

Abstract
Mature retinal ganglion cells (RGCs) cannot normally regenerate axons into the injured optic nerve but can do so after lens injury. Astrocyte-derived ciliary neurotrophic factor and leukemia inhibitory factor have been identified as essential key factors mediating this effect. However, the outcome of this regeneration is still limited by inhibitors associated with the CNS myelin and the glial scar. The current study demonstrates that Taxol markedly enhanced neurite extension of mature RGCs and PC12 cells by stabilization of microtubules and desensitized axons toward myelin and chondroitin sulfate proteoglycan (CSPG) inhibition in vitro without reducing RhoA activation. In vivo, the local application of Taxol at the injury site of the optic nerve of rats enabled axons to regenerate beyond the lesion site but did not affect the intrinsic regenerative state of RGCs. Furthermore, Taxol treatment markedly increased lens injury-mediated axon regeneration in vivo, delayed glial scar formation, suppressed CSPG expression, and transiently reduced the infiltration of macrophages at the injury site. Thus, microtubule-stabilizing compounds such as Taxol might be promising candidates as adjuvant drugs in the treatment of CNS injuries particularly when combined with interventions stimulating the intrinsic regenerative state of neurons.
Introduction
In contrast to the peripheral nervous system, neurons of the adult CNS do not normally display axonal regeneration after injury. Failure of axonal regeneration is explained in part by the presence of number of molecules that act as barriers to axon growth. These include inhibitors that are associated with CNS myelin as well as components of the glial scar (Wong et al., 2003; Silver and Miller, 2004; Harel and Strittmatter, 2006; Yiu and He, 2006). Recently, macrophages infiltrating the injured CNS and accumulating at the lesion site have been proposed as an additional significant barrier for axonal growth in vivo (Horn et al., 2008; Busch et al., 2009). Another important reason for the regenerative failure of the CNS is the insufficient intrinsic capacity of mature neurons to regrow axons (Goldberg, 2004; Teng and Tang, 2006; Park et al., 2008; Moore et al., 2009; Smith et al., 2009). For instance, mature retinal ganglion cells (RGCs) only extend a few axons on a growth-permissive substrate in culture or into a peripheral nerve graft in vivo (David and Aguayo, 1981; Bähr et al., 1988; Aguayo et al., 1991; Cui and Harvey, 2000; Fischer et al., 2000; Yin et al., 2003; Müller et al., 2009). However, RGCs can be transformed into an active regenerative state after lens injury (Fischer et al., 2000, 2001; Leon et al., 2000; Lorber et al., 2005; Pernet and Di Polo, 2006). In the regenerative state, RGCs survive axotomy and can extend axons beyond the optic nerve injury site. Ciliary neurotrophic factor (CNTF) and leukemia inhibitory factor (LIF) have been identified as essential key factors mediating both the neuroprotective and axon growth-promoting effects (Müller et al., 2007; Fischer, 2008; Leibinger et al., 2009; Hauk et al., 2010). Regeneration of RGCs that have been stimulated by lens injury in vivo is enhanced further, when inhibitory signaling is compromised, for instance by the expression of a dominant-negative form of the Nogo-receptor or the RhoA inactivator ADP-ribosyl-transferase in RGCs, in which most inhibitory signals converge. However, overcoming inhibitory signaling alone without stimulating the axonal growth state enabled only little regeneration (Fischer et al., 2004a,b). These findings suggest that combinatorial treatments are required to attain extensive regeneration in vivo. Thus, the identification of new compounds that help overcoming the inhibitory signaling or modulate the inhibitory environment may open new avenues to treat CNS injuries.
Paclitaxel (Taxol) is a clinically approved anti-cancer drug. Taxol stabilizes microtubules and as a result interferes with the normal breakdown of microtubules during cell division (Vyas and Kadow, 1995). At low concentrations, Taxol alters microtubule dynamics and promotes polymerization at the plus end (Chuckowree and Vickers, 2003). Taxol can prevent the formation of axon retraction bulbs after spinal cord injury, and, in culture, it can overcome myelin inhibition of neurite outgrowth (Ertürk et al., 2007). However, whether Taxol can be used to enhance axon regeneration after CNS injury in vivo has not yet been shown.
The present study investigates whether microtubules stabilization by Taxol affects axon extension of mature primary neurons in the presence of myelin as well as chondroitin sulfate proteoglycan (CSPGs) and whether a local application of Taxol at the lesion site of the injured CNS alone or in combination with measures that increase the intrinsic growth state of neurons can be used to promote axon regeneration in the CNS. Using the optic nerve as an established model for studying regenerative failure of axons in the CNS, we show that locally applied Taxol promotes axon regeneration in the injured optic nerve, in particular when combined with an additional stimulation of the axonal growth state of RGCs by lens injury. These findings may be useful for the development of novel therapeutic strategies for CNS repair.
Materials and Methods
Optic nerve crush, lens injury, and Taxol treatment of the optic nerve.
Surgical procedures were approved by the local authorities (Regierungspräsidium Tübingen). Adult female Sprague Dawley rats (weighing 200–230 g) were anesthetized by intraperitoneal injections of ketamine (60–80 mg/kg) and xylazine (10–15 mg/kg), and a 1–1.5 cm incision was made in the skin above the right orbit. The optic nerve was surgically exposed under an operating microscope. The dural sheath was longitudinally opened. The nerve was crushed 1 mm behind the eye for 10 s using a jeweler's forceps, avoiding injury to the retinal artery. The vascular integrity of the retina was verified by fundoscopic examination after surgery. Lens injury was induced by a retrolenticular approach, puncturing the lens capsule with the tip of a microcapillary tube. For the application of PBS or Taxol into the vitreous body, we injected 5 μl of either PBS or Taxol (1 μm) into the vitreous body after retrieving the same volume from the anterior chamber. For the application of PBS or Taxol at the lesion site of the optic nerve, we used a rectangular formed piece of Gelfoam with side lengths of ∼2 mm, which was longitudinally opened and completely soaked with a solution of either PBS or different concentrations of Taxol (1, 10, 100, and 1000 μm). These pieces of Gelfoam were then carefully placed around the lesion site of the optic nerve in a way that parts of the proximal optic nerve stump and ∼1 mm of the distal nerve were covered (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
Dissociated retinal cell cultures.
