

Abstract
EAAC1 is a neuronal glutamate and cysteine transporter. EAAC1 uptake of cysteine provides substrate for neuronal glutathione synthesis, which plays a key role in both antioxidant defenses and intracellular zinc binding. Here we evaluated the role of EAAC1 in neuronal resistance to ischemia. EAAC1−/− mice subjected to transient cerebral ischemia exhibited twice as much hippocampal neuronal death as wild-type mice and a corresponding increase in microglial activation. EAAC1−/− mice also had elevated vesicular and cytosolic zinc concentrations in hippocampal CA1 neurons and an increased zinc translocation to postsynaptic neurons after ischemia. Treatment of the EAAC1−/− mice with N-acetyl cysteine restored neuronal glutathione concentrations and normalized basal zinc levels in the EAAC1−/− mice. Treatment of the EAAC1−/− mice with either N-acetyl cysteine or with zinc chelators reduced ischemia-induced zinc translocation, superoxide production, and neuron death. These findings suggest that cysteine uptake by EAAC1 is important for zinc homeostasis and neuronal antioxidant function under ischemic conditions.
Introduction
Excitatory amino acid carrier type 1 (EAAC1) (also termed EAAT3 and SLC1A1) was originally described as a neuronal high-affinity glutamate transporter (Kanai and Hediger, 1992). However, several studies have shown that EAAC1 has a negligible effect on glutamate clearance from brain extracellular space and that this function is performed primarily by the astrocyte glutamate transporters GLT1 and GLAST (glutamate–aspartate transporter) (Tanaka et al., 1997; Watase et al., 1998). Recent studies indicate a role for EAAC1 in regulating synaptic glutamate uptake (Scimemi et al., 2009); however, EAAC1 also functions as a high-affinity, concentrative cysteine transporter (Zerangue and Kavanaugh, 1996; Chen and Swanson, 2003; Himi et al., 2003; Watabe et al., 2008), and it is diffusely distributed over nonsynaptic neuronal membranes (Coco et al., 1997; Shashidharan et al., 1997). EAAC1 uptake of cysteine into neurons provides cysteine substrate for the synthesis of glutathione, the major thiol antioxidant (Aoyama et al., 2008; Li and Maret, 2009).
Cysteine and glutathione (GSH) also influence the cellular mobility of zinc ions. Zinc ions bind to intracellular thiols and undergo fast ligand exchange from these binding sites. These thiol binding sites are in turn influenced by the cellular redox state, such that changes in thiol oxidation produce changes in zinc buffering capacity (Maret, 2009). When thiol oxidation reduces zinc buffering, resulting increases in intracellular zinc concentrations can lead to cell injury (Maret, 2009). In neurons, reduced cysteine and GSH levels have been shown to increase free zinc levels in the cytoplasm and presynaptic terminals (Ryu et al., 2002; Cho et al., 2003). Release of this presynaptic zinc can in turn potentiate oxidative damage in postsynaptic neurons, because postsynaptic neuronal zinc accumulation promotes neuronal oxidative injury and death (Frederickson et al., 1989; Koh et al., 1996; Sensi et al., 1999; Suh et al., 2000, 2004).
In view of these associations, we evaluated the role of EAAC1 in neuronal zinc homeostasis and neuronal responses ischemia–reperfusion. We addressed four questions. Are free zinc levels elevated in EAAC1−/− neurons? Is zinc translocation increased in EAAC1−/− mice after ischemia? Is ischemia-induced reactive oxygen species production and neuronal death increased by EAAC1−/− deficiency? Can zinc chelation or N-acetylcysteine (NAC), an exogenous source of cysteine, negate the effects of EAAC1 gene deletion in neuronal response to ischemia–reperfusion?
Materials and Methods
Mouse colonies.
EAAC1−/− mice were descendents of the strain established by Peghini et al. (1997), in which exon 1 is disrupted by a neomycin resistance (NEO) cassette. The mice were obtained from Miltenyi Biotec GmbH and subsequently outbred to wild-type CD-1 mice for more than 10 generations. A wild-type CD-1 colony was maintained using the wild-type offspring from the latter outcrosses. Wild-type breeding stock and EAAC1−/− mice were intercrossed at least once every eight generations to prevent genetic drift, in accordance with the Banbury Conference recommendations (Banbury Conference, 1997). Genotyping was accomplished with a multiplex PCR reaction using three primers to generate up to two product bands, one for the wild-type allele and one for the disrupted EAAC1 allele. Both PCR products shared a forward primer that binds near the start of exon 1 (5′-ACGAGCTCGGGATGTGACT-3′). The wild-type band (63 bp) was a result of binding the EAAC1 reverse primer to the uninterrupted EAAC1 exon 1 sequence (5′-CACGGTGGAGAGCAGCAG-3′), whereas the disrupted EAAC1 band (1050 bp) resulted from binding the reverse primer for the neomycin resistance cassette that interrupts exon 1 in the EAAC1−/− mice (5′-GCTCTTCGTCCAGATCATCC-3′).
Transient ischemia.
