

Abstract
Death receptor 3 is a proinflammatory member of the immunomodulatory tumor necrosis factor receptor superfamily, which has been implicated in several inflammatory diseases such as arthritis and inflammatory bowel disease. Intriguingly however, constitutive DR3 expression has been detected in the brains of mice, rats, and humans, although its neurological function remains unknown. By mapping the normal brain expression pattern of DR3, we found that DR3 is expressed specifically by cells of the neuron lineage in a developmentally regulated and region-specific pattern. Behavioral studies on DR3-deficient (DR3ko) mice showed that constitutive neuronal DR3 expression was required for stable motor control function in the aging adult. DR3ko mice progressively developed behavioral defects characterized by altered gait, dyskinesia, and hyperactivity, which were associated with elevated dopamine and lower serotonin levels in the striatum. Importantly, retrograde tracing showed that absence of DR3 expression led to the loss of corticostriatal innervation without significant neuronal loss in aged DR3ko mice. These studies indicate that DR3 plays a key nonredundant role in the retention of normal motor control function during aging in mice and implicate DR3 in progressive neurological disease.
Introduction
The process of aging in the brain is complex and characterized by progressive alterations to neural signaling pathways affecting behavior (O'Sullivan et al., 2001; Andrews-Hanna et al., 2007). Age-related declines in motor control function appear to reflect environmental and genetic components altering neuron lifespan and function (Mattson et al., 2002; Heuninckx et al., 2005; Yankner et al., 2008). Continued motor control function and spatial awareness are critically reliant upon neuronal signaling between the cortex, which translates sensory input, and the striatum (Goldman-Rakic, 1987; Rakic, 1988), which mediates motor and cognitive processes (Goldman-Rakic, 1987; Dubé et al., 1988; Rakic, 1988; Albin et al., 1989; Smith et al., 1994; Groenewegen, 2003; Grahn et al., 2008). The neurotransmitter dopamine plays a primary role in the conduction of all motor control signaling within the striatum (Di Matteo et al., 2008; Palmiter, 2008; Yao et al., 2008). The processes that govern the homeostatic maintenance of these neural pathways following their formation in the aging adult are poorly understood.
Death receptor 3 (DR3, Wsl-1, Apo3, LARD, TRAMP, TNFRSF25, TR3) (Chinnaiyan et al., 1996; Kitson et al., 1996; Marsters et al., 1996; Bodmer et al., 1997; Screaton et al., 1997) is a member of the tumor necrosis factor receptor superfamily (TNFRSF), a group of structurally related immunomodulatory cytokine receptors capable of triggering NF-κB induction and/or caspase activation (Ashkenazi and Dixit, 1998; Mackay and Kalled, 2002). DR3 along with its ligand TNF-like factor 1A (TL1A) has been described as proinflammatory in several murine disease models (Al-Lamki et al., 2003; Bamias et al., 2006; Bull et al., 2008; Fang et al., 2008; Meylan et al., 2008; Pappu et al., 2008; Takedatsu et al., 2008) and in chronic human diseases such as inflammatory bowel disease and rheumatoid arthritis (Osawa et al., 2004; Papadakis et al., 2005; Bamias et al., 2006; Borysenko et al., 2006; Cassatella et al., 2007). In the brain, increased DR3 expression has been reported in ischemic rats (Harrison et al., 2000) and human Alzheimer' disease (Newman et al., 2000), suggesting this receptor contributes to neuropathology. Intriguingly however, endogenous DR3 expression is also observed in the brain of normal mice, rats, and humans (Marsters et al., 1996; Wang et al., 2001b; O'Keeffe et al., 2008). Furthermore, such basal expression may be neurologically important as some TNFRSF members appear to facilitate normal neuronal differentiation and survival (Kojima et al., 2000; Nieoullon and Coquerel, 2003; Graybiel, 2005; Ishiguro et al., 2007; O'Keeffe et al., 2008). Collectively, these observations suggest basal DR3 brain expression may play an important role in normal neuronal function.
In this study, we addressed the physiological role of DR3 in the adult brain by examining the behavior of DR3ko mice during aging. We report that adult DR3ko mice spontaneously develop severe behavioral defects characterized by a progressive decline in higher motor control functions with advancing age. Importantly, adult neuronal DR3 expression was found to be critical for continued maintenance of striatal neurotransmitter expression and for stable neuronal contact between the cortex and the striatum.
Materials and Methods
Mice.
DR3ko mice on a C57BL/6 background were previously generated at, and colony-founding animals supplied by, Cancer Research UK (Wang et al., 2001a). All United Kingdom (UK)-based animal experiments were subject to local ethical review and conducted according to personal, project, and institutional licenses under the UK Animals (Scientific Procedures) Act 1986. For experiments performed in Spain, animal-related procedures were in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and approved by the local animal care committee of the Universitat de Barcelona and by the Generalitat (Autonomous Government) of Catalonia.
Histological analysis.
Tissue preparation, tyrosine hydroxylase (TH) and NeuN immunohistochemistry on free-floating sections, and subsequent morphometry were performed as described previously (Kelly et al., 2007). Brain region volume and cell number were based on NeuN/TH staining and determined using the Olympus CAST grid stereology system on a 1:12 series using previously described methods. At high magnification, brain region volume, neuron number, and the density of TH immunostaining were measured on four anatomically matched sections per animal using the ×40 objective (Leica DMRB microscope, Leica Microsystems) and Scion image system.