The retinal cell cultures were prepared as described previously (Grozdanov et al., 2010). In brief, tissue culture plates (four-well plates; Nalge Nunc International) were coated with poly-d-lysine (0.1 mg/ml; molecular weight, <300,000 Da; Sigma), rinsed with distilled water, and then air dried. Wells were then coated with laminin (20 μg/ml; Sigma). To prepare retinal cell cultures, rats (weighing 200–230 g) were killed by an overdose of chloral hydrate solution (14%). Retinas were rapidly dissected from the eyecups and incubated at 37°C for 30 min in a digestion solution containing papain (16.4 U/ml) (Worthington) and l-cysteine (0.3 μg/ml; Sigma) in DMEM (Invitrogen). Retinas were then rinsed with DMEM and triturated in 2 ml of DMEM. To remove cell fragments or factors released from the cells, the cell suspension of one retina was immediately adjusted with DMEM to a volume of 50 ml. Cells were then centrifuged at 9000 × g for 5 min, and the pellet was carefully resuspended in 6 ml of DMEM containing B27 supplement (1:50; Invitrogen) and penicillin/streptomycin (1:50; Biochrom). Dissociated cells were then passed through a cell strainer (40 μm; Falcon; BD Biosciences Discovery Labware). Cell suspension, 300 μl, was added into each well, resulting in a low density of cells. Cultures were arranged in a pseudorandomized manner on the plates so that the investigator would not be aware of their identity. Retinal cells of animals that had received pretreatment in vivo were cultured for 24 h and then fixed with a paraformaldehyde solution (4%) and methanol (Sigma). Then, they were processed for immunocytochemical staining with an antibody against βIII-tubulin, which is a phenotypic marker for RGCs (Fournier and McKerracher, 1997; Cui et al., 2003; Fischer et al., 2004a,b; Lorber et al., 2005; Müller et al., 2007). At least three independent experiments were performed to verify the results.
For testing the effects of Taxol alone or Taxol plus CNTF on neurite outgrowth, cultures of untreated retinas were prepared and 0, 0.5, 1, 3, 10, and 50 nm Taxol was added to the medium. CNTF was applied at a concentration of 200 ng/ml. Inhibitory CNS myelin extract (CME) was added to cultures at a preoptimized concentration of ∼10 μg/ml. Neurocan (R & D Systems) was applied at a concentration of 5 μg/ml as described previously (Inatani et al., 2001; Ahmed et al., 2009). Nocodazole (Sigma) was used at a concentration of 45 nm. After 72 h in culture, cells were fixed with a paraformaldehyde solution (4%) and methanol (Sigma). Then they were prepared for immunocytochemical staining with a βIII-tubulin antibody (TUJ-1) (1:2000; Babco). All RGCs with regenerated neurites in wells were photographed under a fluorescent microscope (200×). The neurite length of RGCs was determined using the NIH ImageJ software. Furthermore, the total number of βIII-tubulin-positive RGCs with an intact nucleus (4′,6-diamidino-2-phenylindole staining) per well was quantified to test for potential neurotoxic or neuroprotective effects after each treatment. Values for neurite outgrowth were determined by the sum of the neurite length per well divided by the total number of RGCs per well, resulting in the average neurite length per RGC. Values were then normalized to control groups as indicated. The data are given as the mean ± SEM of four replicate wells. The significances of intergroup differences were evaluated using a one-way ANOVA test, followed by corrections for multiple post hoc tests (Bonferroni–Holm, Tukey's). The results from individual experiments were averaged within each experimental group. Each experiment was repeated at least three times.
PC12 cell cultures.
PC12 cells were cultured in 5% fetal bovine serum plus 10% horse serum in DMEM (Invitrogen) on poly-d-lysine-coated dishes (0.1 mg/ml; molecular weight, <300,000 Da; Sigma). Cells were exposed to 100 ng/ml nerve growth factor (NGF) (Sigma), Taxol (3 nm), and/or CME as indicated in Results. Cultures were fixed after 72 h with a paraformaldehyde solution (4%) and methanol (Sigma). Cells were prepared for immunocytochemical staining with a βIII-tubulin antibody (TUJ-1) (1:2000; Babco). PC12 cells with neurites were photographed under a fluorescent microscope (200×). To measure effects of Taxol and CME on neurite outgrowth, the length of the longest neurite of PC12 cells was determined using the NIH ImageJ software (at least 800 cells per group). The significances of intergroup differences were evaluated using a one-way ANOVA test, followed by corrections for multiple post hoc tests (Bonferroni-Holm, Tukey's). The results from individual experiments were averaged within each experimental group. Each experiment was repeated at least three times.
Rho activity assay.
PC12 cells were cultured in 5% fetal bovine serum plus 10% horse serum in DMEM and differentiated by NGF (100 ng/ml) on poly-d-lysine-coated dishes (0.1 mg/ml; Sigma) for 48 h. Then some cultures were treated with Taxol (3 nm) for another 3 h. After that, CME was added to the medium of some cultures for 10 min. Then cells were rinsed with TBS (50 mm Tris/HCl, pH 7.5, and 150 mm NaCl) and lysed by the addition of 500 μl of Rho–radioimmunoprecipitation assay lysis buffer [50 mm Tris/HCl, pH 7.2, 500 mm NaCl, 1% Triton X-100, 10 mm MgCl2, 0.5% sodium deoxycholate (dissolved in ddH2O), 0.1% SDS, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mm PMSF] and passing them 10 times through a 18 gauge needle. After centrifugation at 15,800 × g for 10 min, the supernatants were incubated with 20–50 μg of Sepharose-bound glutathione S-transferase (GST) proteins containing the RhoGTPase binding domain Rhotekin (RBD) for 45 min at 4°C by gently shaking, followed by five washing steps with Rho–RBD washing buffer (50 mm Tris/HCl, pH 7.2, 150 mm NaCl, 1% Triton X-100, 10 mm MgCl2, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 0.1 mm PMSF). Activated RhoA protein was analyzed by immunoblotting. The films were scanned and the band intensity was measured using the NIH ImageJ software. All values in the histograms have been corrected against the total RhoA band densities in lysates. Data obtained from three independent experiments were normalized and are represented as the mean ± SEM.
Quantification of growth cone sizes.
Dissociated primary RGCs were cultured for 3 d and then treated with Taxol (3 nm) for 3 h. Half of the cultures were then exposed to CME for 10 min. Cultures were fixed with a paraformaldehyde solution (4%) and stained with a βIII-tubulin antibody (TUJ1) (1:2000; Babco), followed by staining of actin cytoskeleton with phalloidin–tetramethylrhodamine isothiocyanate (100 ng/ml diluted in PBS; Sigma) for 40 min at room temperature. The phalloidin staining was performed to improve the visualization of full growth cone (see Fig. 2C). Growth cones (at least 400 per treatment group) were photographed under a fluorescent microscope (1000×) and the area of the phalloidin-stained growth cone was analyzed using NIH Image J software. To confirm the results, experiments were repeated at least two more times. The significances of intergroup differences were evaluated using a one-way ANOVA test, followed by corrections for multiple post hoc tests (Bonferroni-Holm, Turkey's).