Wild-type or EAAC1−/− mice (3 to 5 months old; weighing 30–50 g) were anesthetized with 2% isoflurane in a 75:25 mixture of nitrous oxide and oxygen. Both carotid arteries were exposed through a midline neck skin incision, and the thymus tissue was separated by scissors. The common carotid arteries were encircled with a 4/O silk suture before the start of the occlusion. Small aneurismal clips were applied to both common carotid arteries for 30 min of occlusion (Suh et al., 2008a). Core temperature was kept at 36.5–37.5°C with a homoeothermic blanket control unit (Harvard Apparatus). The EEG was monitored to reduced variability in the ischemic insult, and the time period from an isoelectric EEG (iso EEG) to artery unclipping is referred to as the ischemic period. Onset of iso EEG was defined as the point of the last cortical burst, and termination of the isoelectric period was marked when three spikes appeared within a 60 s interval (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The EEG was monitored with monopolar needle electrodes placed in the cortical surface (BIOPAC System) through biparietal burr holes placed 0.5 mm caudal from bregma and 2.0 mm lateral from the midline. A reference needle was placed in neck muscle. Animals that did not show an iso EEG were excluded from the study. At the end of the 30 min ischemic period, the aneurismal clips were removed and the common carotid arteries were inspected for normal recovery of blood flow. Sham-operated animals received the same neck skin incision and thymus separation but carotid occlusion was not performed. Anesthetics were discontinued after skin closure. When mice showed spontaneous respiration, they were returned to a recovery chamber maintained at 37°C.
Zinc chelators.
To determine the relative contributions of extracellular and intracellular pools of Zn2+ to ischemia-induced neuronal cell death, we used the membrane-permeable chelator clioquinol (5-chloro-7-iodo-8-hydroxyquinoline) (CQ) (Cherny et al., 2001; Adlard et al., 2008) and the cell-impermeable chelator calcium EDTA (CaEDTA) (Koh et al., 1996; Suh et al., 2000, 2004). CQ was dissolved in 100% dimethylsulfoxide (DMSO) and diluted with normal saline to achieve a final concentration of 10% DMSO. CQ (30 mg/kg; Sigma) was administered into the peritoneal space immediately after transient cerebral ischemia and again every 24 h for 3 d (Nitzan et al., 2003; Suh et al., 2009). CaEDTA was dissolved with 0.9% normal saline. Two microliters of CaEDTA (100 mm; Sigma) was slowly injected into the lateral ventricle immediately after transient cerebral ischemia (Koh et al., 1996; Suh et al., 2000).
Neuron death.
Neuronal death after transient cerebral ischemia was evaluated after a 3 d survival period. Mice brains were perfused with 4% formaldehyde (FA) after 0.9% saline perfusion. After overnight postfixation, brains were submerged in 20% sucrose for cryoprotection until they sank to the bottom. Five coronal sections were collected from each animal, spaced 80 μm apart and spanning the hippocampus. Brain sections were stained by the Fluoro-Jade B method (Histo-Chem) (Schmued and Hopkins, 2000). A blinded observer counted the total number of Fluoro-Jade B-positive neurons in each structure of interest, in both hemispheres (Suh et al., 2003). Data from each animal were expressed as the mean number of degenerating neurons per structure of interest.
Measurement of superoxide production.
For the detection of superoxide in the brain, dihydroethidium (dHEt) (Invitrogen) was prepared as a 1 mg/ml solution in 1% DMSO and administered 1 mg/kg into intraperitoneal space at the onset of ischemia. Mice were killed 3 h after termination of ischemia and perfusion fixed with 4% FA. Twenty micrometer cryostat sections were prepared and photographed with a confocal fluorescent microscope with excitation at 510–550 nm and emission >580 nm to detect ethidium (Et) fluorescence (Murakami et al., 1998; Suh et al., 2007, 2008b). Five sections were analyzed from each brain, taken at 80 μm intervals to span the hippocampus. Ethidium signal intensity was expressed as the ratio of the mean fluorescence in neuronal perikaria to fluorescence in the stratum radiatum of hippocampal CA1.
Detection of vesicular and cytoplasmic zinc.
Vesicular and intracellular chelatable zinc was visualized using the 4.5 μm N-(6-methoxy-8-quinolyl)-para-toluenesulfonamide (TSQ) method (Frederickson et al., 1987). Mice were killed, and brains were removed without perfusion. The brains were then frozen on powered dry ice and cut into coronal sections. Five evenly spaced sections were collected through the hippocampal region of each brain and dried. The dried sections were immersed in a solution of TSQ (Invitrogen), 140 mm sodium barbital, and 140 mm sodium acetate, pH 10.5–11 for 60 s and then rinsed for 60 s in 150 mm NaCl. TSQ-zinc binding was imaged and photographed with a fluorescence microscope with 360 nm UV light and a 500 nm long-pass filter. The mean fluorescence intensity within the mossy fiber terminal area and hippocampal CA1 pyramidal area was measured and expressed as arbitrary intensity units after subtraction of background fluorescence as measured in the lateral ventricle. Measurements from the five sections were averaged for each n value. Detection of TSQ-positive (TSQ+) neurons in the hippocampal CA1 area was also evaluated in the same manner.
Evaluation of microglia.
