

Abstract
Although gap junctions regulate essential processes during development and differentiation, the role of gap junctions in cell death is poorly understood. We demonstrate here that the forced expression of connexin 43 (Cx43), the main constituent of astrocytic gap junctions, protected against cell injury with a potency that was comparable with that from the expression of the proto-oncogenebcl2. The expression of two other members of the Cx family, Cx32 and Cx40, also increased the resistance to injury from exposures to calcium overload, oxidative stress, metabolic inhibition, tamoxifen, and UV irradiation, but not against staurosporine- and dexamethasone-mediated death. Surprisingly, the anti-death activity of connexin proteins was independent of gap junction channel function, because physical isolation or the pharmacological inhibition of coupling did not significantly increase cell death. Moreover, cells expressing nonfunctional mutant connexins also acquired a high resistance to injury. These observations identify Cx proteins as active players in cell survival.
Introduction
Gap junctions are a subset of membrane channels that link neighboring cells. They are composed of connexins (Cxs), a highly conserved multigene family (Bennett et al., 1994). At present, 20 Cx genes have been cloned and characterized from rodents, and homologs have been identified in humans, chicks, frogs, and fish (Evans and Martin, 2002). Gap junctions are ubiquitously expressed by cells of many types, and they are believed to play an essential role in diverse processes, including proliferation, differentiation, morphogenesis, and pattern formation (Kumar and Gilula, 1996; Goldberg et al., 2000; Grueterich et al., 2002; Huang et al., 2002; Levin, 2002).
In the brain, astrocytes are the chief expressers of gap junctions. During ischemic injury, astrocytes remain coupled until a very late stage of cell death (Cotrina et al., 1998a). Multiple roles have been ascribed to the persistence of coupling between dying and viable cells. First, the so-called kiss of death, whereby gap junctions facilitate the propagation and amplification of cell injury (Lin et al., 1998), increases neuronal vulnerability to ischemic (Rami et al., 2001) and traumatic injury (Frantseva et al., 2002). Similarly, ganciclovir gene therapy relies on gap junctions to conduct the bystander killing (Mesnil et al., 1996; Carystinos et al., 1999;Andrade-Rozental et al., 2000; Krutovskikh et al., 2002). Second, the so-called Good Samaritan effects, whereby gap junctions serve to stabilize cellular calcium homeostasis and dissipate oxidative stress, decrease neuronal vulnerability to oxidative stress (Blanc et al., 1998).
In general, the effects of Cx expression have been attributed to gap junction coupling and sharing of a common pool of intracellular messengers. However, Cxs play multiple roles other than being an integral constituent of gap junction channels. For example, Cx expression facilitates the release of ATP and oxidized nicotinamide adenine dinucleotide independently of gap junction coupling (Cotrina et al., 1998b; Bruzzone et al., 2001; Arcuino et al., 2002); several lines of work suggest that Cxs regulate cell growth by mechanisms that do not require gap junction communication (Huang et al., 1998; Omori and Yamasaki, 1998; Moorby and Patel, 2001; Qin et al., 2002).
In this study, we set out to define the role of gap junction channels versus other effects of Cx expression in cell injury. We used an array of wild types as well as mutated Cxs with defects in channel function to analyze the role of Cx expression in cellular resistance. We found that Cx expression very significantly enhanced injury resistance, anin vitro finding that echoes the results of thein vivo study by Oguro et al. (2001) that Cx32 contributes to the survival and resistance of GABAergic interneurons in the hippocampus during global ischemia. Three members of the Cx family increased the injury thresholds required to activate the classical pathways of both apoptotic and necrotic cell death. Mechanistic analyses revealed that functional gap junction channels played a minor role in injury protection. Rather, the increased resistance was attributed to Cx-mediated cytoskeletal organization and faster normalization of cytotoxic elevations of calcium, which enabled Cx-expressing cells to survive an otherwise lethal injury.
These studies conclude that Cx expression has a very significant impact on cellular injury resistance by processes independent of gap junction coupling. The fact that Cx-mediated injury resistance does not require functional gap junction channels provides a platform for separating the advantage of Cx expression from the harmful effects of bystander death.
Materials and Methods
Cell cultures, stable transfection, and adenovirus-mediated gene transfer. C6 glioma, HeLa (American Type Culture Collection, Manassas, VA), and N2A (a clone with no electrophysiological detectable gap junction coupling; a gift from D. C. Spray, Albert Einstein College, Bronx, NY) were grown in DMEM supplemented with 10% fetal bovine serum and antibiotics. cDNA for human Bcl2 (a gift from S. Korsmeyer, Harvard Medical School, Boston, MA) was cloned in pCEP4 (Invitrogen, Carlsbad, CA), and its expression driven by a cytomegalovirus promoter. Transfection was performed with Clonfectin (Clontech, Palo Alto, CA) according to the manufacturer's instructions; stable transfectants were selected with 200 U/ml hygromycin. Independent clones were established. Parallel control transfectants were obtained simultaneously using the expression vector without the cDNA insert. The expression of Bcl2 was evaluated by Northern blot analysis, immunostaining (anti-Bcl2; Oncogene Sciences, Cambridge, MA) and resistance to calcium overload, oxidative stress, and metabolic inhibition. Control transfectants, like their parental C6, expressed no detectable Bcl2. Likewise, native C6 glioma expresses little Cx43. Control transfected clones with no detectable Cx43 immunoreactivity, and which exhibited no functional coupling by dye-transfer assays, were used in this study as a control (C6–mock 1–4 cells). cDNA for Cx43 was ligated into the expression vector pcDNA1, and cDNAs for Cx40 and Cx32 were ligated into pBEHpac18. Transfections were performed as described above, and stable transfectants were selected with 2 mg/ml geneticin (for Cx43) or 2 μg/ml puromycin (for Cx32 and Cx40). The chimeric constructs Cx40*43C3 and Cx40*43E2 were generated by exchanging Cx40 domains [C3 is the third cytoplasmic domain (i.e., the C-terminal tail) and E2 is the second extracellular loop] for the corresponding domains of Cx43 by site-directed mutagenesis (Haubrich et al., 1996). The Cx43M1 point mutation (C61S) was generated by site-directed mutagenesis, replacing the cysteine residue in position 61 with a serine residue. All of these cDNA constructs were ligated to the expression vector pBEHpac18 (Lin et al., 2002). The expression of Cx43, Cx40, and Cx32 was determined by immunolabeling; they were related to gap junction coupling by functional dye transfer on a biweekly schedule.
Fusion cDNA of enhanced green fluorescent protein (EGFP; Clontech) and one of the two mutant Cx43 constructs was generated by the PCR overlap extension method (Ho et al., 1989): dominant-negative mutants L160M (residue 160 at the third transmembrane domain, where lysine is replaced with methionine) (Omori and Yamasaki, 1998; Goldberg et al., 2000) and Δ130–137 (residues 130–137 at the second cytoplasmic domain were deleted) (Krutovskikh et al., 2002; Oyamada et al., 2002). The fusion cDNAs were subcloned into the Rous sarcoma virus-driven expression cassette of the pAdlox vector containing the adenovirus type 5′ left long terminal repeat and Ψ5 packaging sequence followed by a loxPsequence at the 3′ end (Hardy et al., 1997). The resultant constructs (e.g., Cx43L160M–EGFP–pAdlox) were each linearized and coinfected into a Cre–recombinase-expressing stable human embryonic kidney 293 cell line along with the Ψ5 DNA, generating an infective but replication-deficient adenovirus [e.g., Ad(Cx43L160M–EGFP)]. These fusion proteins expressed as green fluorescent plaques at cell-to-cell contact (see Fig. 5A). A viral construct for wild-type Cx43 was generated in the same way, except that cDNAs for Cx43 and EGFP were carried on separate cassettes. EGFP expressed throughout cytoplasm, whereas Cx43 expression needed to be visualized via immunocytochemistry (see Fig. 6,inset).