In other experiments, we used PC12 cells that were differentiated with NGF (100 ng/ml) for 3 d. PC12 cells were then treated with vehicle or Taxol (3 nm) for 3 h and then exposed to CME for 10 min. The staining procedure and measurement of growth cone sizes was the same as described above for RGCs.
Quantification of axons in the optic nerve and RGCs in retinal cross-sections.
Regeneration of axons was quantified 14 d after surgery as described previously (Leon et al., 2000; Fischer et al., 2004b; Müller et al., 2007; Hauk et al., 2008; Leibinger et al., 2009). In brief, under 400× magnification, the number of GAP-43-positive axons extending ≥0.5, ≥1, ≥1.5, and ≥2 mm from the injury site in six sections per case were counted, normalized to the cross-sectional width of the optic nerve. These data were used to calculate the total numbers of regenerating axons in each animal (Leon et al., 2000; Müller et al., 2007; Hauk et al., 2008, 2010). For each experimental group, at least five animals were used. Additionally, the length of the longest axon of each section was measured, and the numbers of βIII-tubulin-positive cells per section were counted. Measurements were averaged per case, and then averaged across all similarly treated animals (at least five animals per group) to obtain a group means and SE as described previously (Müller et al., 2007). The significance of intergroup differences was evaluated using a one-way ANOVA test, followed by corrections for multiple post hoc tests (Bonferroni-Holm, Tukey's). To measure axon sprouting 1 and 3 d after optic nerve crush, animals received an intravitreal injection of 2 μl of cholera toxin subunit B (CTB) conjugated with Alexa Fluor 488 (Invitrogen) simultaneously with surgery, because GAP-43 expression in RGCs was still too low to clearly detect axons 3 d after optic nerve crush (Park et al., 2008; Smith et al., 2009). The quantification of axons that had sprouted into the distal part of the injured optic nerve was performed as described before for GAP-43-positive axons 14 d after surgery.
Western blot assays.
For retinal lysate preparation, rat retinas were dissected and collected in lysis buffer (20 mm Tris/HCl, pH 7.5, 10 mm KCl, 250 mm sucrose, 10 mm NaF, 1 mm DTT, 0.1 mm Na3VO4, 1% Triton X-100, and 0.1% SDS) with 1:100 protease inhibitor (Calbiochem). Retinas were homogenized and centrifuged at 5000 rpm for 10 min at 4°C. The supernatants were analyzed by Western blot assay. Separation of proteins was performed by 10% SDS-PAGE, according to standard protocols (Bio-Rad). After SDS-PAGE, proteins were transferred to nitrocellulose membranes (GE Healthcare). The blots were blocked in either 5% dried milk or 2% ECL Advance blocking agent in Tris-buffered saline/Tween-20. They were then processed for immunostaining with an antiserum against rat phospho-STAT3 (Tyr705, 1:5000; Cell Signaling Technology), antibody against CNTF (1:5000; Serotec), LIF (1:6000; Santa Cruz Biotechnology), an antibody against GAP-43 (1:1000), a monoclonal antibody against rat tubulin (1:1000; Babco), or an antibody against RhoA (sc-418, 1:5000; Santa Cruz Biotechnology). Bound antibodies were visualized with anti-rabbit, anti-goat, or anti-mouse IgG secondary antibodies conjugated with horseradish peroxidase diluted at 1:80,000 (all from Sigma). The antigen–antibody complexes were detected by enhanced chemiluminescence (ECL) or ECL advance (ECL; GE Healthcare). Western blots were repeated at least two times to confirm results.
Quantification of macrophages.
Rat optic nerve sections from animals that had undergone optic nerve crush and treated with either Taxol (3 μm) or PBS at the lesion site for 0, 1, and 3 d were stained with a polyclonal antibody against CD68 (ED1, 1:500; Serotec). The number of macrophages around the lesion site was counted for each optic nerve section, averaged per case, and then averaged across all similarly treated animals to obtain a group means and SE. Each experimental group consisted of at least four animals. The significance of intergroup differences was evaluated using a one-way ANOVA test, followed by corrections for multiple post hoc tests (Bonferroni-Holm, Tukey's).
Measurement of GFAP-free gap at the lesion site.
Rat optic nerve sections from animals that had undergone optic nerve crush and either treated with Taxol (1 μm) or PBS at the lesion site for 1, 3, and 14 d were stained with a monoclonal antibody against GFAP (1:100; Santa Cruz Biotechnology), followed by a secondary antibody (Alexa Fluor 594, 1:1000; Invitrogen). The area without appreciable staining for GFAP, both proximal and distal to the lesion site, was measured using the NIH ImageJ software. The measured area was divided through the width of the optic nerve section at the lesion site to obtain the average length of the gap. The obtained values of gap length per section were averaged per case and then averaged across all similarly treated animals (n = 4) to obtain a group means and SE. The significance of intergroup differences was evaluated using a one-way ANOVA test, followed by corrections for multiple post hoc tests (Bonferroni-Holm, Tukey's).
Primary astrocyte cultures.