To detect microglia activation in the wild-type or in the EAAC1−/− mice, mice were killed at 3 d after ischemia. Brain sections were prepared and immunostained as described previously (Kauppinen et al., 2008). Rat anti-mouse F4/80 antibody (Serotec) was used at a 1:200 dilution and visualized with an Alexa Fluor 488-conjugated chicken anti-rat IgG (Invitrogen). Negative controls were prepared by omitting the primary antibodies. Digital images were acquired with a confocal laser-scanning microscope and processed identically. A grading scale for microglia activation was based on morphology and F4/80 staining (Kauppinen et al., 2008). Images were scored by an investigator who was blinded to the identity of the brain sections.
N-acetylcysteine administration.
NAC was dissolved with 0.9% saline and was injected into the peritoneal space (150 mg/kg; Sigma) once per day for 4 consecutive days. Controls were injected with saline only. Mice subjected to cerebral ischemia underwent ischemia on the day after the last NAC injection.
Reactive thiol determination.
For in situ evaluation of reactive thiols (of which GSH is the dominant species), brain sections were fluorescently stained with a maleimide derivative (Lantz et al., 2001; Aoyama et al., 2006). After incubation under the designated conditions, hippocampal sections were washed with PBS and incubated overnight at 4°C with 10 μm Alexa Fluor 488 C5 maleimide (Invitrogen) in PBS containing 0.2% Triton X-100 and 0.1% bovine serum albumin. The sections were imaged with a confocal microscope, and fluorescent intensity was analyzed using Photoshop.
Studies with acute brain slices.
One-month-old male mice were anesthetized with a mixture of ketamine and xylazine and decapitated. Brains were rapidly removed and placed in ice-cold isotonic cutting solution (Brennan et al., 2006). Coronal sections (350 μm) were prepared on a vibratome (Technical Products International), and slices were subsequently transferred to oxygenated room temperature artificial CSF (ACSF). Cutting and recording solutions were both 300–305 mOsm/L. After warming to 34°C for 1 h, the ACSF was exchanged again, and slices were then held at room temperature. For oxygen glucose deprivation (OGD), half of the slices were transferred to ACSF prepared without glucose and bubbled with mixed gas composed of 95% CO2 and 5% nitrogen for 30 min at room temperature. After 30 min of OGD, slices were transferred back into oxygenated ACSF with glucose in which they were allowed to recover for an additional 30 min. All slices were then placed into 4% FA for 24 h at 4°C. After fixation, the slices were washed and blocked with a buffer containing 10% goat serum and 0.15% Triton X-100 (Hoskison et al., 2007). Slices treated with the control and OGD conditions were then incubated with antibody to 4-hydroxynonenol (1:500; Alpha Diagnostic) and MAP2 (1:500; Millipore Bioscience Research Reagents) or primary antiserum for nitrotyrosine (1:500; Alpha Diagnostic) and MAP2 for 48 h at 4°C. Slices were washed, and antibody binding was visualized using secondary antibodies conjugated to fluorescent indicators. The n values denote the number of independent studies, each using slices from different mice.
Statistical analyses.
All data were expressed as the mean ± SEM. Microglia activation scores were evaluated with the Kruskal–Wallis test, followed by the Dunn's test for multiple group comparisons. All other were compared with ANOVA, followed by the Bonferroni's test for multiple group comparisons.
Results
Increased neuronal death in EAAC1−/− mice after ischemia
To determine whether the ischemia-induced neuronal death was aggravated by EAAC1 gene deletion, wild-type and EAAC1−/− mice were subjected to 30 min of transient cerebral ischemia. Neuronal death was assessed by Fluoro-Jade B staining at 3 d after ischemia. Thirty minutes of ischemia induced substantial neuronal death in the hippocampus. The pattern and the degree of neuronal death in the wild-type mice are similar to that reported previously (Suh et al., 2008a). Hippocampal neuronal death was significantly higher in the EAAC1−/− mice in the CA1, subiculum, dentate gyrus, and hilus (Fig. 1A).