Immunohistochemical detection and colocalization of NeuN, GFAP, and β-Gal in DR3het and DR3ko mice.
Indirect fluorescent immunocytochemistry was performed using standard protocols with primary antibodies directed against neuronal nuclei (NeuN, PharMingen), β-galactosidase (β-Gal, Millipore Bioscience Research Reagents), and glial fibrillary acidic protein (GFAP, DAKO). Sections were counterstained with Hoechst 33342 (2 μg/ml; Sigma) to label nuclei before visualization by confocal microscopy on an Olympus IX70 microscope linked into an Ultraview system (Perkin and Elmer).
Brain mapping of β-Gal expression in DR3het and DR3ko mice.
Mice were generated by replacement of the DR3 gene with a cassette including an internal ribosome entry site (IRES) and lacZ poly(A), allowing visualization using X-gal staining. Brains from DR3het and DR3ko mice were removed, flash frozen in liquid nitrogen (LN2), coronally sectioned at 10 μm thickness, and stored at −80°C. Whole mouse brains being assayed for β-Gal were removed, fixed in 4% PFA, and then washed extensively in PBS. Sections being assayed for β-Gal were prepared as described above, immersed in X-gal staining solution for 18–24 h at 37°C, counterstained with Nuclear Fast Red (Vector Laboratories), and hard mounted using standard techniques. Images were taken using a Nikon light microscope, Kodak EPY-64T color slide film, or a Nikon-based digital camera imaging system.
Reverse transcriptase-PCR.
DR3 and TL1A expression in the brains of 8- to 12-week-old mice was examined by reverse transcriptase (RT)-PCR. Brains from 8- to 12-week-old mice were microdissected on ice into the hippocampus, basal ganglia, cortex, cerebellum, dentate gyrus, and colliculi. Total RNA was extracted from tissues or cultured cells using RNeasy (Qiagen) and cDNA prepared using Superscript II (Invitrogen) according to the manufacturer's instructions. PCR was performed according to the following cycle preceded with Taq activation for 15 min at 95°C: step 1, 94°C for 45 s, an annealing step (temperature dependent upon primers) for 30 s, and 72°C for 45 s. The reaction was terminated by 10 min at 72°C and then kept at 4°C until required. PCR primers, annealing temperature, and expected product size for the amplification of TNFRSF members and TL1A are shown in supplemental Table 1 (available at www.jneurosci.org as supplemental material). The amount of cDNA in each PCR was assessed by additionally amplifying a fragment of the GAPDH cDNA. Reaction products were resolved on a 1.2% agarose gel and stained with ethidium bromide before UV visualization. Band intensities were measured using Labworks analysis software.
Gait, balance, and open-field analysis.
Gait and balance analysis was performed using footprint pattern analysis as previously described in the presence or absence of a 1 cm rod balance beam elevated by 3 cm (Dunnett et al., 1998). Open-field testing was performed as previously described (Torres et al., 2007). Open-field analysis was conducted on scaled drawings of animal movement produced by frame-to-frame examination of captured digital footage. Crossovers were determined by counting the combined number of times mice crossed their own paths during observation per min during a 10 min observation period.
Neurochemistry.
Male DR3het and DR3ko mice 3 and 18 months of age were killed, and their brains were removed and microdissected on ice. Brain tissues were weighed, snap frozen using LN2, and sent for commercial analysis (RenaSci Consultancy) by HPLC to determine the level of 5-hydroxyindoleacetic acid (5-HIAA), serotonin (5-HT), 3,4-dihydroxyphenylacetic acid (DOPAC), dopamine, homovanillic acid (HVA), and noradrenaline (NA).
Retrograde tracing.
Intracerebral injection of the retrograde tracer Fluorogold (FG) (Fluorochrome) was performed in 3- and 18-month-old mice as previously described (Pineda et al., 2005). In brief, animals were anesthetized and placed in a stereotaxic apparatus (Stoelting), and Fluorogold solution (1% in PBS) was injected into the striatum at the following coordinates: A/P +0.5 mm, L −2 mm from bregma; and D/V 2.7 mm from dura; incisor bar set at −3 mm. After 2 d, mice were perfused transcardially with 4% paraformaldehyde in PBS, cryoprotected with 30% sucrose, and frozen in dry-ice-cooled isopentane. Coronal sections (30 μm) were cut on a cryostat and mounted with Mowiol (Calbiochem).
Statistical analysis.
Statistical analyses were made with either Student's t tests or t tests assuming unequal variance if normal distributions could not be assumed. A χ2 test was used to compare frequency of run completion data. Unless stated otherwise, plotted data represent mean ± SEM; p values of <0.05 were considered significant.