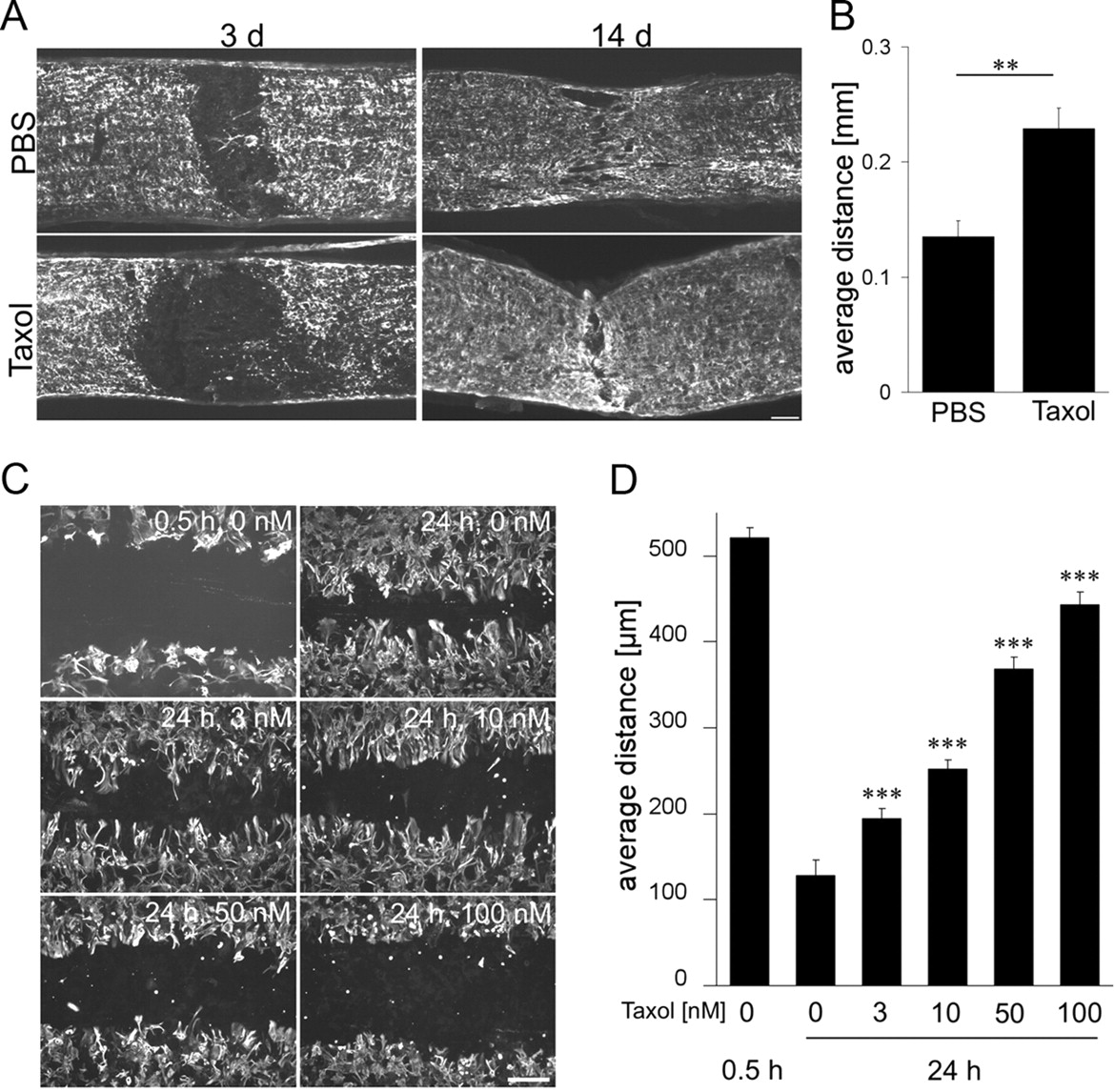
Primary cell cultures of astrocytes were prepared from postnatal (4-d-old) C57BL/6 mice. Cerebra of the pups were dissected aseptically, and the meninges were removed. Cortices were homogenized in ice-cold 1× PBS with Pasteur pipette. The homogenate was washed two to three times in PBS by centrifugation at 2000 × g for 5 min. The cell pellet was resuspended in DMEM containing 10% fetal bovine serum, 1% glutamine, and 1% penicillin–streptomycin solution and cultured in poly-d-lysine-coated 75 cm2 culture flasks for 14 d until monolayers reached confluence. Confluent cultures were vigorously agitated to dislodge oligodendrocytes and microglia. The astrocytes remain attached to the culture flask and were trypsinized and replated in poly-d-lysine-coated four-well plates until they reached confluency. Scratch injury was performed on confluent primary astrocyte cultures using a sterile plastic tip with a diameter of 0.5 mm. Cultures were exposed to different concentrations of Taxol (0, 3, 10, 50, and 100 nm) and incubated for another 24 h. Cells were fixed and stained with monoclonal antibody against GFAP (Santa Cruz Biotechnology). Images were taken at different sites of lesion for all the culture conditions, and the width of the unclosed lesion site was measured using NIH ImageJ software.
Measurement of CSP G expression.
After an optic nerve crush, animals were treated with either Taxol (1 μm) or PBS at the lesion site for 0, 1, or 3 d. Rat optic nerve sections from these animals were taken and stained with a monoclonal antibody against the glycosaminoglycan portion of native CSPGs (1:100; Sigma). Optic nerve sections from the animals of the different groups (n = 4 per group) were stained simultaneously, and images were taken in a single session at the same exposure time. The staining intensity for CSPGs of equal area distal to the lesion site was measured in all optic nerve sections using NIH ImageJ software and normalized to untreated controls. The measured intensities were averaged per case and then averaged across all similarly treated animals (n = 4) to obtain a group means and SE. The significance of intergroup differences was evaluated using a one-way ANOVA test, followed by corrections for multiple post hoc tests (Bonferroni-Holm, Tukey's). To verify the specificity of the CSPG staining, sections were exposed to chondroitinase ABC (Sigma) at 0.3 U for 10 h at 37°C and then washed and stained for CSPGs.
Results
Taxol facilitates neurite elongation of RGCs in culture
To test whether Taxol affects neurite extension of mature, primary neurons, we prepared dissociated retinal cultures from untreated adult rats and cultured these cells in the absence or presence of increasing concentrations of Taxol (0–50 nm). In the absence of Taxol, RGCs showed moderate neurite outgrowth after 72 h in culture. Neurite extension was significantly increased in the presence of low concentrations of Taxol (0.5–3 nm) (Fig. 1A,B). Strongest effects were measured at a concentration of 3 nm. However, no positive effect of Taxol was measured at a concentration of 10 nm, and, at a concentration of 50 nm, the average neurite length was reduced compared with untreated controls (Fig. 1B). The survival of RGCs was not altered between groups, demonstrating that the Taxol concentrations used were not toxic to RGCs (Fig. 1C). To test whether the increased neurite growth measured in the presence of Taxol (3 nm) was mediated by the stabilization of microtubules, we cultured mixed retinal cells in the presence of nocodazole, which moderately increases the catastrophe rate of microtubules at low concentrations (Vasquez et al., 1997; Grosheva et al., 2008). The presence of nocodazole (45 nm) decreased neurite outgrowth and compromised the beneficial effect of Taxol (3 nm) (Fig. 1D) but did not affect the survival of RGCs in culture (Fig. 1E). These data suggest that Taxol indeed enhanced neurite growth by microtubule stabilization.

Low concentrations of Taxol enhance neurite growth of primary RGCs. A, βIII-Tubulin-positive RGCs with regenerated neurites that were cultured in the absence (con) or presence of 3 nm Taxol for 3 d. Scale bar, 50 μm. B, Quantification of neurite outgrowth of mature RGCs that were cultured in the absence or presence of either 0.5, 1, 3, 10, or 50 nm Taxol for 3 d. At low concentrations, Taxol increased neurite outgrowth significantly, whereas at a concentration of 50 nm, Taxol compromised neurite extension. Treatment effects compared with untreated cultures (con): ***p < 0.001. C, Taxol did not affect the survival of RGCs at any concentration tested in culture. D, Quantification of neurite outgrowth of mature RGCs that were cultured in the presence of either 3 nm Taxol or nocodazole (Noco) at 45 nm for 3 d. Nocodazole compromises the neurite growth-promoting effect of Taxol. Treatment effect: ***p < 0.001. E, Number of RGCs per well of experiments shown in D. F, Influence of Taxol on in vivo growth-stimulated RGCs. RGCs were transformed into a regenerative state by optic nerve crush (ONC) and lens injury (LI) in vivo. After 5 d, RGCs were cultured in the absence or presence of different concentrations (3, 10, and 50 nm) of Taxol for 24 h to measure spontaneous neurite extension. At 3 nm, Taxol significantly enhanced axon elongation of growth-stimulated RGCs, whereas 50 nm Taxol reduced outgrowth. Treatment effect: ***p < 0.001. G, Quantification of RGCs per well of groups shown in F. Taxol did not affect the survival of RGCs at any concentration tested in culture.