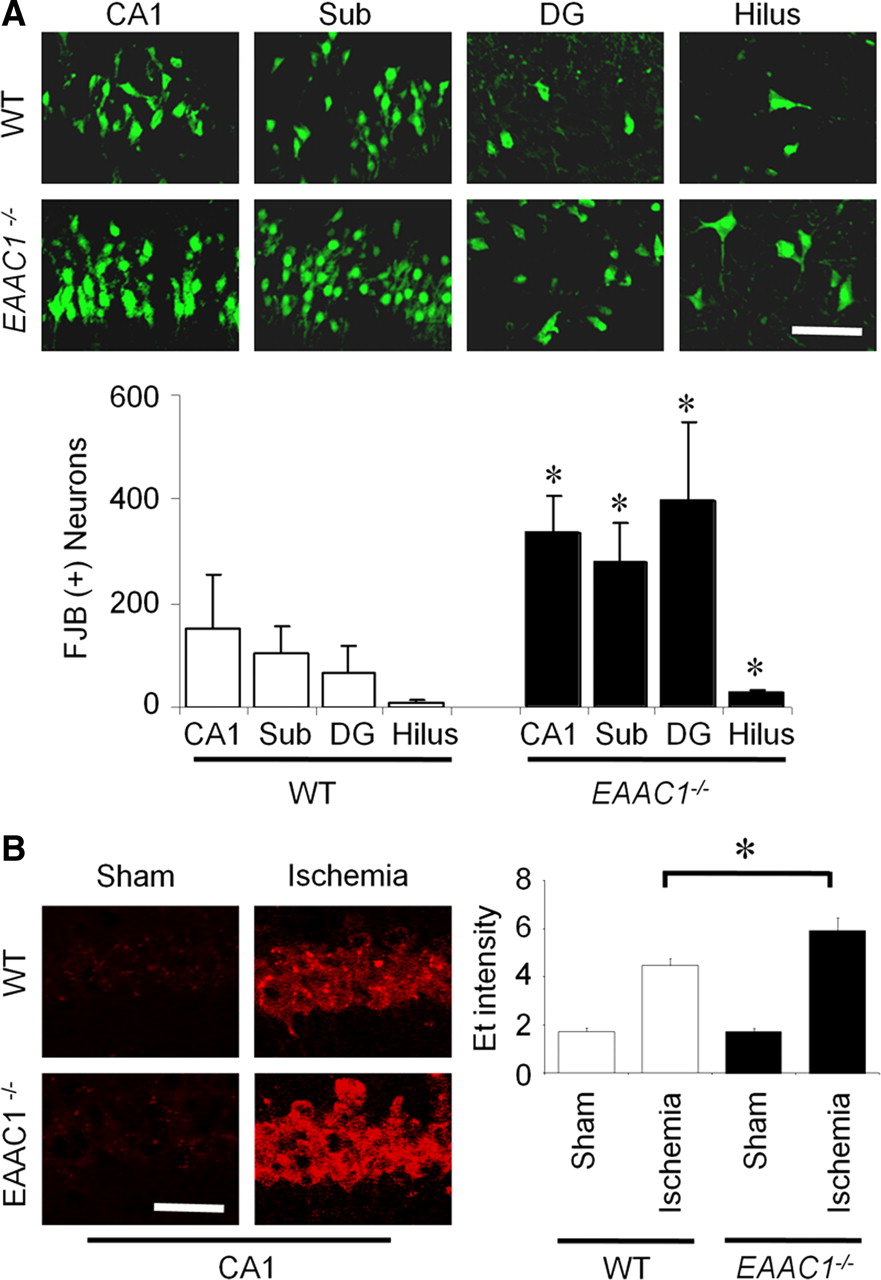
Increased ischemia-induced neuronal death and superoxide production in EAAC1−/− mice. A, EAAC1−/− mice show more neuronal death than wild-type (WT) mice after ischemia. Degenerating neurons are identified by Fluoro-Jade B (FJB) staining (green). Confocal fluorescence images show neuronal death in hippocampal CA1, subiculum (Sub), dentate gyrus (DG), and hilus at 3 d after transient ischemia. Scale bar, 100 μm. Bar graph shows quantified cell counts. Data are mean + SEM; n = 5–6, *p < 0.05. B, Ischemia-induced neuronal superoxide production is increased in EAAC1−/− mice. Superoxide is detected by Et fluorescence (red). Images show Et fluorescence in the CA1 hippocampal neurons 3 h after ischemia–reperfusion. Sham-operated mice received surgery without ischemia. Scale bar, 100 μm. Bar graph shows quantified Et fluorescence. n = 4–5, *p < 0.05.
Increased neuronal superoxide production in EAAC1−/− mice after ischemia
Superoxide production in hippocampal CA1 neurons was evaluated by dHEt method in wild-type and EAAC1−/− mice after transient cerebral ischemia. Ischemia–reperfusion increased Et fluorescent intensity several-fold relative to sham ischemia. The intensity of the Et signal after transient ischemia was significantly higher in EAAC1−/− mice than in wild-type mice (Fig. 1B). Without ischemia (sham), the intensity of Et in the hippocampal CA1 was similar in wild-type and EAAC1−/− mice.
Ischemia-induced microglia activation is increased in EAAC1−/− mice
Ischemia promotes microglia activation, as evidenced by a stereotyped change in morphology from highly ramified to amoeboid shape (Morioka et al., 1991; Stence et al., 2001; Kauppinen et al., 2008). We evaluated the degree of microglia activation 3 d after ischemia in wild-type and EAAC1−/− mice. EAAC1−/− mice showed a significantly higher degree of microglia activation in hippocampus (Fig. 2), corresponding to the increase in neuronal death.

Increased ischemia-induced microglial activation in EAAC1−/− mice. Sections were harvested at 3 d after ischemia and immunostained with F4/80 (green). Changes in microglia morphological and F4/80 expression level occur in the hippocampus (CA1 and hilus) and cortex (Ctx). EAAC1−/− mice show increased microglia activation compared with wild-type (WT) mice. Scale bar, 100 μm. Bar graph shows quantified microglia activation in the CA1. Data are mean + SEM; n = 5, *p < 0.05.