For killing studies, viruses were added to dissociated N2A, HeLa, or wild-type C6 cells at ∼300 pfu per cell during seeding. Viral expression was monitored by confocal microscopy of EGFP. Peak expression (>98% efficiency) usually occurred at ∼48 hr, at which time cells were exposed to various concentrations of tamoxifen. The cell survival rate was evaluated 24 hr later by Alamar Blue assay (Biosource, Camarillo, CA) (Farinelli and Greene, 1996).
Of note, the Cx- as well as the mock-transfected clones selected for this study all exhibited a proliferation rate that did not differ significantly from C6 wild type. Typically, the clones doubled in cell number every 26–28 hr (data not shown).
Mice with a null-mutation of Cx43. Heterozygotes of the Cx43 knock-out (KO) line were obtained from The Jackson Laboratory (Bar Harbor, ME). Pregnant females were killed at 18–20 d of gestation, and the embryonic brains were cultured as described previously (Nedergaard, 1994; Cotrina et al., 1998a). To identify Cx43 null homozygotes, heterozygotes, and wild types, PCR for amplifying tail-blood genomic DNA flanking the null deletion was used, as per The Jackson Laboratory protocol. Also, immunohistochemical mapping of the extent of Cx43 expression was performed in conjunction with dye-transfer assays. Astrocytes from the Cx43 null homozygote and wild-type mice used in this study were from three different litters. Cultures were grown 2–6 weeks in vitro before use.
Immunocytochemistry and functional coupling assay.Astrocytes and C6 cells were plated on 12 mm uncoated coverslips (2–4 × 104 cells), grown in 24 well plates to near confluence, and fixed with 4% paraformaldehyde. Cultures were permeabilized with 0.1% Triton X-100, blocked with 10% normal goat serum (Cotrina et al., 1998a), and immunoreacted with one of the following: polyclonal antibodies directed against amino acid residues 302–319 of the C-terminal tail of Cx43 (Bruce Nicholson, State University of New York, Buffalo, NY) (De Sousa et al., 1997), polyclonal antibodies directed against residues 337–358 of the C-terminal tail of Cx40 (Otto Traub, Universität Bonn, Bonn, Germany) (Traub et al., 1994), and monoclonal antibodies against residues 95–125 in the central cytoplasmic loop of Cx32 (David Paul, Harvard University, Boston, MA) (Meda et al., 1993).
The dye-transfer technique was adapted from Goldberg et al. (1995). Cells were loaded with 5 (and 6)-carboxy-2′,7′-dichlorofluorescein diacetate (CDCF diacetate; Molecular Probes, Eugene, OR) for 5 min, washed, and trypsinized. After resuspension, the cells were labeled with 10 μm1,1′-dioctadecyl-3,3,3′,3′-[tetramethylindocarbocyanine perchlorate (DiIC18) (Molecular Probes) for 10 min and mixed with unlabeled cells at a 1:250 ratio. One hour after plating on polylysine-coated dishes, dye transfer from the CDCF/DiIC18-labeled (donor) cells to unlabeled (recipient) cells was evaluated using confocal scanning microscopy. Counts of both the labeled donor cells and their recipients were performed manually. The coupling index was defined as the fraction of donor cells that transferred dye to surrounding cells, multiplied by the mean number of receiving cells.
Cell killing. Seven different paradigms of cell killing were studied. (1) Exposure to the calcium ionophore lasalocid (Lin et al., 1998) (40 μm lasalocid for 20–90 min); (2) oxidative stress, as induced by the free radical generator menadione (200 μm for 10–35 min) (Zhong et al., 1993); (3) metabolic inhibition, accomplished using a combination of 1 mm KCN (an electron transport inhibitor) and 0.02 mm iodoacetate (a blocker of glycolysis) for 2–8 hr (Cotrina et al., 1998a); (4) tamoxifen (10–25 μm for 24 hr) (Zhang et al., 2000); (5) dexamethasone (0.1–5 mm for 24 hr) (Simard et al., 1999); (6) UV exposure (30 W, 15 cm for 10–50 min) (Billecke et al., 2002; Zeng et al., 2002); and (7) staurosporine (0–3 μm) (Kabir et al., 2002; Rabkin and Kong, 2002). In each run, six cultures were included at a minimum: one set as a control and the remaining five to adjust exposure duration (e.g., 10, 12, 14, 16, and 18 min of menadione treatment) or concentrations, to generate a dose–response curve. Cultures were confluent 1 d after seeding. Exposures to the first three insults were performed in HBSS at 37°C. The cultures were washed before and after with HBSS and returned to the incubator in fresh serum-free DMEM/F12 medium. Control cultures were exposed to HBSS and processed identically. Twenty-four hours later, the viability was assessed either by quantifying the number of apoptotic cells [Hoechst (2 μm) or terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) stain (Lin et al., 1998)] or by alamar blue assay (Biosource) (Farinelli and Greene, 1996). Each data point represented the average of at least five different runs (range, 5–177).
Cell labeling and calcium imaging. In cocultures, C6–Cx43 cells were prelabeled with a 2 μm concentration of the red fluorescent cell tracker dye 5-(and-6)-(((4-chloromethyl)benzoyl)-amino)tetramethylrhodamine (CMTMR) (Molecular Probes), according to the manufacturer's instructions. CMTMR contains a thiol-reactive chloromethyl group that, after reaction with intracellular thiols, becomes membrane- and gap junction-impermeable; the labeling does not alter the sensitivity to injury (Lin et al., 1998). The mixed cultures were loaded with 5 μm fura-2 AM for 1 hr. Cytosolic Ca2+ levels were quantified using Image-1 software (Universal Imaging Corporation, West Chester, PA) and an SIT camera (Dage-MTI, Michigan City, IN) as described previously (Lin et al., 1998).
Results
Exogenous expression of Cx43 increases the resistance of C6 cells to injury
The C6 glioma cell line was originally cloned from a rat glial tumor induced by N-nitrosomethylurea (Benda et al., 1968). Wild-type C6 cells are poorly coupled and display very low levels of transfer of gap junction-permeable dyes (Cotrina et al., 2000). C6 cells have been extensively used as a model of astrocytes, because the expression of many receptor types, ion channels, and transport systems mimics that of astrocytes (Brismar, 1995). To study the role of gap junctions in cellular responses to injury, we have established 10 stable clones of C6 cells expressing the predominant astrocytic gap junction protein connexin 43 (C6–Cx43) (Lin et al., 1998). The data presented here were from four representative clones compared with four mock-transfected clones. The expression of Cx43 was associated with a significant increase in intercellular coupling. A gap junction-permeable dye, CDCF, was transferred to a mean of 0.8 ± 0.2 and 7.0 ± 0.07 C6–mock 1 versus C6–Cx43 cells, respectively (p < 0.001). The expression of Cx43 was associated with a substantial increase in the cellular resistance to injury. Control C6 cells are highly sensitive to injury, and most cells died uniformly if exposed for >25 min to the calcium ionophore lasalocid (40 μm). Half-maximal death occurred after 12 ± 5 min (LD50, in the range of 10–25 min) of C6–mock 1 cell exposure to ionophore, but after 47 ± 6 min (in the range of 40–65 min) for C6–Cx43 cells (Fig. 1). In comparison, the stable expression of Bcl2 (C6–Bcl2) increased the LD50to 42 ± 7 min, as reported previously (Lin et al., 1998). Thus, Cx43 expression provided the same extent of protection against calcium ionophore-induced injury as bcl2, a proto-oncogene widely studied for its anti-apoptotic action (Fig. 1).