Neurite outgrowth of RGCs is significantly augmented in the presence of CNTF in the culture medium (Jo et al., 1999; Lorber et al., 2005; Müller et al., 2007, 2009; Lingor et al., 2008; Leibinger et al., 2009; Hauk et al., 2010). Consistently, CNTF alone increased the neurite outgrowth to a similar proportion as Taxol at a concentration of 3 nm in our cultures (Fig. 2A,B). A combinatorial exposure to CNTF and Taxol increased neurite outgrowth of RGCs stronger than each treatment alone, suggesting that both treatments functioned synergistically (Fig. 2A,B).

Taxol compromises myelin and neurocan inhibition of mature RGCs in culture. A, βIII-Tubulin-positive RGCs with regenerated neurites in culture 3 d after exposure to vehicle (−), CNTF (200 ng/ml), Taxol (3 nm), or Taxol plus CNTF. In addition, half of the cultures were also exposed to CNS myelin extract (Myelin). Scale bar, 50 μm. B, Quantification of neurite outgrowth of RGCs in cultures shown in A. Treatment effects: ***p < 0.001. C, Growth cones of RGCs stained with phalloidin (red) and antibodies against βIII-tubulin (green). RGCs were cultured for 3 d, then some of the cells were treated with Taxol for 3 h, and afterward half the cultures were exposed to CNS myelin extract for 10 min as indicated. Myelin-treated growth cones are also depicted at higher exposure times, indicating that microtubules (green) are also located in the extensions (arrows) of the growth cones that were exposed to Taxol. Scale bar, 2 μm. D, Quantification of growth cone sizes of groups treated as described in C. Treatment effects: ***p < 0.001. E, Quantification of neurite outgrowth of RGCs in cultures 3 d after exposure to vehicle (−) or Taxol (3 nm). In addition, half of the cultures were also exposed to the proteoglycan neurocan (NC) (5 μg/ml). Treatment effects: ***p < 0.001.
To test whether Taxol can also enhance neurite growth of RGCs that were previously transformed in vivo into a regenerative state, rats received an optic nerve crush and a lens injury. After 5 d, RGCs were cultured in the absence or presence of different concentrations of Taxol for 24 h. Consistent with previous reports, lens injury and axotomy-treated RGCs showed spontaneous neurite outgrowth in culture (Müller et al., 2007, 2009; Leibinger et al., 2009). At a concentration of 3 nm, Taxol increased the average neurite length of spontaneously regenerating RGCs 2.5-fold compared with controls, demonstrating that Taxol can also markedly enhance spontaneous neurite extension of in vivo growth-stimulated RGCs (Fig. 1F). These effects on growth-stimulated RGCs were not seen at a concentration of 10 nm, and neurite outgrowth was reduced at 50 nm Taxol in the culture medium (Fig. 1F). The survival of RGCs in culture was not affected by any treatment (Fig. 1G). These data indicate that Taxol facilitates neurite extension within specific concentration ranges by microtubule stabilization and suggest that the beneficial effects of Taxol act synergistically with measures that activate the intrinsic growth state of neurons.
Taxol treatment desensitized axons toward inhibitory myelin extract and neurocan
We then tested whether Taxol affects neurite growth inhibition of RGCs by myelin. To this end, we cultured mixed retinal cells in the presence of CME. Consistent with previous reports (Ahmed et al., 2006, 2009; Logan et al., 2006), CME significantly reduced neurite extension of both untreated and CNTF-treated RGCs (Fig. 2A,B). The application of Taxol (3 nm) completely abolished the inhibitory effects of CME, and cultures showed a similar average neurite length as control groups that were cultured in the presence of Taxol but without CME (Fig. 2A,B). The number of RGCs per well of each experimental group remained unchanged over the observation time of 3 d (data not shown). Because myelin inhibitors induce growth cone collapse/shrinkage, we tested whether Taxol might stabilize the growth cones of primary RGCs. To this end, we cultured mixed retinal cells for 3 d and then treated half of the cultures with Taxol (3 nm) for 3 h. Untreated and Taxol-treated cultures were then exposed to CME for 10 min. The average size of growth cones was significantly reduced in RGCs after exposure to CME compared with untreated controls (Fig. 2C,D). However, Taxol significantly alleviated the effects of myelin, and growth cones showed almost similar sizes as untreated controls, demonstrating a Taxol-mediated stabilization (Fig. 2C,D).
We next tested whether the disinhibiting effect of Taxol was restricted to myelin inhibitors or also applicable to other inhibiting molecules such as CSPGs. To this end, we cultured primary RGCs in the absence and presence of neurocan and exposed these cells either to Taxol (3 nm) or vehicle. As reported previously, neurocan significantly reduced neurite growth of RGCs (Inatani et al., 2001). These inhibitory effects were completely overcome in the presence of Taxol, suggesting that the disinhibitory effect of Taxol occurred downstream of specific inhibitory receptors (Fig. 2E).
To extend the results obtained in primary RGCs, we exposed Taxol (3 nm) to PC12 cells, which are also responsive to myelin (Lehmann et al., 1999; Fournier et al., 2003), and primed these cells with NGF. Consistent with our findings in primary RGCs, Taxol significantly enhanced neurite extension of PC12 cells compared with non-Taxol-treated controls (Fig. 3A,B). Moreover, the presence of Taxol also overcame the inhibitory effect of myelin on neurite growth (Fig. 3A,B) and significantly compromised the destabilizing effect of myelin on growth cones (Fig. 3C,D). Because myelin activates the RhoA/Rho kinase (ROCK) signaling pathway in PC12 cells and blocking of RhoA activity overcomes myelin inhibition (Lehmann et al., 1999; Fournier et al., 2003), we tested the possibility as to whether Taxol interferes with myelin-induced RhoA activation. In myelin-treated cultures, RhoA GTP levels were significantly increased compared with non-myelin treated controls (Fig. 3E,F). However, additional treatment with Taxol did not measurably affect the activation of RhoA in the absence or presence of myelin extract (Fig. 3E,F). These data suggest that Taxol desensitized growing neurites toward inhibitory molecules without influencing RhoA activation.