Increased neuronal free zinc and ischemia-induced zinc translocation in EAAC1−/− mice
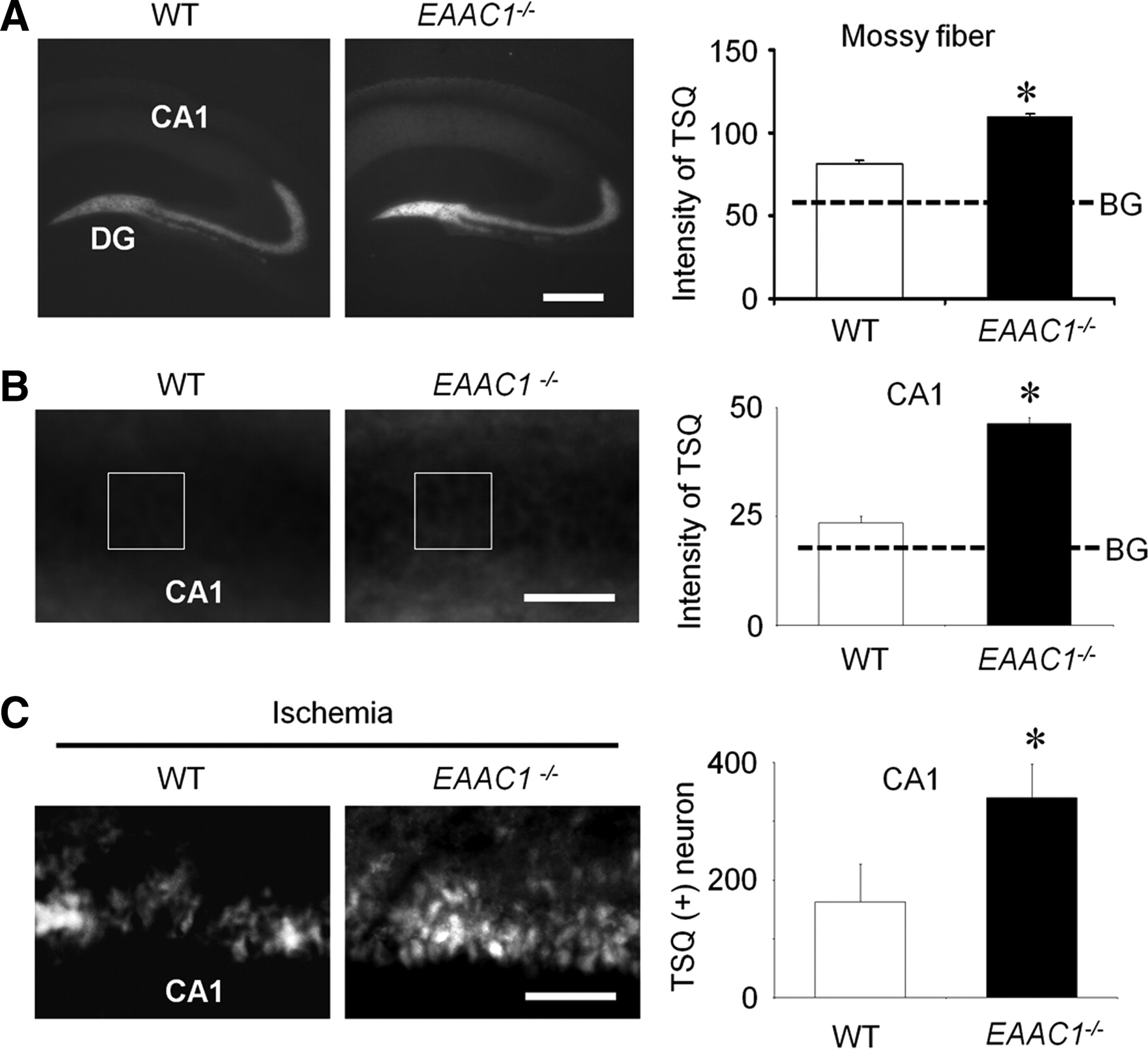
Hippocampal vesicular zinc was measured by TSQ staining. In wild-type mice, intense TSQ fluorescence was detected in the hippocampal hilus mossy fiber area, and a less intense fluorescence was detected in the stratum radiatum of the hippocampal CA1. This same distribution was observed in the EAAC1−/− mice, but intensity of TSQ fluorescence in the hippocampal mossy fiber area was significantly increased (Fig. 3A).

Basal free zinc and ischemia-induced zinc translocation is elevated in EAAC1−/− mice. A, Images show TSQ zinc fluorescence signals from wild-type (WT) and EAAC1−/− mouse hippocampus. EAAC1−/− mice exhibit an increased TSQ signal in the hilus and stratum radiatum. Scale bar, 500 μm. Graph shows measured TSQ intensity, with BG indicating the background signal measured from the lateral ventricle. B, Higher-magnification images of TSQ-stained hippocampus. Scale bar, 100 μm. The CA1 pyramidal layer shows a brighter signal in EAAC1−/− mice than wild-type mice. Box indicates area of TSQ signal measurement. Graph shows quantified TSQ measurements from CA1. C, Higher-magnification images of TSQ-stained hippocampus from mice 3 d after ischemia shows zinc accumulation in the CA1 pyramidal neurons is greater in EAAC1−/− mice. Higher background signal in the low-power image (A) is attributable to increased image exposure time. Scale bar, 100 μm. Bar graph shows the number of TSQ-positive neurons in CA1 of EAAC1−/− and wild-type mice. Data are mean + SEM; n = 5–6, *p < 0.05.