Cx43 expression confers resistance to injury.A–C, Confocal microscopic images showing double-immunofluorescence labeling for Bcl2 (fluorescein-tagged secondary antibody) and Cx43 (Texas Red-tagged secondary antibody). The nuclei were counterstained with Hoechst (blue).A, C6–mock 1 cells express undetectable levels of either Cx43 or Bcl2. B, C6 cells stably expressing Bcl2 (C6–Bcl2 cells) are strongly immunoreactive. C, Cx43 immunoreactive plaques in C6 cells stably expressing Cx43 (C6–Cx43 cells). Scale bar, 10 μm. D, TUNEL-positive C6–mock 1 cells fixed 24 hr after a 10 min exposure to the calcium ionophore lasalocid (40 μm). E, Percentage of cell death as a function of exposure time to lasalocid (40 μm) in C6–mock 1, C6–Cx43, and C6–Bcl2 cells. Cx43 expression and Bcl2 expression are both associated with a substantial increase in cellular resistance to the calcium ionophore. Error bars indicate SEM.
Three different members of the connexin family possess the anti-death activity
To determine whether anti-death activity is restricted to Cx43, or alternatively, is a more general feature of gap junction coupling, we generated C6 clones that stably expressed Cx32 (C6–Cx32). Cx32 is not endogenously expressed by C6 glioma or astrocytes, but it is the prominent gap junction protein among Schwann cells (Abrams et al., 2002). Seven of the C6–Cx32 clones displayed extensive dye coupling and transferred CDCF to an average of 11 ± 1 neighboring cells. C6–Cx32 cells were also highly resistant to ionophore, with an LD50 of 55 ± 6 min (Fig.2). The expression of connexin 40 (C6–Cx40) also increased coupling and ionophore resistance in parallel. In comparison, the LD50 values of four mock-transfected clones with low coupling were in the range of 12–16 min (Fig. 2). Plotting LD50 as a function of the coupling index (Fig. 2B, inset) showed that injury resistance was a linear function of the coupling index (r = 0.97). Collectively, these observations indicate that several different Cx proteins robustly protect against ionophore-induced injury.

Anti-death activity in Cx43-, Cx40-, and Cx32-expressing clones. A, LD50 (dosage causing half-maximal death) for the calcium ionophore lasalocid (40 μm) is significantly higher for Cx43, Cx40, and Cx32 cells than for four mock-transfected sister clones, C6–mock 1, C6–mock 2, C6–mock 3, and C6–mock 4. *p < 0.01. Error bars indicate SEM. B, Coupling index in the same clones as in A. Inset, LD50 is a direct function of the coupling index (r = 0.97). *p < 0.01; ANOVA and post hoc Bonferroni t test. Error bars indicate SEM. C, An example of the dye-transfer assay. C6–Cx43 cells were preloaded with DiIC18(red) and the gap junction-permeable tracer CDCF (green). Labeled C6–Cx43 cells were cocultured with unlabeled C6–Cx43 cells for 1 hr; gap junctional coupling was quantified by the transfer of CDCF from DiIC18-labeled cells to unlabeled cells. Donor cells appear yellowbecause of the merge of red and greenlabeling. Clusters of green receiving cells surroundyellow donors when the donor cell establishes a gap junction with adjacent cells (white arrowheads), whereas single yellow–red cells represent donor cells that fail to establish gap junctions (red arrowheads). The coupling index is defined as the mean number of receiving (green) cells per donor (yellow) cell.
Connexin expression increases resistance to a variety of injury paradigms
Transient exposure to the calcium ionophore lasalocid triggers a cell-death process that in its time course and progression shares many features with necrosis. Similar patterns of cell death can be evoked by exposure to the producer of free radicals, menadione, or by “chemical ischemia,” combined treatment with KCN and iodoacetate. We found here that expression of Cx43, Cx32, and Cx40 was associated with a substantial increase in resistance to menadione and KCN/iodoacetate exposure (Fig. 3). Likewise, the proto-oncogene Bcl2 also afforded protection against both of these insults in accordance with previous reports (Zhong et al., 1993; Kane et al., 1995; Myers et al., 1995). Several injury paradigms commonly used in the study of apoptotic injury, including UV irradiation, tamoxifen, staurosporine, and dexamethasone, were studied next. Cx43 expression robustly protected against tamoxifen and UV irradiation but not against dexamethasone- and staurosporine-induced injury. Both C6–mock and Cx-expressing cells died if exposed to dexamethasone at a concentration of >3 mm. Likewise, staurosporine killed all cell types at concentrations of >2 μm. Together, these observations indicate that gap junction-coupled cells display high resistance to most but not all types of cellular stress. A similar observation was made that Bcl2 does not protect against several injury paradigms (Takayama et al., 1995; Reed et al., 1996). We compared gap junction coupling during the process of tamoxifen- and dexamethasone-induced cell death. Tamoxifen (20 μm) and dexamethasone (2.5 mm) both reduced coupling (15.2 ± 3 vs 22.1 ± 0.6% of vehicle-treated control cultures, respectively). Thus, both injury paradigms decreased but did not abolish gap junction coupling during the active process of cell death. Because Cx43 expression afforded protection against tamoxifen-induced injury but not against dexamethasone-induced injury, the extent of coupling during the process of cell death may not be a significant factor in Cx-mediated injury resistance.

Cx43 and Cx32 confer resistance to several but not all injury paradigms. The LD50 of C6–mock 1, C6–Cx43, and C6–Cx32 cells exposed to menadione (200 μm), KCN (1 mm) and iodoacetate (IA; 0.02 mm), tamoxifen (10–25 μm), UV irradiation (0–60 min), dexamethasone (0–6 mm), and staurosporine (0–3 μm) is shown. The expression of Cx43 and Cx32 protects C6 cells against menadione, KCN and IA, tamoxifen, and UV irradiation but not against dexamethasone and staurosporine. *p < 0.01; ANOVA and post hoc Bonferroni t test. Error bars indicate SEM.
Of note, the level of Cx expression appeared to be an important determinant of injury resistance. We compared several clones with low Cx32 expression (coupling indices in the range of 2–4) with C6–Cx32 with high expression (coupling indices in the range of 8–13). High expressers of Cx32 were consistently more resistant to injury evoked by calcium ionophore, ATP depletion, and menadione exposure, compared with low-expressing clones. Figure4A–C illustrates results from studies of the mock 1 control versus one representative clone of each of the two expression levels. In addition, C6–Cx43 retained the transfected cDNA well. Repeated passaging did not diminish either the level of Cx43 expression or the corresponding injury resistance (Fig. 4D).