Results
DR3 expression in the brain is neuron specific
Analysis of DR3 mRNA transcript levels in mouse (Wang et al., 2001b; O'Keeffe et al., 2008) and rat (Harrison et al., 2000) tissue has previously shown that DR3 is expressed using alternate length transcripts in the brain compared to lymphoid tissue. In the absence of a range of antibodies that are confirmed to recognize all variants of murine DR3 protein in histological sections, we used the β-Gal gene expressed under the control of the murine DR3 promoter (Wang et al., 2001a), to investigate the regional and developmental pattern of putative DR3 protein expression in the brains of mice. DR3 gene expression was first detected neonatally after d5 after birth and was rapidly upregulated by d9 after parturition (Fig. 1). This expression pattern was retained through early to late adulthood (Fig. 2 A–C). To explore the expression pattern of DR3 in adult DR3het and DR3ko mice, matched β-Gal- and H&E-stained coronal brain serial sections (10 μm thickness) were compared, allowing construction of DR3 promoter expression maps. Positionally, β-Gal expression was detected between the piriform cortex throughout the brain of transgenic mice to the caudal mesencephalon, disappearing at the transition to the cerebellum. In mice, the density and pattern of β-Gal staining was region specific: substantial numbers of β-Gal+ cells were present within the hippocampal and cortical regions, but expression was undetectable in the striatum, substantia nigra, and cerebellum (Fig. 2 A–C). In the hippocampus, nearly all granular (dentate gyrus) and pyramidal cells (CA1–CA3 fields) stained densely for β-Gal, whereas coloration between these layers was less frequent. In the cortex, β-Gal+ cells were diffuse but present in all layers (Fig. 2 A,B). The β-Gal+ staining pattern found in the brains of DR3ko mice was largely mirrored in age-matched DR3het mice, although weaker in intensity. Intriguingly, however, a severalfold greater number of β-Gal+ cells were found throughout the cortex of DR3ko than in that of DR3het mice, suggesting an attempt to express the DR3 gene in its absence (Fig. 2 A).

Developmental DR3 expression in the early postnatal murine brain. Shown is DR3 promoter-driven β-Gal expression (X-Gal, blue) in the brains of early postnatal DR3ko mice. Low magnification is shown in left-hand panels; high magnification in the right-hand panels at postnatal days 1 to 9 (P1 to P9) as indicated. Scale bars: 500 μm for left-hand panels; 100 μm for right-hand panels. The dentate gyrus is shown in each case with images representative of 2 animals per age.

DR3 and TL1A expression in the adult brain. A , B , Representative images of β-Gal staining in adult DR3ko and DR3het mice in different areas of the brain. Number of β-Gal-stained cells increased in the cortex of DR3ko mice, particularly in prefrontal areas such as the anterior olfactory nuclei ( Ai , Aii ) and generally across other areas such as the primary motor cortex ( Biv , Bv ), but remained high and unchanged in the dentate gyrus ( Bvi , Bvii ). Comparative expression is shown for the prefrontal ( Aiii ) and motor cortex ( Bviii ). Areas in which β-Gal expression was detected are shown for DR3ko in yellow and DR3het in blue. Filled areas show high-density β-Gal staining. Areas from which high-magnification sections are shown ( Ai , Aii , Biv–Bvii ) are indicated by numbers with red squares for DR3ko and green squares for DR3het animals. C , DR3 expression in DR3het brain as detected using the β-Gal reporter. BG, Basal Ganglia; Co, cortex; Cb, cerebellum; DG, dentate gyrus; Hf, hippocampal formation. D , Expression of DR3 and TL1A mRNA in the indicated areas of the brain as measured by RT-PCR. GAPDH is shown as a loading control. E , Microscopy showing neuron-specific DR3 promoter-driven β-Gal expression in the dentate gyrus using immunofluoromicroscopy. Signals from β-Gal overlap with NeuN ( Ei–Eiii ), but not GFAP ( Eiv–Evi ).
To confirm the basal regional pattern of DR3 expression in the brain revealed by β-Gal, an adult DR3wt brain was microdissected into regions and DR3 mRNA expression assessed by RT-PCR. DR3 transcripts were expressed at high levels in the cortex, hippocampus, and dentate gyrus but remained undetectable in the cerebellum of adult DR3wt mice, consistent with DR3 localization visualized by β-Gal expression studies (Fig. 2 D; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Some message was obtained from the DR3wt basal ganglia, but β-Gal staining was not observed in this region in DR3het and DR3ko mice. As expected, no expression of DR3 mRNA transcript was detected in DR3ko mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We next used immunohistochemical studies to define the lineage of cells within the brain. In DR3het mice, analysis revealed that β-Gal expression colocalized almost exclusively with the neuron-specific marker, NeuN, and not with the astrocyte and glial cell marker, GFAP (Fig. 2 E). These results indicate that DR3 expression is neuron-lineage specific, developmentally regulated, and regionally defined in the adult mouse brain.