Nonvesicular cytoplasmic zinc levels were also compared in wild-type and EAAC1−/− mice. In the wild-type mice, intense TSQ fluorescence was detected in the hippocampal stratum radiatum and a less intense fluorescence was detected in hippocampal CA1 pyramidal neurons. The intensity of TSQ fluorescence in the pyramidal neurons in the EAAC1−/− mice was again significantly higher than wild-type mice (Fig. 3B), indicating that the hippocampal pyramidal neurons of EAAC1−/− mice contain a higher concentration of chelatable (free) zinc in the synaptic vesicles and cytoplasm of CA1 pyramidal neurons. Ischemia-induced zinc translocation was evaluated by counting TSQ+ neurons in the hippocampal CA1 3 d after ischemia. EAAC1−/− mice showed significantly more TSQ+ neurons than wild-type mice (Fig. 3C).
The zinc chelator CQ reduces ischemia-induced zinc translocation, superoxide production, microglia activation, and neuronal death
The results shown in Figure 3 suggest the possibility that increased zinc accumulation in postsynaptic zinc might contribute to the increased neuron death observed in the EAAC1−/− mice. We therefore tested whether the membrane-permeable zinc chelator CQ would reduce neuronal death in EAAC1−/− mice. CQ has been used previously to reduce the formation of β-amyloid plaques in mouse brain (Cherny et al., 2001; Adlard et al., 2008). We found that CQ substantially decreased ischemia-induced zinc translocation into hippocampal CA1 pyramidal neurons in EAAC1−/− mice after ischemia (Fig. 4A). CQ also decreased ischemia-induced superoxide production, microglial activation, and neuronal death in the EAAC1−/− mice (Fig. 4B–D). The reduction in ischemic neuronal death observed with CQ in EAAC1−/− mice was greater, in absolute numbers, than that observed in wild-type mice (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material); however, this difference is difficult to interpret because the number of neurons killed in EAAC1−/− mice not receiving CQ was also much higher than in wild-type mice.

The zinc chelation reduces ischemia-induced zinc translocation, superoxide production, microglia activation, and neuronal death in EAAC1−/− mice. Brains were analyzed at 3 d after ischemia. A, The zinc chelator CQ reduced the number of CA1 TSQ+. Scale bar, 50 μm. B, CQ treatment reduced superoxide production (Et fluorescence) in the hippocampal CA1 area in EAAC1−/− mice. Scale bar, 50 μm. C, CQ treatment reduced microglia activation in the hippocampal CA1 area in EAAC1−/− mice. Scale bar, 50 μm. D, CQ treatment reduced neuron death (FJB+ neurons) in the hippocampal CA1. Scale bar, 100 μm. Data are mean + SEM; n = 5–8, *p < 0.05.
CQ binds to zinc in both the intracellular and extracellular compartments. Studies were also performed using intracerebroventricular injection of CaEDTA, a zinc chelator that is restricted to the extracellular space (Perrin, 1979; Koh et al., 1996). CaEDTA was found to reduce neuronal death in both wild-type and EAAC1−/− mice (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material), and CaEDTA also decreased ischemia-induced zinc translocation into hippocampal pyramidal neurons (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material).
NAC normalizes thiol content and free zinc levels in EAAC1−/− neurons
Reactive thiol content was lower in EAAC1−/− hippocampal neurons than in wild-type neurons (Fig. 5), as reported previously (Aoyama et al., 2006). NAC provides cysteine to cells, probably by uptake and subsequent deacetylation (De Vries and De Flora, 1993). NAC has been shown previously to normalize free thiol content in EAAC1−/− neurons (Aoyama et al., 2006), and here NAC given for 4 consecutive days was similarly shown to normalize free thiol content (Fig. 5). Importantly, NAC also normalized free zinc levels in EAAC1−/− pyramidal neurons (Fig. 6A) and reduced the zinc TSQ signal observed in postsynaptic EAAC1−/− neurons after ischemia (Fig. 6B). Wild-type mice treated with NAC showed similar but smaller effects on both neuronal free zinc and ischemia-induced zinc accumulation (Fig. 6A,B).

Neuronal GSH deficiency in EAAC1−/− mice is reversed by NAC treatment. Reactive thiol content was evaluated in mouse brain using C5 maleimide fluorescence. A, Reduced fluorescence in the neurons of EAAC1−/− hippocampus sections relative to those in the wild-type (WT) slices. This signal was increased in hippocampal sections from mice treated with the cell-permeable cysteine precursor NAC in both wild-type and EAAC1−/− mice. B, Higher-magnification images represent C5 maleimide-stained CA1 pyramidal neurons. Box indicates area of C5 maleimide fluorescence measurement. Bar graph shows the quantified C5 maleimide intensity. Data are mean + SEM; n = 3 in each group, *p < 0.05.

NAC reduces vesicular zinc levels and ischemia-induced zinc translocation. A, NAC reduces basal (without ischemia) TSQ intensity in the hippocampal mossy fiber area of both wild-type and EAAC1−/− mice. Scale bar, 500 μm. Data are mean + SEM; n = 3–5, *p < 0.05. B, NAC treatment reduces ischemia-induced zinc translocation in hippocampal CA1. Scale bar, 50 μm. Data are mean + SEM.; n = 5–6, *p < 0.05.