Effects of Taxol on PC12 cell growth cones. A, PC12 cells were cultured for 3 d in the presence of NGF, CNS myelin extract (Myelin), and/or Taxol (3 nm) as indicated. Cells were then stained with an antibody against βIII-tubulin. B, Quantification of neurite outgrowth of PC12 cells that were treated as described in A. Treatment effects: ***p < 0.001. C, Representative growth cones of PC12 cells that were cultured for 3 d in the presence of NGF, then exposed to Taxol or vehicle for 3 h, and then treated either with vehicle or myelin extract for 10 min. Growth cones were stained with phalloidin (red) and βIII-tubulin (green). The presence of Taxol compromised the destabilizing effect of myelin extract on growth cones. Scale bar, 10 μm. D, Quantification of growth cone sizes of groups treated as described in C. Treatment effects: ***p < 0.001. E, PC12 cells that were primed with NGF and treated with Taxol. Cells were exposed to myelin for 10 min, and cell lysates were affinity precipitated with GST–RBD to detect GTP-bound Rho (RhoA–GTP). Rho–GTP levels were significantly enhanced after exposure to myelin extract. Total Rho protein levels remained unchanged in cell lysates. Taxol treatment did not affect RhoA activation by myelin. F, Quantification of average RhoA–GTP levels in PC12 cells from three independent experiments as described in E.
Taxol treatment promotes optic nerve regeneration in vivo
In additional experiments, we tested whether the application of Taxol at the injury site of the optic nerve was sufficient to promote axon regeneration in vivo. We applied Gelfoam soaked with either PBS or increasing concentrations of a solution of Taxol (1, 10, 100, and 1000 μm) directly around the lesion site of the nerve (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Because RGCs do not normally show significant regeneration in the injured optic nerve, we additionally transformed RGCs into an active regenerative state by inducing a lens injury. The application of the lowest concentration of Taxol solution (1 μm) improved regeneration in the optic nerve strongest. At this concentration, the number of axons that had regenerated 1 mm beyond the lesion site was increased 15-fold compared with PBS-treated controls (Fig. 4A,B). Significant numbers of regenerating axons were found even at 1.5 and 2 mm beyond the lesion site in Taxol-treated animals, whereas almost no measurable regeneration was observed in PBS-treated groups at these distances. Moreover, in the Taxol with lens injury-treated rats, the average length of the longest axons per optic nerve section reached up to 2.17 ± 0.18 mm, 14 d after surgery, whereas PBS and lens injury-treated controls showed an average length of 1.1 ± 0.05 mm (Fig. 4D). In both groups, the number of surviving RGCs in the retina was very similar and significantly higher than in groups that had not received lens injury (Fig. 4C). However, with increasing concentrations of Taxol, the number and length of regenerating axons decreased significantly (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), although the application of the highest concentration of Taxol solution (1000 μm) still slightly improved regeneration in the optic nerve compared with PBS-treated controls (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) (Fig. 4A,B). These data demonstrate that the Taxol treatment enhanced axon regeneration in the injured optic nerve but did not significantly influence the neuroprotective effects of lens injury on RGCs (Fig. 4C).

Taxol enhances axonal regeneration in the crushed optic nerve but does not affect the survival of RGCs. A, Longitudinal sections through the optic nerve were stained with an antibody against GAP-43, 2 weeks after optic nerve crush (ONC). Additional local treatment of the optic nerve with PBS or Taxol, or ONC with lens injury (LI) plus additional treatment with PBS or Taxol is indicated. The asterisks mark the lesion site. Arrows point to the longest axons. Scale bar, 100 μm. B, Quantitative analysis of axon regeneration in the optic nerve at 500–2000 μm beyond the lesion site of groups described in A. Treatment effects: ***p < 0.001, **p < 0.01 compared with group treated with PBS; †††p < 0.001 compared with group treated with PBS with lens injury. C, Quantification of surviving βIII-tubulin-positive RGCs in sections of the same eyes as described in A and B. Untreated eyes, which are not shown in this diagram, displayed a number of 212 ± 5 RGCs per retinal section. Treatment effects: ***p < 0.001 compared with group treated with PBS. D, Quantification of longest axons per section of groups described in A. Treatment effects: ***p < 0.001 compared with group treated with PBS; †††p < 0.001 compared with group treated with PBS plus lens injury.
We also tested whether Taxol treatment alone (without lens injury) is sufficient to enhance axon growth in the crushed optic nerve. In contrast to PBS-treated animals, which displayed very little regeneration, Taxol-treated rats showed a moderate but significantly increased growth of fibers beyond the injury site. Longest axons in this group reached on average an appreciable length of 0.89 ± 0.12 mm 14 d after optic nerve crush (Fig. 4A,D). The number of RGCs in Taxol-treated rats was similar to that seen in PBS-treated controls, confirming that the local Taxol application at the optic nerve did not affect the survival of RGCs (Fig. 4C). In additional experiments, we also tested whether injections of Taxol into the vitreous body may also influence axon regeneration 14 d after optic nerve crush. However, intravitreal application of Taxol did not measurably affect axon growth beyond the lesion site compared with PBS-treated controls (data not shown).
We next examined axon sprouting 1 and 3 d after optic nerve crush. At 1 d after crush, injured nerve fibers terminated at the proximal end of the crush site in animals that had received either local PBS or Taxol treatment or intravitreal injection of Taxol. In all cases, no axons had grown beyond the lesion site (Fig. 5A). In contrast, 3 d after optic nerve crush with the local Taxol treatment, most CTB-positive axons had already penetrated the lesion site (Fig. 5A,B). The longest axonal sprouts showed a notable length of up to 400 μm, whereas minimal axonal sprouting was found in the PBS-treated control animals or cases that had received an intravitreal injection of Taxol (Fig. 5C). These data demonstrate that only local, but not intravitreal, treatment with Taxol significantly enhances initial axon sprouting beyond the lesion site of the optic nerve and that in this case, regeneration occurs even without stimulation of the intrinsic growth state.

Taxol enhances initial axonal sprouting in the injured optic nerve. A, Longitudinal sections of the optic nerve (ON) 1 and 3 d after optic nerve crush and either treatment with Gelfoam soaked with PBS or Taxol (1 μm) or intravitreal injection (ivit) of Taxol as indicated. Axons (green) were anterogradely labeled with CTB. White asterisks indicate the injury site, and arrows point to axons that sprouted beyond the injury site of the optic nerve. Scale bar, 100 μm. B, Quantitative analysis of axonal sprouting at 50 and 100 μm beyond the lesion site after local PBS or Taxol treatment as indicated 3 d after surgery. Treatment effect: ***p < 0.001. C, Quantification of the longest axons per section of groups described in A and B. Treatment effect: ***p < 0.001.