We next sought to determine the level and pattern of TL1A mRNA expression, the only substantiated ligand for DR3 (Kaptein et al., 2000; Migone et al., 2002; Bossen et al., 2006), in the brains of adult mice. Interestingly, expression of transcripts for TL1A mirrored that of DR3 with the highest expression found in the cortex, and the lowest expression detected in the cerebellum (Fig. 2 D). More moderate TL1A mRNA expression was found in the hippocampus, colliculi, basal ganglia, and dentate gyrus (Fig. 2 D). The survival, structure, and function of neurons in the brain is known to be critically dependent on glial cells (Nave and Trapp, 2008; Fiacco et al., 2009). Several studies have identified astrocytes as being responsible for intensive production of several TNF family ligands following a range of stimuli in vitro and in vivo (Sawada et al., 1989; Badie et al., 2001; Shin et al., 2002; Yan et al., 2007). We therefore purified astrocytes from the cortex of DR3wt mice and examined whether TL1A mRNA was expressed by these cells ex vivo. Cultured astrocytes expressed low levels of transcript for murine TL1A, which was upregulated rapidly by TNFα, suggesting that these cells may be responsible for constitutive low level provision of TL1A to local neurons within the cortex (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We also examined whether the expression levels of a range of other TNFRSF members (TNFR1, FAS, DR4/6, DR5, and p75NTF) or the DR3 modulator, silencer of death domain (SODD), might be altered in the absence of DR3 expression. Expression of mRNA for these proteins was comparable between DR3het and DR3ko mice by RT-PCR, indicating that loss of neuronal DR3 expression is not compensated for by upregulation of other death domain TNFRSF members (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Collectively, these studies suggest that TL1A is expressed by glial cell populations in the absence of inflammation, while expression levels of other TNF family members in the cortex of DR3ko mice remain unaltered. The presence of constitutive cortical TL1A expression in the absence of inflammation suggests that TL1A/DR3 signaling may play an important role in normal neuronal function or maintenance in the cortex.
Progressive loss of motor control in aging DR3ko mice
To assess the role of constitutive DR3 expression in normal brain function, the behavior of DR3wt, DR3het, and DR3ko mice was monitored and assessed during aging. Initial observations revealed that DR3ko mice developed a progressive behavioral disorder characterized by abnormal gait, rapid head movement, apparent disorientation, dyskinesia, and body tremor that was triggered by environmental change such as handling, none of which were seen in DR3wt or DR3het mice. Occasionally, early aged founder mice were also found to suffer from seizures. Behavioral abnormalities were detectable in a proportion of DR3ko mice as young as 4 months, and the sex-independent disorder was 100% penetrant by 18 months of age (Fig. 3). To quantify this defect, mice were timed traversing a Perspex corridor, and measurements of stride length, width, and distance between hindpaw and forepaw placement (overlap) were made for mice aged 3 (young) and 24 (old) months. While run times between young DR3het and DR3ko and old DR3het mice did not differ, old DR3ko mice took significantly longer to complete their runs (DR3ko 6.8 ± 1.3 s vs DR3wt 3.4 ± 0.7 s; F (1,10) = 5.84, p < 0.04) (Fig. 4 A). An age-dependent difference was also detected in the gait measurements of DR3ko mice, a representation of which is shown in Figure 4 B. Only old DR3ko mice exhibited significantly shortened forepaw stride lengths (DR3ko 5.1 ± 1.1 cm vs DR3wt 6.3 ± 0.5 cm; F (1,10) = 6.17, p < 0.04), broader hindpaw stride width (DR3ko 3.0 ± 0.3 cm vs DR3wt 2.6 ± 0.2 cm; F (1,10) = 9.84, p < 0.02), and decreased overlap relative to matched controls (DR3ko 1.4 ± 0.6 cm vs DR3wt 0.7 ± 0.2 cm; F (1,10) = 8.11, p < 0.02) (Fig. 4 C). We also explored whether the balance of DR3ko mice might also be impaired by defective motor control as found for some models of neurological disease (Fernagut et al., 2004; Truong et al., 2006). Aged DR3het mice all traversed the 60 cm long balance beam on all three runs in an average time of 11.4 ± 0.6 s. In contrast, the DR3ko mice had much greater difficulty completing the task: only two of the eight mice succeeded in crossing the beam within the 120 s observation period on all three runs, three failed on one or two occasions to cross, and three failed on all three occasions (Fig. 5 A). For purposes of analysis, failed runs were allocated a maximum crossing time of 120 s, yielding a group mean latency to cross of 83.0 ± 15.7 s, which was significantly longer than that taken by the DR3het group (F (1,13) = 18.09, p < 0.003) (Fig. 5 B). While loss of footing by well balanced adult DR3het mice was rare, movement by DR3ko mice was marked by frequent imbalance, foot slippage, and stationary behavior (Fig. 5 C).

Time course of development of observable gait disorders in DR3ko mice. Proportion of DR3ko and DR3het mice showing observable gait disorder with age. Numbers under the graph represent numbers of mice per age group assessed.

Defects in gait in aged DR3ko mice. Gait defects in 3-month- or 2-year-old mice were compared using timed footprint and movement analysis. A , Average run time for young and old mice. B , Representative footprints of DR3ko and DR3het mice are shown. C , Footprint analysis showing stride length ( i , ii ) and stride width and overlap ( iii , iv ) of young ( i , iii ) and old ( ii , iv ) mice. *Student's t tests showed significant differences as indicated.

Defects in balance in aged DR3ko mice. Balance was compared in 2-year-old DR3het and DR3ko mice by timing of runs along a raised balance beam within a 60 cm run. A , Completion frequency for DR3het and DR3ko mice. χ2 test showed significant difference as indicated. B , Average run times for DR3het and DR3ko mice, with animals not completing given a maximum time of 120 s. Each point represents the average from 3 runs from an individual animal. *Student's t test assuming unequal variance showed significant differences as indicated. C , Representative footprints of DR3ko and DR3het mice are shown.