Anti-death activity is highest in cells with high Cx expression and is not affected by multiple replating. Comparison of injury resistance of C6–mock 1 cells (coupling index, 0.2), a low Cx32-expressing clone (coupling index, 2.3 ± 0.5), and a high Cx32-expressing clone (coupling index, 11 ± 1). Resistance to various insults is expressed as LD50 for lasalocid (40 μm) (A), menadione (200 μm) (C), and KCN (1 mm) and iodoacetate (0.02 mm) (B). The high-expression clone is consistently more resistant than the low-expression clone. *p < 0.01; ANOVA and post hoc Bonferroni ttest compared with C6–mock 1 cells. D, Multiple replating of C6–Cx43 cells does not decrease the LD50 of lasalocid. Error bars indicate SEM.
Approaching from the opposite direction, we tested the effect of two dominant-negative Cx43 mutants, L160M and Δ130–137, on injury resistance. We chose C6 wild-type cells because these cells display a low level of Cx43 expression and a minor degree of coupling (Fig. 2) (Cotrina et al., 2000). As shown in Figure5A, these dominant-negative Cx43 proteins succeeded in trafficking to the cell-to-cell junction and exerted their inhibitory actions, resulting in a significant increase in the sensitivity of their host cells to tamoxifen. Resistance of both L160M- and Δ130–137-expressing cultures was significantly increased compared with EGFP-expressing sister cells (Fig.5B). Thus, interference of Cx43 functions increased tamoxifen sensitivity, which is consistent with the idea that Cx expression positively regulates cellular resistance.

Dominant-negative Cx43 mutants reduce the resistance of wild-type C6 cells. A, The expression of the mutant Cx43 L160M as plaques at cell-to-cell contact was visualized via the fluorescence of EGFP (white arrows), whereas C6 wild type transfected with Ad(EGFP) displays diffuse fluorescence. Scale bar, 10 μm. B, C6 wild-type cells endogenously express a low level of Cx43, and their resistance to tamoxifen is significantly reduced after the expression of the Cx43 dominant-negative mutants L160M and Δ130–137 compared with cultures of C6 wild type expressing EGFP only. *p < 0.01; ANOVA and post hoc Bonferroni t test. Error bars indicate SEM.
Connexin expression also increases the resistance of N2A and HeLa cells to injury
Interestingly, the anti-death activity of Cx43 was not restricted to C6 glioma cells. Treatment of N2A neuroblastoma cells with Ad(Cx43–EGFP) increased the level of resistance to tamoxifen relative to controls that were treated with Ad(EGFP) (Fig.6). Both Cx43 and GFP expression (Fig. 6,inset) and the protection persisted for at least three additional passages. HeLa cells, a human-derived cervical tumor cell line with low endogenous Cx expression (Elfgang et al., 1995), also displayed a significant increase in cellular resistance to tamoxifen after the AdCx43-EGFP treatment. Control Ad(EGFP)-HeLa cells died uniformly if exposed to >22.5 μm tamoxifen, whereas AdCx43-EGFP-transfected sister cells tolerated 30 μm tamoxifen without a sign of cellular deterioration. In all, these data support the idea that Cx expression protects against cell injury in a variety of cell lines.
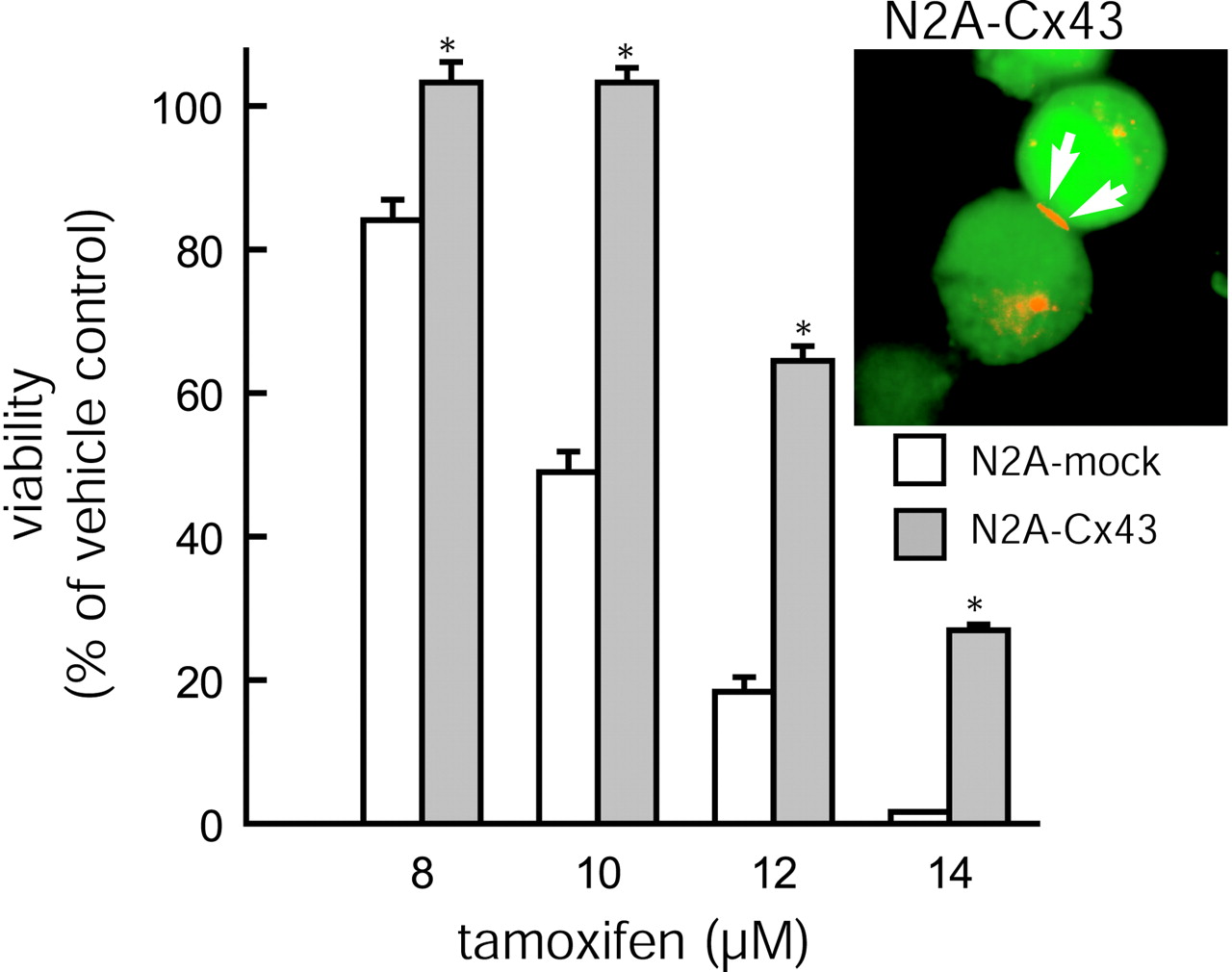
Resistance of N2A neuroblastoma cells to tamoxifen is increased with Cx43 expression. Cellular resistance to tamoxifen is increased in N2A cells transfected with an adenoviral construct, Ad(Cx43–EGFP), relative to mock controls that have been transfected with Ad(EGFP). *p < 0.01; ANOVA and post hoc Bonferroni t test. Error bars indicate SEM.Inset, Expression of Cx43 was visualized via immunocytochemistry with Cy3-tetramethylrhodamine isothiocyanate-tagged secondary antibodies. Because cDNAs for Cx43 and EGFP are located on separate cassettes, EGFP fluorescence is diffuse, whereas Cx43 immunoreactivity is restricted to a large plaque (red) at a region of cell-to-cell contact (white arrows).