Local Taxol treatment does not alter the intrinsic regenerative state of RGCs
We next investigated whether the local application of Taxol may have direct or indirect influences on the intrinsic regenerative state of RGCs. The regenerative state of RGCs 5 d after surgery can be functionally evaluated by culturing these neurons and measuring their spontaneous neurite outgrowth (Lorber et al., 2005; Müller et al., 2007, 2009; Leibinger et al., 2009; Hauk et al., 2010). For the in vivo treatment, we applied Gelfoam soaked with either PBS or Taxol (3 μm) at the lesion site of the optic nerve and stimulated the axonal growth program in half of the rats by performing a lens injury. In contrast to lens injury, which caused a marked increase in spontaneous neurite outgrowth in RGCs of both PBS- and Taxol-treated groups to a comparable extent, a local Taxol treatment per se did not alter the outgrowth (Fig. 6A). These data suggest that, in contrast to lens injury, the local application of Taxol does not affect the intrinsic regenerative state of RGCs. Consistently, the expression levels of retinal GAP-43, CNTF, LIF, and activated STAT3 (phospho-STAT3), which are normally upregulated after optic nerve crush combined with lens injury (Müller et al., 2007; Leibinger et al., 2009; Hauk et al., 2010), were not changed by Taxol treatment compared with similarly treated PBS controls (Fig. 6B).

Local Taxol application does not influence the intrinsic regenerative state of RGCs. A, Quantification of spontaneous neurite outgrowth of RGCs in dissociated retinal cell cultures. Rats had either received an intraorbital optic nerve crush (ONC) alone or optic nerve crush combined with lens injury (LI). The optic nerves of animals were treated with Gelfoam soaked with either PBS or Taxol (1 μm). Treatment effect: ***p < 0.001. B, Western blot analysis of retinal lysates from rats after optic nerve crush alone or optic nerve crush with lens injury, locally treated at the optic nerve with either PBS or Taxol (1 μm) using specific antibodies against GAP-43, phospho-STAT3 (pSTAT3), LIF, or CNTF. Tubulin served as a loading control.
Taxol treatment delays cellular responses at the lesion site of the optic nerve
Because Taxol reduces cell proliferation and cell migration (Ogasawara et al., 2001; Wiskirchen et al., 2004; Maranhão et al., 2008), we speculated whether the local Taxol treatment may have also affected the glial scar formation. We therefore tested the influence of Taxol on the cellular responses in the optic nerve after injury and initially focused on the infiltration/activation of CD68-positive cells (macrophages/microglia) in the optic nerve. Two hours after optic nerve crush and PBS treatment, no CD68-positive cells were detected around the lesion site. This number was dramatically increased 1 and 3 d after optic nerve crush (Fig. 7A,B). In contrast, Taxol-treated animals showed a greatly reduced number of macrophages in the optic nerve after 1 d. The Taxol-induced reduction of macrophage infiltration was still detectable after 3 d, although it was less pronounced at this time point (Fig. 7A,B). We also stained adjacent sections for GFAP to investigate the responses of astrocytes to Taxol after optic nerve crush. Taxol- and PBS-treated animals revealed a GFAP-free area of similar size 1 d after optic nerve crush (data not shown). However, when analyzed at 3 d after crush, the GFAP/astrocyte-free gap at the lesion site of Taxol-treated animals was significantly larger than that of PBS-treated controls (Fig. 8A,B). After 14 d, the GFAP-free area in Taxol- and PBS-treated optic nerves were not detectable anymore (Fig. 8A,B), suggesting that the proliferation of astrocytes was transiently delayed by Taxol treatment. To confirm this hypothesis, we prepared primary astrocytic cultures. When astrocytes were confluent, we performed a scratch injury, which resulted in a gap of ∼0.5 mm in the astrocyte cell layer. After 24 h, this gap was strongly reduced (Fig. 8C,D) and completely filled again by astrocytes after 72 h (data not shown). The presence of Taxol significantly delayed this process in a concentration-dependent manner, with Taxol already being effective at a concentration of 3 nm (Fig. 8C,D).

Taxol delays macrophage activation in the optic nerve. A, Longitudinal sections of optic nerves stained for CD68-positive cells 2, 24, and 72 h after optic nerve crush and additional local treatment with either PBS or Taxol (1 μm). Asterisks indicate the injury site. Scale bar, 100 μm. B, Quantification of CD68-positive cells per optic nerve section of animals described in A. ***p < 0.001.

Taxol delays the gliosis formation at the injury site of the optic nerve. A, Longitudinal sections of optic nerves stained for GFAP 3 and 14 d after optic nerve crush and additional local treatment with either PBS or Taxol (1 μm). Scale bar, 100 μm. B, Quantification of the average width of the GFAP-free area at the lesion site of the optic nerve 3 d after surgery. **p < 0.01. C, Cultured GFAP-positive primary astrocytes after scratch injury and exposure to various concentrations of Taxol (as indicated) for 24 h. Scratch injury of confluent astrocytes caused a defined gap in the astrocytic cell layer of a width of ∼0.5 mm. The width of this gap was strongly reduced in untreated cultures after 24 h. The presence of Taxol significantly delayed this process in a concentration-dependent manner. Taxol at 3 nm was sufficient to compromise this process significantly. Scale bar, 200 μm. D, Quantification of the average width of the gap in the astrocytic cell layer 0.5 and 24 h after scratch injury and exposure to increasing concentrations of Taxol. Treatment effect: ***p < 0.001.
Because Taxol affected astrocytic responses to injury in vivo, we also analyzed whether Taxol might influence the induction of CSPG expression normally seen after optic nerve crush. No CSPG staining was found in optic nerves that had received an optic nerve crush 2 h earlier, whereas little staining was observed after 1 d and significant staining 3 d after optic nerve crush and PBS treatment. This staining was strongly reduced when sections were pretreated with chondroitinase ABC, verifying the specificity for CSPGs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). However, the induction of CSPG expression at the injury site was significantly reduced in animals that had received optic nerve crush and been treated with Taxol compared with PBS-treated controls (Fig. 9A,B), suggesting that the Taxol application compromised the upregulation of CSPG expression in vivo.

Taxol compromises expression of CSPGs at the injury site of the optic nerve. A, Longitudinal sections of optic nerves stained for CSPGs 2, 24, and 72 h after optic nerve crush and additional local treatment with either PBS or Taxol (1 μm). Scale bar, 100 μm. B, Quantification of pixel intensity (normalized toward untreated optic nerve sections) of CSPG staining of sections described in A. **p < 0.01.