We next examined the movement and exploratory behavior of DR3ko mice by comparing the activity of transgenic mice during open-field tests using a graduated 1 m Perspex square. Compared to 24-month-old DR3het controls, similarly aged DR3ko mice exhibited hyperactivity and increased complexity of movement characterized by rapid, abrupt changes in forward direction and path crossing with some animals showing strong circling behavior (supplemental Videos 1–3, available at www.jneurosci.org as supplemental material) (Fig. 6 A). In a 10 min observation period, DR3ko mice traversed an average of 250% more accumulative open space, covering 89 ± 17 m relative to DR3het controls, which traversed 35 ± 5 m (F (1,10) = 8.87, p < 0.02) (Fig. 6 B). The relative complexity of DR3ko mice open-field activity also increased compared to DR3het mice as determined by the frequency mice crossed their own path, with DR3ko mice averaging this readout 286 ± 86 times over 10 min, ∼14 times more frequently than their DR3het counterparts who averaged this 20 ± 6 times (F (1,10) = 9.47, p < 0.02) (Fig. 6 B). This increased locomotor complexity was not due to their hyperactivity as the average distance traveled before a crossover event was significantly shorter in DR3ko mice than in DR3het controls (DR3ko 0.41 ± 0.07 m vs DR3wt 2.5 ± 0.5 m; F (1,10) = 16.83, p < 0.003) (Fig. 6 C). No directional preference was observed in the circling behavior of individual mice during open-field tests regardless of age or sex. Similar results were obtained with DR3ko mice that had been crossed with CD1 (outbred) mice for two generations (data not shown), demonstrating that the dysfunction was not due to a specific genetic background. Collectively, these results confirm that DR3ko mice develop significant motor coordination problems, hyperactivity, and abnormal exploratory behavior in an age-dependent manner.

Hyperkinesia and increased movement complexity in DR3ko mice. A , Representative plots of the movement of digitally tracked mice in a 1 m square open field over 1 min (n = 6–8 per group). S, Start; F, finish. B , Accumulative distance traversed ( i , ii ) and accumulative frequency of path-crossing events ( iii , iv ) displayed by young ( i , iii ) and old ( ii , iv ) DR3ko and DR3het mice during 10 min. C , Average distance traversed before mice crossed their own paths. Each group included 4–8 mice, and data are representative of four experiments. Asterisks in relevant panels indicate significance of Student's t tests. Lines and error bars represent mean ± SEM.
Dysregulated dopamine and serotonin expression in the striatum of aged DR3ko mice
We next determined whether the behavioral abnormalities exhibited by aged DR3ko mice were associated with altered levels of neurotransmitters in the brain. Brains from 3- and 18-month-old mice were microdissected into the cortex, striatum, hippocampus, and cerebellum, and the levels of neurotransmitters assessed by HPLC. Brains from DR3ko mice appeared histologically normal being devoid of apparent cellular infiltrates, lesions/plaques, upregulated inflammatory markers, and apparent inflammation as assessed by immunohistochemistry (unpublished observations). Weights of whole and individually dissected regions of brain were also similar between DR3ko and DR3het animals (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Young DR3ko and DR3wt mice (3 months of age) showed no significant differences in neurotransmitter levels in the brain, but strikingly, we found that dopamine levels within the striatum of 18-month-old DR3ko mice were 60% higher than their DR3het counterparts (DR3ko 15,546 ± 1163 ng/g tissue vs DR3het 9768 ± 2383 ng/g tissue; F (1,9) = 5.32, p < 0.05), while serotonin was fourfold lower (DR3ko 77 ± 21 ng/g tissue vs DR3het 328 ± 81 ng/g tissue; F (1,9) = 10.63, p < 0.01) (Table 1). These changes could not be explained by a failure of dopamine breakdown, as the ratios of dopamine to dopamine metabolites (DOPAC, HVA) were unchanged (Table 1; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Furthermore, it could not be explained by upregulation of TH, the primary enzyme responsible for dopamine production, as TH levels throughout the brain were comparable between DR3ko and DR3wt controls as assessed by immunohistochemistry on coronally matched sections (Fig. 7 A,B). These data demonstrate that despite an absence of overt neurological pathology, aged DR3ko mice have profound differences in striatal neurochemistry.
Concentration of neurochemicals in the DR3ko and DR3het brain

Expression of tyrosine hydroxylase in the brain. A , Immunohistochemistry showing TH expression in DR3ko ( i ) and DR3wt ( ii ) mice. B , Summary data showing no difference in TH expression at all matched levels throughout the striatum between DR3ko and DR3wt mice.
Corticostriatal innervation is DR3 dependent in aging DR3ko mice
Current literature suggests that DR3 can control cell fate by initiating either proliferative or apoptotic signaling (Chinnaiyan et al., 1996; Kitson et al., 1996; Marsters et al., 1996; Migone et al., 2002). We therefore examined whether neuronal lifespan might be altered in the absence of DR3 expression in vivo by examining neuron numbers in aged mice. The number of NeuN+ cells present in aligned sections from the brains of aged DR3het and DR3ko mice showed no significant difference following two-dimensional histological topographic analysis (Fig. 8 A). The area and thickness of individual layers or regions of the brain as calculated by computer topography was also found to be similar between DR3het mice and their controls (supplemental Table 2, available at www.jneurosci.org as supplemental material). No difference was also found in the number of apoptotic neurons in the brains of adult DR3ko mice, compared to controls, which were barely detectable (unpublished observations) in agreement with others (Yang et al., 2008). Together, these data suggest that DR3 plays no overt role in the normal determination of neuron lifespan in adult mice in vivo.