Gap junction function is not required for connexin-mediated injury resistance
We subsequently assessed the effect of nonfunctional Cx mutations on cell resistance. Exchanging the cysteine residue of position 61 of the Cx43 sequence with serine by site-directed mutagenesis results in the loss of functional channel formation in cell lines expressing the mutant connexins (Elfgang et al., 1995; Haubrich et al., 1996; Lin et al., 2002). Three high-expressing clones were selected for the killing study. Despite a high level of C61S–Cx43 expression, Cx43 immunoreactive plaques were not found at cellular interfaces. Instead, a diffuse increase in cytosolic Cx43 immunoreactivity was evident (Fig.7A). Dye coupling was 0.5 ± 0.2, not different from that of mock-transfected clones; similarly, cellular resistance to injury was no better than mock-transfected controls (Fig. 7C). Thus, the expression of a mutant connexin that is not translocated to the membrane did not increase cellular tolerance to injury.

Plaque formation, but not functional gap junction channels, is required for Cx-mediated injury resistance.A, Diffuse Cx43 immunoreactivity in the cytosol of C6 cells transfected with mutant C61S–Cx43, which harbors a cysteine-to-serine point mutation at position 61. The C61S–Cx43 proteins do not form gap junction plaques, and correspondingly, do not increase the injury resistance. B, C6 cells transfected with a chimeric construct, Cx40*43C3, display immunoreactive plaques in cell membrane (arrows) and an increase in injury resistance. The expression of the Cx40*43 chimeras was visualized by an anti-Cx40 antibody, because the Cx43 antibodies target the C-terminal tail, which is not present in the chimeric constructs. Scale bar: (inB) A, B, 35 μm.C, Histograms summarizing the LD50 for ionophore (40 μm) exposure. *p < 0.01; ANOVA and post hoc Bonferroni ttest compared with C6–mock 1 cells. Error bars indicate SEM.
Our next question was whether gap junction function is required for connexin-mediated injury resistance or the docking of two hemichannels in the absence of channel activity is sufficient for the resistance. To this end, chimeric constructs that had been produced by swapping corresponding domains of Cx40 with those of Cx43 were expressed in C6 cells. Two of the constructs, Cx40*43E2 and Cx40*43C3, formed abundant Cx40 immunoreactive plaques that were nonetheless composed of nonfunctional channels in which neither dye transfer nor electrical coupling persisted (Haubrich et al., 1996; Lin et al., 2002) (Fig. 7B). Importantly, the injury resistance of C6 cells increased substantially after the expression of each of these plaque-forming yet functionally incompetent connexin chimeras: the LD50 of Cx40*43E2- and Cx40*43C3-expressing C6 to calcium ionophore exposure was not significantly different from the highly resistant C6–Cx43 cells (Fig.7C). Together, these results suggest that plaque formation is a critical element in connexin-dependent injury resistance, but that functional gap junction channels are not required.
High resistance of isolated Cx43-expressing cells
Another approach to test the role of intercellular coupling in injury resistance is to generate low-density cultures in which the lack of physical contact prevents gap junction assembly. Several plating densities were examined, and it was clear that the resistance to tamoxifen of both C6–Cx43 and C6–mock 1 cells decreased as a function of plating density in accordance with the general impression that low-density cultures are less resistant to injury (Fig.8). When the cells were plated at or below a density of 10,000 per 24 wells, only a limited number of cells established contact with surrounding cells. Despite their inability to form gap junctions, C6–Cx43 cells still tolerated and survived higher concentrations of tamoxifen than did C6–mock cells. This observation supports the notion that the high injury resistance of C6–Cx43 cells is not a direct result of gap junction coupling (Fig. 8). Of note, low-density cultures were not apt to be exposed to lasalocid, because the repeated medium changes used in the injury paradigm resulted in a significant cell loss.

C6–Cx43 cells remain resistant to injury in the absence of gap junctions. Representative fields of C6–Cx43 (left) and C6–mock 1 (middle) cell cultures in 24 well plates at 7000 (A), 15,000 (B), 23,000 (C), and 115,000 (C6–Cx43) or 200,000 (C6–mock 1) (both were confluent cultures) (D) cells per well. Note that at the lowest plating densities, cells rarely contact each other, thereby physically preventing the formation of gap junction channels. Scale bar, 50 μm. Right, Comparison of viability of C6–Cx43 (○) and C6–mock 1 (●) cells after exposure to increasing concentrations of tamoxifen. The sensitivity to tamoxifen increases inversely with the plating density for both C6–Cx43 and C6–mock cells. C6–Cx43 cells maintain their high resistance at plating densities at which gap junction coupling is prevented by the physical separation of the cells. Data are from a representative set of experiments. Similar results have been obtained from two other independent studies.
Gap junction inhibitors do not decrease the resistance of C6–Cx43 cells
To further confirm the gap junction-independent nature of Cx-mediated resistance, we tested the effect of a relatively nontoxic gap junction inhibitor, 18α-glycyrrhetinic acid (α-GA) on cellular resistance. α-GA (50 μm, 24 hr) decreased coupling among C6–Cx43 cells by 97 ± 4% (Fig. 1B,C) (Cotrina et al., 1998b). The treated cells appeared normal otherwise. α-GA-treated cultures were more sensitive to calcium ionophore than vehicle-exposed controls, although the difference was not significant. Similarly, C6–Cx32 exposed to α-GA displayed an insignificant reduction in resistance to calcium ionophore compared with matching controls. The LD50 of C6–Cx32 cells decreased from 43 ± 12 to 38 ± 9 min when treated with α-GA (p = 0.09). Because C6–mock 1 cells were insensitive to α-GA (Fig. 9), these observations suggest that the decreased resistance of C6–Cx43 and C6–Cx32 cultures in the presence of α-GA was linked specifically to a reduction in intercellular coupling. However, because α-GA changed the injury threshold only marginally while efficiently reducing gap junction coupling, this finding supports and extends the notion that a functional gap junction channel plays only a minor and insignificant role in Cx-mediated injury protection.

Cx-mediated resistance is not affected by gap junction blockage. The gap junction inhibitor α-GA does not significantly (p = 0.09 for LD50of C6–Cx32 ± α-GA) reduce the extent of injury after ionophore exposure. Viability (as percentage of vehicle control) is plotted as a function of increasing exposure time to lasalocid (40 μm) of C6–mock 1, C6–Cx43, and C6–Cx32 cells. Error bars indicate SEM.
