 1.80 and 12.8 ± 1.25 gm, respectively in these groups (Fig.8A). The responses to tactile stimuli returned to baseline values within 90 min of microinjection. In contrast, the microinjection of BIBO 3304 into the n. gracilis did not produce significant changes in paw withdrawal latencies to noxious radiant heat (Fig. 8B). The response latencies did not differ significantly from the post-SNL baseline values of 10.0 ± 0.29 and 10.1 ± 0.24 sec after 15 μg (23.3 nmol) and 30 μg (46.6 nmol), respectively. The microinjection of these doses of BIBO 3304 into the n. gracilis of sham-operated rats did not alter either tactile or thermal responses (Fig. 8A,B). The microinjection of vehicle also did not produce any changes in either behavioral end point in SNL or sham-operated rats (data not shown).

Acute effects of microinjection of a Y1 selective antagonist, BIBO 3304, on the tactile hypersensitivity (A) and thermal hyperalgesia (B) induced by L5 and L6 SNL. Male Sprague Dawley rats were prepared with guide cannulas directed at the n. gracilis. Tactile hypersensitivity (A) and thermal hyperalgesia (B) were confirmed in rats with L5 and L6 SNL. Microinjection of BIBO 3304 (15 and 30 μg, corresponding to 23.3 and 46.6 nmol, respectively) into the n. gracilis produced a time-dependent reversal of tactile hypersensitivity (*p < 0.05). BIBO 3304 administered to the same rats did not alter thermal hyperalgesia (B). Microinjection of BIBO 3304 into the n. gracilis of sham-operated rats did not produce any changes in responses to either tactile (A) or thermal (B) stimuli.
Lack of antagonism of SNL-induced tactile hypersensitivity by n. gracilis NPY Y2 receptor antagonist
The selective Y2 agonist BIIE 0246 was also used in the present studies in an attempt to determine the possible contribution of this receptor type to the manifestation of tactile hypersensitivity. The microinjection of 30 μg (33.5 nmol) of BIIE 0246 into the n. gracilis produced immediate and dramatic behavioral effects, including vocalization, spontaneous motor activity, barrel rolling, and lethality. The microinjection of 5 or 15 μg was without effect against either tactile or thermal hypersensitivity in rats with L5 and L6 SNL (data not shown).
DISCUSSION
The results of the present investigation are consistent with the possibility that upregulated NPY in afferent fibers projecting to the n. gracilis selectively mediates nerve injury-induced tactile but not thermal hypersensitivity. Evidence supporting this conclusion stems from the demonstration that (1) upregulation of NPY after SNL occurs in large fibers projecting to the n. gracilis via the dorsal columns with a time course that is consistent with the expression and persistence of tactile hypersensitivity; (2) microinjection of NPY into the n. gracilis produces reversible tactile but not thermal hypersensitivity of the ipsilateral hindpaw in uninjured rats; (3) microinjection of an anti-NPY antiserum selectively reverses SNL-induced tactile but not thermal hypersensitivity; and (4) microinjection of two NPY receptor antagonists with different physicochemical characteristics and receptor selectivity into n. gracilis selectively reverse SNL-induced tactile but not thermal hypersensitivity. These data are consistent with previous observations demonstrating that ipsilateral disruption of the dorsal columns or microinjection of lidocaine into the n. gracilis selectively blocks SNL-induced tactile hypersensitivity. The findings are also in line with the typical response characteristics of low-threshold myelinated fibers in mediating normally innocuous stimuli.