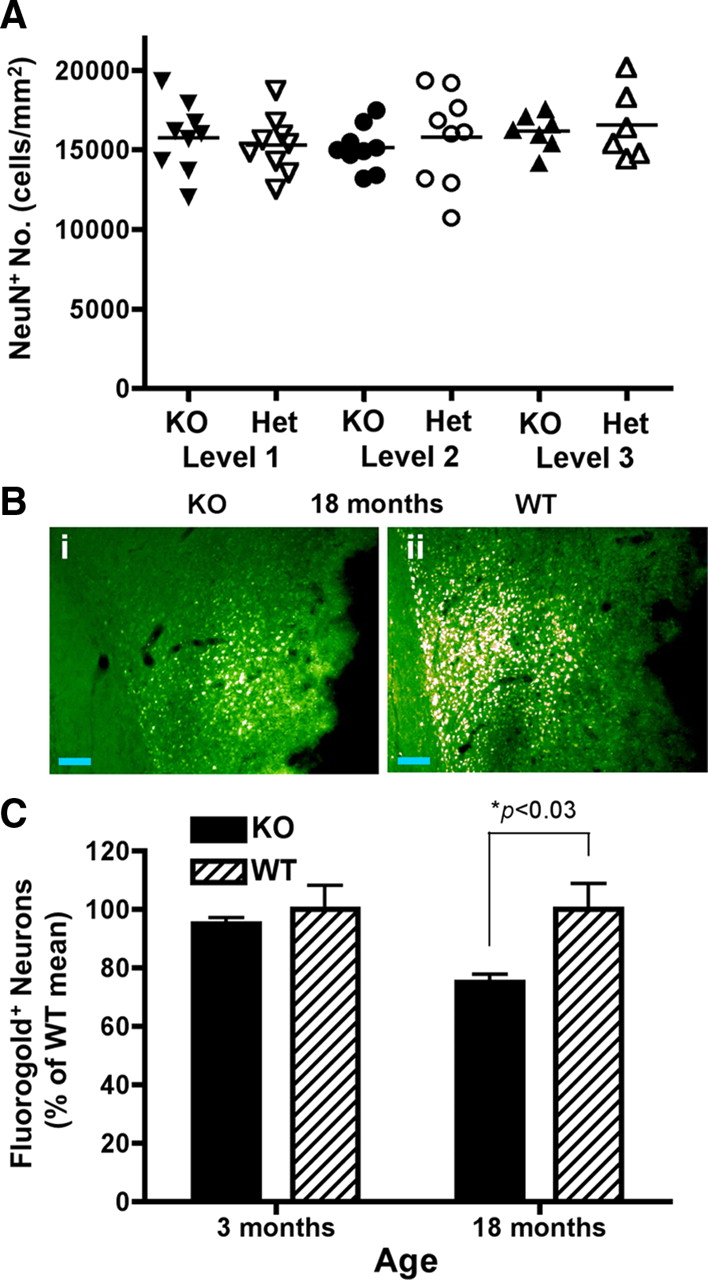
Alterations in corticostriatal innervation in the absence of changes of neuronal number in DR3ko mice. A , Stereological counting of NeuN+ cells showed no difference between DR3ko and DR3het mice at 4 different matched levels in the brain. Data are mean ± SEM from n = 9 mice. B , C , Retrograde tracing of innervation into the striatum. B , Representative images from the cortex of 18-month-old DR3ko ( i ) and DR3wt ( ii ) mice. Scale bars, 100 μm. C , Young (3 months of age) DR3ko and DR3wt mice showed equivalent corticostriatal innervation, while old (18 months of age) DR3ko mice showed a reduction in corticostriatal innervation compared to age-matched DR3wt mice. Data are percentage of the age-matched DR3wt mean ± SEM from n = 4 mice. Statistical significance of *p < 0.03 using a Student's t test.
Given that aged DR3ko mice had normal neuron numbers and DR3 was not expressed in the striatum, we next hypothesized that corticostriatal innervation might be altered by the loss of neuronal DR3 expression, as has been recently observed for another TNFRSF member, GITR (O'Keeffe et al., 2008). This was directly assessed by injection of the retrograde axonal tracer FG into the striatum of mice. The cortex of 18-month-old DR3ko mice had significantly lower numbers of FG-labeled pyramidal cells than similarly treated control animals (Fig. 8 B,C). Overall, >25% of corticostriatal contact had been lost in aging DR3-deficient mice in the absence of continued cortical DR3 expression by neurons as indicated by retrograde tracing (DR3ko 74.7 ± 2.7% vs DR3wt 100 ± 8.5% of normal cortical density; F (1,6) = 8.25, p < 0.03). No significant differences were observed between young (3 months old) DR3wt (100 ± 8%) and DR3ko (95 ± 2%) mice (Fig. 8 C). These results demonstrate that continued neuronal DR3 expression is important for the normal, long-term maintenance of neuronal innervation between the cortex and striatum in adult mice in the absence of appreciable neuron loss. It is less likely that there is a developmental deficit, since there were no differences in innervation density between 3-month-old animals.