NAC reduces ischemia-induced superoxide production and neuronal death
Wild-type and EAAC1−/− mice were treated with NAC or vehicle for 4 d before ischemia, and superoxide production was evaluated by the dihydroethidium method in brains harvested 3 h after ischemia. NAC pretreatment decreased ischemia-induced superoxide in both wild-type and EAAC1−/− mouse hippocampus (Fig. 7A). NAC also reduced both hippocampal neuronal death and microglial activation, evaluated 3 d after ischemia (Figs. 7B, 8). The effect of NAC on neuronal death was numerically greater in EAAC1−/− than wild-type mice (Fig. 7B); however, this difference is difficult to interpret because the number of neurons killed in EAAC1−/− mice not receiving NAC was also higher than in wild-type mice.

NAC reduces ischemia-induced superoxide production and neuronal death in EAAC1−/− mice. A, NAC treatment reduces Et formation in hippocampal CA1 of both wild-type and EAAC1−/− mice, measured 3 h after ischemia. Scale bar, 50 μm. Data are mean + SEM; n = 3–5, *p < 0.05. B, NAC treatment reduces neuron death (FJB+ neurons) in hippocampal CA1 of both wild-type and EAAC1−/− mice, measured 3 d after ischemia. Scale bar, 50 μm. Data are mean + SEM; n = 5–6, *p < 0.05.

NAC reduces ischemia-induced microglia activation. NAC reduces microglia activation in hippocampal CA1 of both wild-type and EAAC1−/− mice. Scale bar, 50 μm. Data are mean + SEM; n = 5–6, *p < 0.05.
NAC prevents superoxide production and oxidative injury in EAAC1−/− brain slice
Given the possibility that EAAC1−/− mice could have systemic metabolic differences or differences in brain vascular anatomy relative to wild-type mice, we also performed ex vivo studies on hippocampal brain slices. Acutely prepared hippocampal slices were subjected to OGD for 30 min, followed by 30 min replacement in normal medium. The lipid and protein oxidation products 4-hydroxynonenal and nitrotyrosine were increased in both wild-type and EAAC1−/− hippocampal neurons (Fig. 9A,B). These increases were greater in the EAAC1−/− slices. Slices prepared from mice of either genotype treated with NAC showed attenuated increases in these markers of oxidative stress (Fig. 9A,B).

NAC prevents OGD-induced nitrotyrosine production and oxidative injury in EAAC1−/− brain slices. A, Confocal fluorescent images show immunostaining for 4-hydroxynoneal (4HNE, green) and MAP2 (red; identifies the CA1 neuronal perikaria and dendrites) in OGD-treated hippocampal slices. 4HNE formation is higher in EAAC1−/− than wild-type slices (#). 4HNE formation was increased by OGD, and this increase was attenuated by NAC in both wild-type or in EAAC1−/− slices. NAC treatment reduced 4HNE formation (*). Scale bar, 100 μm. AFU, Arbitrary fluorescence units. B, Confocal fluorescent images show immunostaining for nitrotyrosine (NT, green) and MAP2 (red) in OGD-treated hippocampal slices. NT formation is higher in EAAC1−/− than wild-type slices (#). NT formation was increased by OGD, and this increase was attenuated by NAC in both wild-type or in EAAC1−/− slices (*). Scale bar, 100 μm. n = 3, *p < 0.05.
Discussion
These results show that EAAC1−/− hippocampal neurons have reduced reactive thiol content and increased fee zinc levels in the cytosol and presynaptic vesicles. EAAC1−/− mice subjected to ischemia–reperfusion showed increased zinc translocation into hippocampal postsynaptic neurons, and this increase was accompanied by an increase in neuronal superoxide production, neuronal death, and microglial activation. These effects were reversed in mice treated with zinc chelators or with the cysteine pro-drug NAC. Together, these results suggest that cysteine is required for normal zinc homeostasis in presynaptic terminals and that cysteine uptake by EAAC1 is important for neuronal responses to oxidative stress in ischemia.
Zinc is concentrated in the presynaptic vesicles of a subset of glutamatergic axon terminals (Beaulieu et al., 1992). These axon terminals are distributed throughout the forebrain and are particularly dense in hippocampus and in cerebral cortex (Danscher et al., 1985; Frederickson, 1989). The vesicular zinc is released into the extracellular space during normal neuronal activity and is massively released during ischemia (Tønder et al., 1990; Koh et al., 1996). Influx of extracellular zinc influx into postsynaptic neurons generates oxidative stress and energy failure (Sensi et al., 1997; Suh et al., 2004, 2008b). In addition, a large fraction of neuronal zinc exists in the form of thiol–zinc complexes, and liberation of zinc from these complexes can also injure neurons (Aizenman et al., 2000; Lee et al., 2000). Here, EAAC1−/− neurons were found to have elevated free zinc in both cytosol and presynaptic vesicles, presumably as a result of the reduced thiol content. The additional finding that both postsynaptic zinc accumulation and ischemic injury are reduced in the EAAC1−/− mice by zinc chelators suggests that the elevated zinc content contributes to the increased ischemic injury observed in these mice; however, we cannot exclude the possibility that the increase in postsynaptic zinc accumulation is not simply a marker of increased neuronal death caused by other factors. Of the zinc chelators used, CQ is membrane permeable and can therefore buffer both intracellular and extracellular free zinc concentrations, whereas CaEDTA is restricted to the extracellular space. The substantial effects of CaEDTA on postsynaptic zinc accumulation and neuronal survival further suggest that zinc release into the extracellular space is a major factor in these events.