Nerve injury-induced pain has been closely linked to activation of spontaneous and persistent abnormal discharges from ectopic foci (Kirk, 1974; Wall and Gutnick, 1974; Devor, 1991, 1994; Koltzenburg et al., 1994). Spontaneous discharge is primarily observed in Aβ fibers after injury (Kajander and Bennett, 1992; Kajander et al., 1992; Boucher et al., 2000; Han et al., 2000; Liu et al., 2000a,b) and A-fibers have been suggested to drive central sensitization (Boucher et al., 2000). Although large myelinated fibers that project in the dorsal columns to the n. gracilis normally transmit sensations of light touch and vibration, these pathways have also been associated with pain (Angaut-Petit, 1976; Berkley and Hubscher, 1995; Willis and Westlund, 1997; Wang et al., 1999). Lesions of the dorsal columns block pelvic cancer pain (Hirshberg et al., 1996; Nauta et al., 1997), pain associated with pancreatitis, and bone pain (Houghton et al., 1997,1999, 2001). The excitation of neurons of the n. gracilis or of the ventroposterolateral nucleus of the thalamus elicited by colorectal distension or a noxious cutaneous pinch was blocked by lesions of the dorsal columns (Al-Chaer et al., 1996a,b). Chemical and physical lesions of afferent fibers or the n. gracilis have also been shown to block nerve injury-induced tactile hypersensitivity (Field et al., 1999; Miki et al., 2000; Sun et al., 2001). Microinjection of lidocaine into the n. gracilis ipsilateral to SNL reversibly blocks tactile but not thermal hypersensitivity (Sun et al., 2001).
The present study confirms and extends previous observations that nerve injury induces an upregulation of NPY in afferent fibers, particularly in medium- and large-diameter cells of the DRG (Zhang et al., 1993b;Persson et al., 1995; Li et al., 1999; Ma and Bisby, 2000). After nerve injury, NPY-IR was seen in the ipsilateral but not contralateral n. gracilis. NPY-IR in the n. gracilis was diffuse and not concentrated in cell bodies, suggesting its presence in nerve terminals. After nerve injury, NPY-IR-positive DRG cells were of a medium to large diameter, consistent with in situ hybridization studies (Wakisaka et al., 1991, 1992; Frisen et al., 1992; Noguchi et al., 1993,Zhang et al., 1993a,b; Marchand et al., 1999; Shi et al., 1999; Ma and Bisby, 2000). Increased spinal NPY-IR occurred in the deeper dorsal horn laminas, corresponding to the innervation field of axonal collaterals of Aβ fibers (Wakisaka et al., 1991; Frisen et al., 1992;Zhang et al., 1993a; Shi et al., 1999). The novel appearance of NPY-IR in deeper spinal laminas has suggested transport of newly synthesized NPY in DRG neurons to axon terminals (Shi et al., 1999). This interpretation is supported by our findings that dorsal rhizotomy abolished the upregulation of NPY-IR in the spinal dorsal horn and by the accumulation of NPY-IR proximal but not distal to the site of injury. The increase in NPY-IR in the spinal cord and DRG was restricted to the ligated segments L5 and L6 and was not increased in sections rostral or caudal to the zone of entry of the ligated spinal nerves. Furthermore, the data indicate that NPY is upregulated after nerve injury within 3 d, a time course consistent with the appearance of tactile hypersensitivity (Burgess et al., 2002). The upregulation of NPY-IR seen in the ipsilateral n. gracilis was abolished by lesions of the dorsal columns, and dorsal rhizotomy blocked the expression of NPY in the n. gracilis, in agreement with previous observations (Li et al., 1999). Collectively, these findings support the conclusion of nerve injury-induced upregulation of NPY in large fiber projections to the n. gracilis, providing an anatomical basis for this transmitter in possible mediation of nerve injury-induced abnormal pain.
Pharmacological studies showed that microinjection of NPY into the n. gracilis of uninjured rats produced relatively short-acting, reversible tactile hypersensitivity of the ipsilateral but not the contralateral hindpaw. The tactile hypersensitivity produced by this dose of NPY was time-related, consistent with the possibility of relatively rapid degradation of the unprotected peptide. The pronociceptive action observed with exogenous NPY was consistent with the finding of a complete, time-related, and reversible blockade of nerve injury-induced tactile but not thermal hypersensitivity by microinjection of an NPY antiserum into the n. gracilis ipsilateral to the side of nerve injury. Administration of a control serum or microinjection of preadsorbed NPY antiserum into the n. gracilis did not alter either tactile or thermal sensitivities in either nerve-injured or sham-operated rats. Furthermore, as with antiserum, the present studies show that microinjection of the nonselective NPY antagonist NPY(18–36) also produced a time-related, reversible block of tactile hypersensitivity but not thermal hyperalgesia in nerve-injured rats.