Discussion
In this study, we demonstrate for the first time that DR3 is critically required for maintaining neural connectivity between the cortex and striatum during aging, which is necessary for normal motor control function. DR3ko mice develop a progressive locomotor disorder defined by deficits in coordinated movement and gait, which emerges during early adulthood, appears sex independent, and is 100% penetrant. The behavioral phenotype of DR3ko mice resembles a milder form of that observed in jerker mice, which carry a recessive point mutation that disrupts the expression of the actin-bundling protein espin in hair cell stereocilia (Zheng et al., 2000). Because espin lies next to DR3 in the murine genome, we studied espin protein expression by Western blotting and found no difference between DR3wt and DR3ko kidney, a tissue in which small espin is found in brush border microvilli (Bartles et al., 1998) (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). This suggests that espin expression is unaffected in DR3ko mice and that the phenotype of the mice is DR3 specific. Importantly, advancing loss of motor control in DR3ko mice occurred through a relatively passive process without the induction of apparent inflammation, alteration to other TNFRSF brain expression levels or radical changes in neuron number. Thus, we propose that low endogenous TL1A stimulation of DR3+ cortical neurons stimulates the maintenance of interneuronal contact with adjoining dopaminergic or serotonergic striatal neurons. Supporting this, we observed in vitro expression of TL1A mRNA from cultured astrocytes isolated from the cortex, the primary site of TL1A transcript expression in vivo. How might TL1A promote DR3-dependent innervation? One possibility is that TL1A may contribute directly to axon growth or dendrite formation in aging neurons such as occurs following FasL treatment of embryonic neurons (Zuliani et al., 2006) and GITRL treatment of neonatal sympathetic neurons (O'Keeffe et al., 2008). Alternatively, TL1A may increase neuron excitability or alter ion channel signaling as has been shown for TNFα (Diem et al., 2001; Ogoshi et al., 2005; Ozaktay et al., 2006; Czeschik et al., 2008). In such cases, more frequent neuronal stimulation would be predicted to drive new synapse/dendrite formation and stabilization (De Roo et al., 2008). Importantly, our results suggests that the maintenance of adult neuronal corticostriatal projections formed earlier during development and critical for motor control is dependent on the continued reception of low level DR3 signaling by neurons involved in these contacts.
The expression of transcripts for TL1A throughout many parts of the brain in a region-specific manner is also a novel finding. Before this study, TL1A has been reported to be found in serum (Bamias et al., 2008), human umbilical vein endothelial cells (Yang et al., 2004), and myeloid and T-cells (Bamias et al., 2003; Cassatella et al., 2007; Prehn et al., 2007; Meylan et al., 2008). TL1A has been reported to have roles in mucosal immunity (Papadakis et al., 2005), inflammatory bowel disease (Bamias et al., 2006; Takedatsu et al., 2008), inflammatory arthritis (Borysenko et al., 2006; Cassatella et al., 2007; Bull et al., 2008), renal inflammation (Al-Lamki et al., 2008), asthma (Fang et al., 2008; Meylan et al., 2008), and autoimmune encephalitis (Meylan et al., 2008; Pappu et al., 2008). Since the brains of DR3-deficient animals have normal cell numbers and appear devoid of any inflammation or apparent neuropathology, this suggests that cerebral mechanisms exist to dampen neuronal DR3 responsiveness, which is likely to be very threshold or age sensitive. This might be expected as TL1A seems to be constitutively produced by cells within the brain in regions where DR3+ neurons cohabit.
Progressive behavioral defects in DR3ko mice paralleled the differential expression of striatal neurotransmitters with increased dopamine and a corresponding serotonin reduction. Dopamine signaling appears essential for motivated behavior as shown by severe defects in feeding, motor control, and reward-based learning apparent in Dopamineko mice (Zhou and Palmiter, 1995; Palmiter, 2008). Serotonin receptor family-deficient mice also display altered locomotion and behavior (Lesch et al., 2003). Moreover, serotonin and dopamine imbalances are prominently associated with human neurological disorders ranging from Parkinson's disease to schizophrenia and circling in mice (Nieoullon and Coquerel, 2003; Ishiguro et al., 2007; Di Matteo et al., 2008). The biosynthesis of dopamine occurs in a two stage process in which l-tyrosine is first converted to l-DOPA, by the enzyme TH, and then l-DOPA is converted to dopamine, by the enzyme DOPA decarboxylase (Zhou and Palmiter, 1995). Striatal dopamine levels appear dependent on production by DA neurons projected from the substantia nigra (SN) as changes in striatal dopamine often reflect alteration to SN neuron number (Kramer et al., 2007). In aged DR3ko mice, however, we show that striatal dopamine levels were elevated to 160% in the absence of any change to brain neuron numbers or alteration in the level or pattern of TH expression. We also found that the levels of two products generated from the breakdown of dopamine, DOPAC and HVA, were found in normal ratios, suggesting that a failure of striatal dopamine metabolism was not responsible for increase levels of this neurotransmitter. It has been suggested that continued dopamine expression is important for the correct maintenance of corticostriatal synapses, as unilateral destruction of dopaminergic striatal input leads to innervation failure (Arbuthnott et al., 2000). However, whether a specific loss of corticostriatal innervation can induce a corresponding upregulation of striatal dopamine remains unclear. Alternatively, neurotransmitter dysregulation in DR3ko mice may reflect the absence of direct DR3-dependent alteration of neurotransmitter production or subsequent synaptic sensitivity as triggered by TNFα (Schafers and Sorkin, 2008). Both models are consistent with the hypothesis that behavioral defects in dopamine-dependent disorders may reflect corticostriatal pathway dysfunction, as found in DR3ko mice, rather than just changes to cortical activity (Costa et al., 2006).