Structural changes associated with the expression of Cx in part explain the increased resistance
We have noted previously that Cx expression is associated with cellular flattening and with the formation of epithelial-like sheets of polygonal cells. C6–mock and C6 wild-type cells contain only few and poorly organized stress fibers, whereas actin is typically organized in parallel arrays of stress fibers in C6–Cx43 and C6–Cx32 cells (Cotrina et al., 1998C) (Fig. 10). Because cell geometry in itself is recognized as a determinant of cellular resistance (Chen et al., 1997; Dike et al., 1999; Huang and Ingber, 2000), we tested the proposition that the Cx-induced phenotypic transformation contributed to the increased cellular resistance beyond the role of connexins in gap junction assembly. To that end, C6–Cx43 cells were raised in spheres by transferring dissociated cells to plates with nonadhesive substrate. The cells clustered within 2–4 hr and formed large aggregates consisting of several hundred cells by 24 hr. Figure 10 illustrates cultures fixed 24 hr after plating and stained with Texas Red–phalloidin. Characteristically, actin organized in aggregates of both C6–Cx43 and C6–mock 1 cells in a rim below the plasma membrane. Phalloidin staining was somewhat weaker in C6–mock 1 cells, in accordance with our previous observations (Cotrina et al., 2000). C6–Cx43 and C6–mock 1 cells raised in spheres were considerably less resistant to injury compared with attached sister cells. LD50 decreased from 67 ± 5 to 40 ± 6 min and from 15 ± 3 to 9 ± 2 min of lasalocid exposure in C6–Cx43 and C6–mock cells, respectively. Thus, the lack of substrate attachment reduced cellular resistance. However, importantly, C6–Cx43 cells remained more resistant than C6–mock cells, indicating that the resistance to injury of C6–Cx43 cells was not simply a result of altered cell morphology.

Cx-mediated resistance persists in suspended cells. A, A sphere of C6–Cx43 cells that grew on a low-attachment plate stained with Texas Red–phalloidin. Actin is organized in a cortical mantle below the plasma membrane and is not in parallel arrays of stress fibers as in attached sister cells (inset). B, Spheres of C6–Cx43 cells exposed for 24 hr to either vehicle (left) or 20 μm tamoxifen (right). The culture was stained with phalloidin (green) and propidium (red). Exposure to tamoxifen does not result in killing C6–Cx43 cells. C, A sphere of C6–mock 1 cells stained with phalloidin. Actin in these cells is organized in a cortical mantle below the plasma membrane both in the sphere and in attached sister cells (inset). Note that phalloidin staining is weaker compared with C6–Cx43 cells (A, B). D, Spheres of C6–mock 1 cells exposed for 24 hr to either vehicle (left) or 20 μm tamoxifen (right). Condensed apoptotic nuclei in the tamoxifen-exposed sphere reveal that C6–mock 1 cells (D, right) remain more sensitive to tamoxifen than C6–Cx43 cells (B, right). Scale bar, 20 μm.
Purinergic receptor antagonists reverse the resistance of Cx-expressing cells by a mechanism requiring structural changes
In addition to its role in energy metabolism, ATP is a transmitter with its own set of receptors, the purinergic receptors. We have noted previously that Cx-expressing cells release 10- to 100-fold more ATP than mock-transfected control cells (Cotrina et al., 1998b, 2000). Because extracellular ATP functions as a differentiation factor that mediates cellular flattening and stress fiber formation (Abbracchio et al., 1995), we tested the effect of purinergic receptor antagonists on injury resistance. Twenty-four hours of pretreatment with either suramin (50 μm) or reactive blue (50 μm) induced cellular compaction and loss of stress fibers, as observed previously, and resulted in a lower threshold to injury than matching vehicle-treated C6–Cx43 cells. LD50 decreased from 14.3 ± 1.2 μm in vehicle-treated cultures to 9.4 ± 0.7 and 8.9 ± 1.1 μm in suramin- and reactive blue-exposed cultures, respectively (Fig.11D). Because both suramin and reactive blue are characterized by a relatively unspecific mode of action, it is important that both antagonists altered the resistance of C6–mock cells only insignificantly. In contrast, if suramin and reactive blue were added immediately before the tamoxifen exposure, the injury sensitivity of either C6–Cx43 or C6–mock cells did not change significantly (data not shown). These observations indicate that purinergic receptors do not play a direct role in injury protection, but that receptor antagonists potentiate cell death indirectly by altering cellular organization to a compact and less-resistant phenotype. Of note, although cultures treated with either suramin or reactive blue exhibited a reduced number of cellular contacts, gap junction coupling was not significantly reduced (Fig.11A–C).
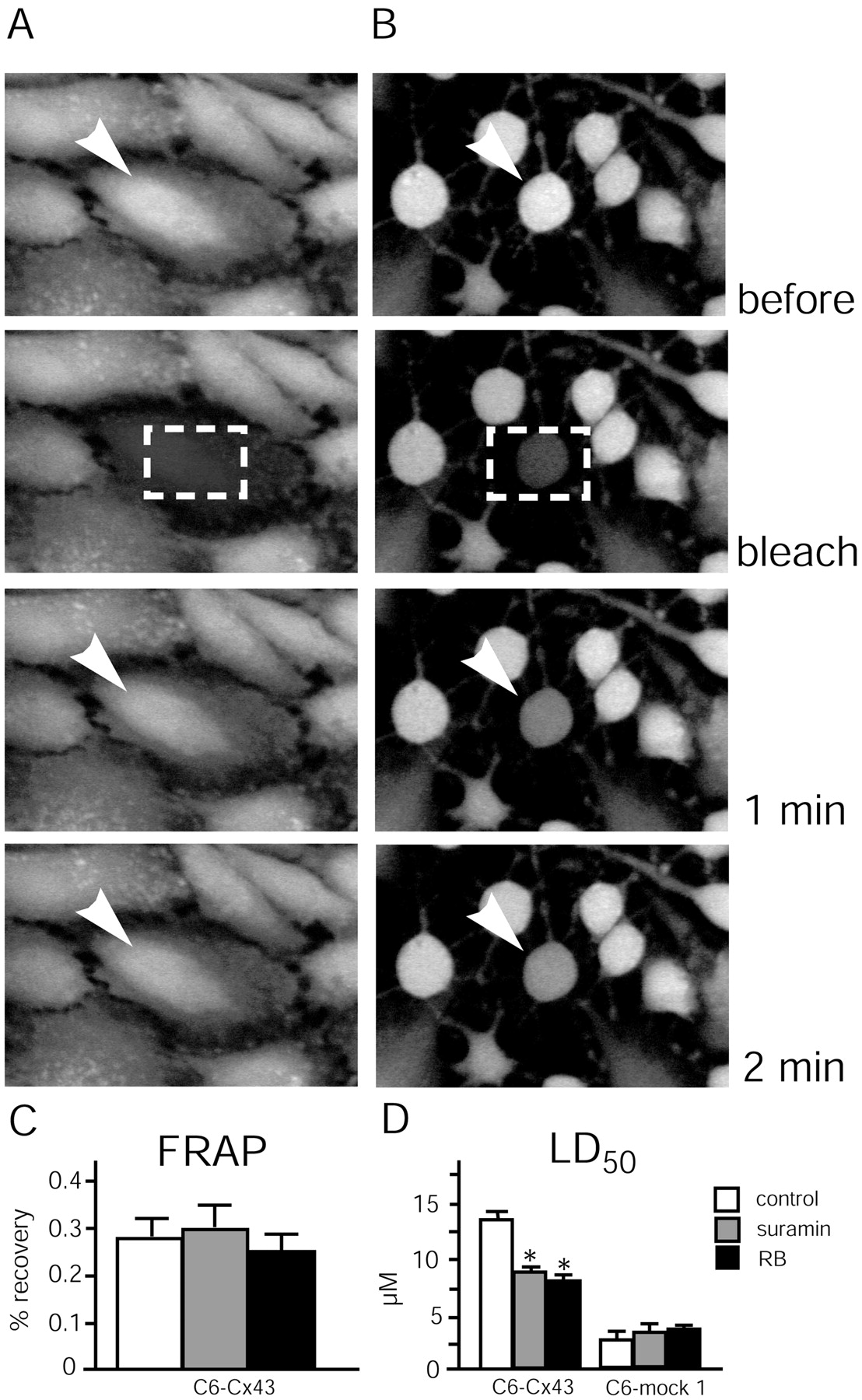
C6–Cx43 cells pre-exposed to purinergic receptor P2Y antagonists remain coupled by gap junctions despite compaction and retraction, but their resistance to tamoxifen is compromised. A, Untreated C6–Cx43 culture loaded with the gap junction-permeable fluorescence indicator CDCF.Top, A field of cells before photobleach.Bottom, A field of cells collected immediately after photobleach, or 1 and 2 min later. The rapid recovery of CDCF fluorescence indicates that the cells are well coupled by gap junctions to neighboring cells. Arrowheads indicate the cell that is subjected to photobleach. Dashed boxes indicate areas of photobleach. B, C6–Cx43 cells exposed to the purinergic receptor antagonist reactive blue (50 μm) for 24 hr. Exposure to reactive blue does not decrease gap junction coupling, despite the reduction in cellular contact. C, Fluorescence recovery after photobleach in vehicle-, reactive blue-, and suramin-treated (50 μm each, 24 hr) C6–Cx43 cultures. D, Comparison of tamoxifen LD50. Reactive blue (RB) and suramin (50 μmeach, 24 hr) significantly reduce LD50 for C6–Cx43 but not the LD50 for C6–mock 1 cells. *p < 0.01; Student's t test.