Results presented here show a correlation between decreased thiol levels on one hand and increased free zinc and vulnerability to ischemic injury on the other. However, it is difficult to ascertain the relative importance of reduced thiol content and elevated free zinc levels to the increased vulnerability of EAAC1−/− neurons to ischemic injury because there are reciprocal relationships between thiol content, free zinc levels, and oxidative stress (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Oxidation of reactive thiol groups leads to zinc release, and elevated zinc levels promotes oxidative stress (Maret, 2009). Conversely, excess zinc influx depletes neuronal glutathione (Kim et al., 1999), and replenishment of glutathione can attenuate zinc toxicity (Chen and Liao, 2003). Oxidative stress may also influence the rate at which zinc enters cells, because zinc influx through both AMPA receptors and TRMP7 receptors is influenced by their redox state (Sensi et al., 1999; Weiss and Sensi, 2000; Aarts et al., 2003).
The influence of EAAC1−/− deletion on ischemic brain injury has not been reported previously, but a previous study evaluated the effect of antisense DNA knockdown of EAAC1 expression in a rat model of focal ischemia. The antisense approach (Rao et al., 2001) produced an ∼50% reduction in EAAC1 protein expression. Infarct size was increased, but this increase did not reach statistical significance. Given our present findings, it is possible that a significant increase in infarct size might have been achieved if the antisense approach more completely suppressed EAAC1 expression. Alternatively, the greater effect observed in the present study may stem from the very high abundance of zinc in hippocampal pyramidal neurons, which are particularly affected by transient ischemia, or from the more dominant role of oxidative stress in transient ischemia–reperfusion injury.
Although C5-maleimide binds to many thiols, previous studies have shown that the increase in neuronal C5 maleimide binding resulting from NAC administration to EAAC1−/− mice is prevented by coadministration of buthionine sulfoximine, an irreversible inhibitor of GSH synthesis (Aoyama et al., 2006). This suggests that the increased binding is primarily attributable to increased GSH but does not exclude other possible mechanisms by which NAC might influence free zinc levels or oxidant production during ischemia. NAC itself has antioxidant effects (Armstead et al., 1988; Milde, 1989; Hussain et al., 1996), and as a sulfhydryl compound it may also directly bind zinc (Jones, 2008). In addition, it is possible that cysteine derived from NAC may support production of metallothioneine, in addition to GSH, in neurons that have impaired cysteine uptake.
Microglial activation is induced by brain ischemia, and this acute inflammatory response can contribute to ischemic neuronal injury (Barone and Feuerstein, 1999; Kauppinen et al., 2009). Activated microglia release several neurotoxic substances, including superoxide, nitric oxide, and metalloproteinases (Chao et al., 1992; Giulian et al., 1993; Hanisch, 2002; Vilhardt, 2005), and microglia activation is morphologically characterized by change to an amoeboid shape (Morioka et al., 1992; Lund et al., 1994; Kreutzberg, 1996). Several intercellular signals have been found to mediate microglial activation in ischemia, including zinc (Kauppinen et al., 2008). A major role for zinc in this process is indicated by near-complete suppression of microglial activation in mice injected with the zinc chelator CaEDTA after ischemia–reperfusion (Kauppinen et al., 2008). Here, we found that ischemia-induced microglia activation was significantly increased in EAAC1−/− mice. The observation that neurons in EAAC1−/− mice have increased levels of neuronal free zinc suggests the possibility that the increased microglial activation in these mice is attributable to increased zinc release; however, we cannot exclude the alternative possibility that the increase in microglial activation is attributable to increased neuronal death in the EAAC1−/− mice.
Previous studies have shown that ischemic injury is exacerbated by reduced expression of either of the major astrocyte excitatory amino acid transporters GLAST or GLT-1 (Tanaka et al., 1997; Watase et al., 1998; Rao et al., 2001), which together are almost exclusively responsible for extracellular glutamate uptake in brain. The present results show that reduced expression of the neuronal excitatory amino acid transporter EAAC1 likewise exacerbates ischemic injury but by a different mechanism; EAAC1−/− mice exhibit reduced thiol content, impaired neuronal zinc homeostasis, and increased oxidative stress after ischemia–reperfusion as shown in supplemental Figure 3 (available at www.jneurosci.org as supplemental material). These findings are concordant with previous reports of impaired antioxidant capacity in neurons with impaired EAAC1 expression or function and identify a novel role for EAAC1 in neuronal zinc homeostasis.
Footnotes
-
This work was supported by Juvenile Diabetes Research Foundation Grant 2-2006-113 (S.W.S.), the United States Department of Veterans Affairs, and Korea Science and Engineering Foundation Grant 2009-0078399 (S.W.S). We thank Colleen Hefner for expert technical assistance and Dr. Midori Yenari for critical comments.
-
The authors declare no conflict of interest.
- Correspondence should be addressed to Dr. Sang Won Suh, Department of Physiology, Hallym University, College of Medicine, Chuncheon 200-702, Korea. sang.suh{at}ucsf.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}