Striatal serotonin levels were also reduced by 73% in adult DR3ko mice. There is evidence to suggest that excessive dopamine production can inhibit serotonin production by serotonergic neurons in the striatum in vivo leading to behavioral defects. Many Parkinson's disease patients receiving long-term l-DOPA treatment suffer motor control loss associated with dopamine dysregulation (Cenci and Lindgren, 2007). Similarly, dopamine uptake, production, storage, and release can supplant and deplete serotonin in striatal serotonin neurons following chronic l-DOPA treatment in several models (Kannari et al., 2006; Yamada et al., 2007; Carta et al., 2008). We therefore suggest that excessive striatal dopamine production in DR3ko mice is downregulating serotonin expression and that neuronal DR3 signaling is likely to be a major inhibitory dampener suppressing dopamine induction in vivo.
The role of basal TNFRSF member brain expression and function is an emerging field with few published studies. In vitro, select TNFSRF members have been shown to promote nerve growth and function (Shao et al., 2005; Czeschik et al., 2008; Hayashi et al., 2008; Hou et al., 2008; O'Keeffe et al., 2008). Constitutive brain expression of some TNFRSF receptors has also been reported (Yan and Johnson, 1988; Pan et al., 1998; Eby et al., 2000; Kojima et al., 2000; Tan et al., 2002; Pispa et al., 2003; Shao et al., 2005; Zuliani et al., 2006; Hamill et al., 2007; Catts et al., 2008; Harry et al., 2008; Hou et al., 2008; O'Keeffe et al., 2008), but the neurological function of most members remains unknown. The extensive behavioral and neurochemical defects exhibited by DR3ko mice are unique among TNFRSFko mice. For example, TNFR1ko mice have normal striatal dopamine/metabolite levels (Leng et al., 2005) and locomotor activity, exhibiting standard behavioral responses in several tests (e.g., open field and Porsholt swim) (Simen et al., 2006; Quintana et al., 2007; Baracchi and Opp, 2008). Why DR3ko mice, among TNFRSF-deficient animals, uniquely develop severe and progressive neurological defects requires further study but probably reflects receptor differences in function and endogenous age-related expression. In this respect, we first show that continued neuronal DR3 signaling functions by uniquely promoting region-specific (corticostriatal) brain innervation. Second and regardless of the mechanism, basal neuronal DR3 appears to functionally regulate the expression of two key neurotransmitters required for movement and behavior, dopamine and serotonin. Thirdly, we note that stable postnatal neuronal DR3 expression appears more intense and widespread in the cortex and hippocampus than most TNFRSF members in brain regions with motor control pathway function. No other TNFRSF member has been similarly reported to functionally promote the stabilization of preexisting adult neural pathways and regulate striatal neurotransmitter expression under normal conditions in vivo. TNFα, though, has been shown to act as a gliotransmitter, altering synaptic transmission (Stellwagen et al., 2005) and controlling synaptic strength (Beattie et al., 2002) and scaling (Stellwagen and Malenka, 2006), while other proinflammatory cytokines such as IL-1 can influence synaptic plasticity (Ross et al., 2003). The interactions between TL1A and these cytokines in the brain are poorly understood and remain an important area of further investigation. Our data are consistent with the suggestion that the key neurological function of individual TNFRSF receptors is likely to be developmentally, functionally, and regionally compartmentalized within the brain.
In this report, we identify neuronal DR3 signaling as being mandatory for normal striatal neurotransmitter expression and maintenance of corticostriatal neural pathways, which are essential for normal motor control function in the aging adult. How DR3 signaling provides pathway stabilization cues to these neural networks remains an important issue to be resolved; however, the observation that aging DR3ko mice acquire defective neurotransmitter expression and progressive motor control loss provides a unique insight into this proinflammatory receptor. Importantly, this study demonstrates that DR3 plays a key nonredundant role in behavior and normal adult brain homeostasis, further expanding upon the essential roles that TNFSFR members play within the mammalian body. As such, we identify DR3 as a primary molecular target for future neuromodulatory and neurodegenerative disease research.
Footnotes
-
This work was supported by Medical Research Council Career Establishment (G0300180) and Collaboration (G0500617) Grants awarded to E.C.Y.W. M.I.R. was supported by a Biotechnology and Biological Sciences Research Council CASE studentship (C/05060) in collaboration with Glaxo Smith Kline (GSK). A.M.T. was supported by a Wellcome Trust programme grant (064232). We thank Peter Maycox and Isabel Benzel at GSK for helpful discussion and support.
-
The authors declare no competing financial interests.
- Correspondence should be addressed to Eddie C. Y. Wang at the above address. wangec{at}cf.ac.uk





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}