Astrocytes from Cx43 knock-out mice and wild-type astrocytes are equally resistant to ionophore
Several groups have shown previously that astrocytes from Cx43–KO mice maintain 5% residual coupling compared with astrocytes from matching wild-type controls, likely reflecting that astrocytes in addition to Cx43 also express low levels of Cx30, Cx40, and Cx45 (Dermietzel and Spray, 1998; Cotrina et al., 2000). The phenotypic characteristics of Cx43–KO astrocytes are indistinguishable from astrocytes prepared from wild-type littermates, despite the severe reduction in gap junction coupling (Cotrina et al., 1998c; Scemes et al., 2000). Likewise, we observed that astrocytes prepared from Cx43–KO mice displayed only an insignificant reduction in injury resistance (Fig. 12). The LD50 of ionophore-induced cell death was 6.4 ± 0.6 min in astrocytes harvested from Cx43–KO mice versus 7.3 ± 1.1 min in astrocytes prepared from the matching wild-type control. Thus, the loss of Cx43 did not significantly reduce injury resistance. Activation of compensatory mechanisms during development is common in studies of KO mice, and the fact that the phenotype (e.g., cell morphology and calcium wave propagation) of astrocytes from Cx43–KO was indistinguishable from that of the wild-type suggests that compensatory mechanisms are indeed activated. Because the expression of other members of the Cx family is not upregulated and gap junction coupling is reduced in astrocytic cultures prepared from Cx43–KO mice, this set of observations adds additional support to the conclusion that gap junction channel formation plays a minor role in cellular resistance to injury.

No difference in ionophore sensitivity between wild-type and Cx43–KO astrocytes. Confluent cultures were treated with 40 μm lasalocid for the times indicated. Viability was evaluated by alamar blue assay and expressed as a percentage of vehicle control. The LD50 for wild-type versus Cx43–KO astrocytes is not significantly different. Error bars indicate SEM.
Impact of Cx expression on calcium regulation
To determine whether changes in cytosolic calcium concentration, [Ca2+]i, during injury predict the extent of cell death, we measured [Ca2+]i during and after exposure to lasalocid (40 μm) in fura-2-loaded cultures (Lin et al., 1998). Resting [Ca2+]i was in the range of 80–120 nm in all clones studied, including C6–mock 1, C6–Cx43, and C6–Cx32 cells. When exposed to the calcium ionophore, C6–Cx43 cells displayed an initial increase in [Ca2+]i, which peaked at 600 nm, followed by a slow normalization of [Ca2+]i ∼10 min later, and [Ca2+]ireturned to 200–300 nm and remained at this level during the rest of the experiment. C6–Cx32 cells responded to the ionophore in a similar manner, as did C6–Cx43 cells (data not shown). In contrast, the ionophore-induced [Ca2+]i elevation peaked at ∼1000 nm in C6–mock 1 cells; the initial attempt to restore [Ca2+]i levels was followed by a delayed and irreversible increase in [Ca2+]i. This second increase in [Ca2+]i rose slowly and did not normalize: After ionophore washout, [Ca2+]i levels decreased but remained elevated in the majority of the C6–mock 1 cells. To compare the different cellular [Ca2+]i responses directly, we exposed cocultures of C6–Cx43 and C6–mock 1 cells to lasalocid. Figure 13 illustrates that the cell-specific changes in [Ca2+]i were maintained in cocultures, and that C6–Cx43 cells displayed minor and shorter-lasting increments in [Ca2+]i compared with C6–mock 1 cells.

Ionophore-induced increases in cytosolic [Ca2+]i are suppressed by Cx expression. A, Resting [Ca2+]i levels in a mixed culture of C6–Cx43 (white arrows) and C6–mock 1 cells loaded with the calcium indicator fura-2. B, Peak [Ca2+]i increments during exposure to the calcium ionophore lasalocid (40 μm). Note the lower amplitude of [Ca2+]i increments in C6–Cx43 cells (white arrows) compared with C6–mock 1 cells. C, After 40 min of ionophore exposure, [Ca2+]i in all four C6–Cx43 cells had normalized to a level somewhat higher than resting [Ca2+]i, whereas [Ca2+]i remained elevated in the majority of C6–mock 1 cells. D, Phase contrast micrograph of the same field. The C6–Cx43 cells were prelabeled with the cell tracker CMTMR. The CMTMR labeling was digitally superimposed to visualize C6–Cx43 cells. Note the flat morphology of C6–Cx43 cells compared with the elongated C6–mock 1 cells that typically exhibit less cellular contact. E, [Ca2+]i as a function of time in the cultures shown in A–C. Peak [Ca2+]i increments in C6–Cx43 cells during ionophore exposure are lower than those in surrounding C6–mock cells (left; p < 0.001; Student'st test), normalize faster, and do not show a delayed secondary increase in calcium. Scale bar: (in D)A–D, 50 μm.
To further compare [Ca2+]i responses in Cx-expressing versus Cx-deficient cells over time, we integrated [Ca2+]i increments above resting levels over the course of the experiment (60 min). The integral of [Ca2+]i increase was significantly higher in Cx-deficient compared with Cx-expressing cells. The [Ca2+]iintegral averaged 12 ± 0.8 and 15 ± 1 μm in C6–Cx43 and C6–Cx32 cells, respectively, compared with 40 ± 6 μm in C6–mock 1 cells (n = 10;p < 0.01). Thus, [Ca2+]i increased significantly less over time in the Cx-expressing clones compared with the mock-transfected clones. In other words, Cx-expressing cells were able to restore and maintain [Ca2+]ihomeostasis in the presence of ionophore, whereas C6–mock 1 cells failed to do so.