BIBO 3304 is a highly selective antagonist with subnanomolar affinity for the rat Y1 receptor and micromolar affinities for rat Y2, Y4, and Y5 receptors (Wieland et al., 1998), whereas BIIE 0246 shows nanomolar affinity for the rat Y2 receptor without competing for Y1, Y4, and Y5 receptors (Doods et al., 1999). BIBO 3304 blocked nerve injury-induced tactile hypersensitivity but not thermal hyperalgesia, whereas BIIE 0246 had no effect up to doses that produced severe behavioral toxicity, making conclusions of possible Y2 receptor contribution impossible. The doses of BIBO 3304 used in the present studies are comparable with those reportedly used to achieve a pharmacologic effect in feeding-related studies (Kask and Harro, 2000; Polidori et al., 2000) but are nevertheless high compared with its binding affinity for the Y1 receptor. At this concentration, BIBO 3304 may not act exclusively at the Y1 receptor to block nerve injury-induced tactile hypersensitivity. Although other NPY receptors may be differentially expressed in the post-nerve injury state, however, there are as yet no selective Y4 or Y5 antagonists that are suitable for studies in vivo (Dumont et al., 2000; Kask et al., 2001). Additional studies will be required to establish the nature of the NPY receptor in the n. gracilis, which may mediate the actions of NPY in the nerve injured condition.
The mechanism by which NPY neurotransmission in the n. gracilis mediates tactile hypersensitivity with regard to rostral projections remains uncertain. The present study indicates that NPY is upregulated in nerve fibers and not neuronal cell bodies within the n. gracilis after SNL. In the n. gracilis, NPY might directly activate rostral projections, or the enhanced output might be mediated through disinhibition of such projections. In this regard, it is noted that NPY has been shown to decrease or increase the amplitude of high-threshold Ca2+ currents of different neuronal populations (Wiley et al., 1993). In DRG neurons obtained from rats with sciatic axotomy, Y2-mediated excitation was enhanced, and Y1-mediated inhibition was suppressed (Abdulla and Smith, 1999). The lemniscal projections from the n. gracilis converge with projections of the spinothalamic tract to innervate neurons of the ventrobasal thalamus, providing for a potential merging of tactile and noxious inputs (Ma et al., 1987). Such convergence might provide for the activation of a descending cascade via spinopetal facilitatory and inhibitory mechanisms occurring downstream from the ventrobasal thalamus, perhaps including structures of the rostroventral medial medulla (Rees and Roberts, 1989, 1993).
The present study extends previous work showing that manipulations that block neuronal transmission in the n. gracilis selectively prevent expression of nerve injury-induced tactile hypersensitivity. Here we show that the pharmacological block of SNL-induced NPY neurotransmission within this nucleus achieves the same result. This conclusion rests on the use of multiple approaches, including the use of specific antiserum to NPY, peptidic and nonpeptidic NPY receptor antagonists, the time course and persistence of upregulation of NPY, the selective blockade of tactile but not thermal hypersensitivity only in the paw ipsilateral to the nerve injury, and the pharmacological mimicking of the tactile hypersensitivity induced by microinjection of NPY into the n. gracilis of uninjured rats. These data support the view that tactile and thermal aspects of experimental neuropathic pain are differentially mediated and identify NPY as a novel mediator of tactile hypersensitivity. The data suggest the possible application of NPY receptor antagonists for therapeutic treatment of nerve injury-induced tactile allodynia.
Footnotes
This work was supported by National Institute on Drug Abuse Grant DA11823.
Correspondence should be addressed to Dr. Frank Porreca, Department of Pharmacology, College of Medicine, University of Arizona Health Sciences Center, Tucson, AZ 85724. E-mail: frankp{at}u.arizona.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}