DISCUSSION
The new observation in this report is that the expression of Cxs, the integral proteins of gap junctions, is associated with an increased resistance of a variety of cells to most but not all types of injury. The magnitude of the resistance provided by several members of the Cx family was comparable with the protection afforded by the widely known proto-oncogene Bcl2. Blockage of Cx43 by its dominant-negative mutants suppressed cellular resistance to tamoxifen. Surprisingly, gap junction coupling per se appeared to play a minor role in injury protection. Several lines of observations supported the notion that Cx-mediated injury resistance did not require the physical formation of gap junction channels. First, Cx-expressing cells retained their resistance in low-density cultures with few cell contacts. Second, pharmacological inhibition of gap junction channels only insignificantly reduced the injury resistance of Cx-expressing clones. Third, the expression of two Cx constructs that failed to establish functional gap junctions efficiently protected the cells against injury. The mechanism by which Cx proteins enhance cell resistance was not clearly defined, but cell flattening and stress fiber assembly contributed to injury resistance in Cx-expressing cells (Chen et al., 1997; Dike et al., 1999; Cotrina et al., 2000; Huang and Ingber, 2000).
In this study we compared Cx-mediated injury resistance with the effects of Bcl2 expression. Bcl2 was first found to permit the survival of cytokine-dependent hematopoietic cells (Vaux et al., 1988), and its anti-apoptotic action was later verified in several other cell types (Chao et al., 1995). Bcl2 is located primarily on the cytoplasmic face of the mitochondrial outer membrane (Hockenbery et al., 1990), where it inhibits the release of cytochromec to cytosol (Kluck et al., 1997; Yang et al., 1997; Shimizu et al., 1999). In addition, Bcl2 has a variety of other functions (Green and Reed, 1998). Several mitochondrial events have been linked to Bcl2, including regulation of the ATPase mitochondrial proton pump, increases in mitochondrial Ca2+ buffering capacity, and prevention of mitochondrial permeability transition (Adams and Cory, 1998; Green and Reed, 1998). It is of interest to note that Bcl2, like several members of the Cx family, forms channels in lipid bilayers and modulates cell-cycle progression and proliferation (Chao and Korsmeyer, 1998). In addition to their anti-apoptotic actions, both connexins andBcl2 prevent necrotic cell death (Zhong et al., 1993; Kane et al., 1995; Myers et al., 1995).
For decades it was assumed that cells uncouple during the process of death. We recently challenged this view by demonstrating functional coupling among ischemic dying astrocytes both in vivo andin vitro (Cotrina et al., 1998a). Although coupling decreases, astrocytes remain interconnected during the process of ischemic cell death. In fact, subsequent experiments have documented that gap junctions can propagate or amplify focal injury to include otherwise viable neighboring cells, so-called bystander death. Bystander death has been observed in a variety of settings, including focal experimental ischemia (Rawanduzy et al., 1997), in cultured cells (Lin et al., 1998), after the traumatic injury of cultured hippocampal slices (Frantseva et al., 2002), and in a model of transient global ischemia (Rami et al., 2001). In essence, the gap junction can increase local injury by expanding death to neighboring cells. However, it is important to note that the Cx-mediated injury resistance observed in this study does not contradict the existence of bystander death (Lin et al., 1998). The two phenomena, bystander death and Cx-mediated injury resistance, differ fundamentally. Bystander death is triggered by gap junction-mediated diffusion of the intracellular messenger and is attenuated by gap junction inhibitors (Rawanduzy et al., 1997). In contrast, Cx-mediated injury resistance does not require gap junction formation and is insensitive to uncoupling agents (Fig. 9). The ability to distinguish clearly between bystander death and Cx-mediated injury resistance in this study may aid future analyses of the role of Cxs in cell injury.
The mechanism of Cx-mediated injury protection is not clearly defined, but there is no doubt that a monolayer of cells fights stressful conditions more efficiently than isolated cells. It is possible that sharing a common pool of metabolites and intracellular messengers improves survival. Our data support this conclusion by demonstrating that cellular resistance decreased in proportion with plating density (Fig. 8). The surprising and less intuitive observation is that Cx-mediated resistance for the most part was not a result of gap junction channel formation. Physically isolated Cx-expressing cells remained highly resistant, despite their inability to form gap junctions. Another observation was that the expression of nonfunctional Cx chimeras gave rise to a highly resistant but poorly coupled cellular phenotype. Additional support for a relatively minor role of gap junction coupling in injury protection derives from the inability of gap junction inhibitors to reduce cellular resistance. We speculate that Cx-induced cellular flattening and stress fiber formation play a significant protective role, but that other factors, in particular a more efficient apparatus for calcium homeostasis, are important. This conclusion is based on the notion that Cx-expressing cells raised in spheres on low-attachment plates remained resistant compared with Cx-deficient sister cells, although these culture conditions prevented stress fiber formation. In this regard, Cx proteins have been implicated in several gap junction-independent processes. For example, Cx expression potentiates ATP release (and long-distance calcium waves), decreases cellular proliferation (Huang et al., 1998), and regulates the invasive capacity of malignant glioma cells by gap junction-independent pathways (Lin et al., 2002). Thus, yet-to-be-determined properties of hemichannels may well be crucial to the survival of Cx-expressing cells. In fact, Cx43 hemichannels have been shown recently to play a role in the anti-apoptotic actions of bisphosphonates (Plotkin et al., 2002). Bisphosphonates are stable analogs of pyrophosphates, which prevent osteocyte and osteoblast apoptosis induced by glucocorticoids. Bisphosphonates are widely used in the treatment of bone diseases and require activation of the extracellular signal-regulated kinases (ERKs). Plotkin et al. (2002) proposed that bisphosphonates induced openings of Cx43 hemichannels, resulting in a conformational change of Cx43, which through a sequence of steps culminated in ERK phosphorylation and cell survival. The difference in the expression of glucocorticoid receptors and thereby in intracellular signal transduction varies among different cell types and may explain why Cx43 failed to protect against dexamethasone-induced death in C6 cells. Nevertheless, it is intriguing that Cx43 hemichannels have been shown to transduce survival signals in response to extracellular cues in another cell type. As for the unaltered sensitivity of astrocytes prepared from Cx43–KO mice, hemichannels assembled by other members of the Cx family may contribute to the retaining of astrocytic resistance, because primary astrocytes in cultures express an abundance of functional Cx43 hemichannels (Hofer and Dermietzel, 1998). Unopposed Cx43 hemichannels are induced to open during chemical ischemia (Contreras et al., 2002). Some of these hemichannel properties may be crucial to the survival of Cx-expressing cells in suspension.
Additional studies are required to establish the mechanism underlying Cx-dependent injury resistance, but it should not be surprising that a protein known to participate in essentially all of the vital cellular processes, including cell-to-cell signaling, proliferation, differentiation, and anaplastic transformation, also has a significant impact on the process of cell death.
Footnotes
This work was supported by American Heart Association Grant 99-50994T (J.H.-C.L.), by National Institute of Neurological Disorders and Stroke/National Institutes of Health Grants NS30007 and NS38073 (M.N.), and by the German Research Association (K.W.). We thank Earl Bueno for excellent technical assistance.
Correspondence should be addressed to Dr. Jane Lin, Department of Pathology, Basic Science Building, New York Medical College, Valhalla, NY 10595. E-mail: jane_lin{at}nymc.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}