Discussion
One widely studied model of regenerative failure of axons in the CNS is the optic nerve (Berry et al., 2008). Although mature RGCs do not normally regenerate axons, they enter into an active regenerative state after lens injury and subsequently regrow axons over considerable distances into the lesioned optic nerve (Fischer et al., 2000, 2008; Leon et al., 2000; Lorber et al., 2005; Pernet and Di Polo, 2006). Nevertheless, this regeneration is still limited by inhibitors associated with the CNS myelin as well as the glial scar that forms at the lesion site. Therefore, combinatorial treatments overcoming inhibitory signaling together with measures that stimulate the regenerative state of RGCs result in markedly stronger regeneration than each treatment alone (Cui et al., 2003; Fischer et al., 2004b; Müller et al., 2007; Lingor et al., 2008; Ahmed et al., 2009). One common strategy to overcome inhibitory signaling is the manipulation of the RhoA/ROCK signaling pathway, which lies downstream of several inhibitory receptors and primarily affects actin and only indirectly microtubules polymerization in the growth cone. In contrast, the current study demonstrates that direct microtubule stabilization by Taxol promotes axon extension per se and is sufficient to overcome additionally myelin, as well as CSPG, inhibition in mature neurons. When locally applied in vivo, Taxol allowed axon regeneration in the injured optic nerve. The extent of regeneration achieved by Taxol and lens injury treatment was higher than that reported in our previous studies in which lens injury treatment was combined with the expression of a dominant-negative form of the Nogo receptor or the RhoA-inactivator ADP-ribosyl-transferase in RGCs to overcome inhibitory signaling (Fischer et al., 2004a,b). These data lend encouragement to the possibility that the clinically established drug Taxol or related compounds that stabilize microtubules may be used as adjuvant drugs for CNS repair in the future.
How does Taxol improve axon regeneration?
In contrast to lens injury, the local Taxol treatment at the injured optic nerve did not affect the endogenous retinal CNTF/LIF expression or STAT3 activation, all of which are essential for the transformation of RGCs into a regenerative state by lens injury (Müller et al., 2007; Leibinger et al., 2009; Fischer, 2010). Accordingly, Taxol did not influence the survival of RGCs or their intrinsic regenerative state. The latter aspect was proven by the fact that the local Taxol treatment in vivo affected neither the expression levels of GAP-43 nor the spontaneous neurite outgrowth of RGCs in culture measured 5 d after surgery in vivo. Furthermore, only the local application of Taxol at the lesion site of the optic nerve, but not its intravitreal injection, promoted axon regeneration. These data suggest that the beneficial effects of Taxol treatment were mainly caused by the enhancement of axon elongation by microtubule stabilization in the growth cone and desensitizing axons toward myelin inhibitors at the lesion site. Moreover, the observed delay in the formation of the inhibitory glial scar could have also partially contributed.
Taxol enhances axon elongation at low concentrations
The current study demonstrates that Taxol not only overcomes myelin inhibition but also significantly enhances neurite elongation of mature primary neurons in the presence of CSPGs and on a growth permissive substrate in culture, when used at a concentration of 3 nm. Although we cannot totally rule out the possibility that indirect effects via other retinal cells may have also contributed to the observed neurite growth-promoting effects of Taxol on RGCs in vitro, we consider a direct effect of Taxol on RGCs as very likely. This is because Taxol enhanced neurite outgrowth of PC12 cells on a growth permissive and inhibitory substrate as well. The observation that the presence of nocodazole, which moderately increases the catastrophe rate of microtubules (Vasquez et al., 1997; Grosheva et al., 2008), abolished the neurite growth-promoting effect of Taxol suggests that Taxol mediated this effect via microtubule stabilization and enhancing their polymerization at plus ends. However, these effects were not detectable at a concentration of 10 nm, and neurite outgrowth was even reduced at higher concentrations (≥50 nm) (Fig. 1B). These findings are in accordance with a previous report demonstrating that, at high concentrations, Taxol blocks neurite extension of cortical neurons in vitro (Chuckowree and Vickers, 2003). This dependence on a specific and low concentration range can be explained by the dynamic instability of microtubules and the influence of Taxol on the growing and shrinking rates of microtubules. At higher concentrations, Taxol hyperstabilizes microtubules and blocks microtubule dynamics completely, whereas at lower concentrations, Taxol selectively compromises catastrophe events, thereby favoring the overall polymerization of microtubules at plus ends (Derry et al., 1995, 1997). Similarly, the concentration-dependent effect of Taxol was also found in vivo. In these cases, the effective concentrations we locally applied via the Gelfoam approach were higher (300×) than used in the culture medium. This is plausible because high-concentration gradients are necessary for a drug to diffuse from the Gelfoam into the tissue to reach sufficient concentrations in the optic nerve. Moreover, most of the drug may be released in the intraorbital space and therefore not reach the optic nerve. This may also explain why the effective concentration range of locally applied Taxol at the injury site in vivo was much more expanded (1–100 μm) compared with the situation in culture. Although the concentrations of Taxol that reached the axons in the optic nerve are unknown, it is quite possible that these were in a similar range as those we applied in vitro. Nevertheless, the finding that the lowest concentration of Taxol solution used in our study improved axon regeneration in vivo the most, whereas the highest tested concentration showed only a little effect (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), is consistent with the results determined in cell culture.
Taxol overcomes myelin and CSPG inhibition
After disruption at the lesion site of the optic nerve, inhibitory factors from the myelin activate RhoA (Mukhopadhyay et al., 1994; Chen et al., 2000; GrandPre et al., 2000; Prinjha et al., 2000; Wang et al., 2002; Atwal et al., 2008) and further downstream cascades lead to a local arrest of axonal microtubule polymerization and axon extension (Mimura et al., 2006; Geraldo and Gordon-Weeks, 2009). The current study demonstrates that neurite growth inhibition of primary RGCs and PC12 cells by CNS myelin in culture is abolished in the presence of Taxol (3 nm). These effects are consistent with the previous report that Taxol improves axon growth of neonatal neurons on myelin substrates, although this group used Taxol at a higher concentration and did not report an enhanced axon extension in the absence of myelin inhibitors (Ertürk et al., 2007). Our data indicate that Taxol treatment does not affect RhoA activation in cells after exposure to myelin, suggesting that the “disinhibitory” effect of Taxol occurs downstream of RhoA. This possibility is also supported by the observation that Taxol overcame neurite growth inhibition of neurocan, which mediates its inhibitory effect via protein tyrosine phosphatase σ (Shen et al., 2009) and also RhoA (Monnier et al., 2003; Jain et al., 2004). Thus, one likely explanation for this general disinhibitory effect of Taxol might be the disentanglement of the coupled interaction of actin and microtubules in the growth cone. This makes the microtubule polymerization at the plus ends independent of the state of actin polymerization and thereby stabilizes the overall cytoskeleton structure of the growth cone, including the microtubules in the filopodial extensions (Geraldo and Gordon-Weeks, 2009). Although the detailed mechanism still needs to be unraveled, it is conceivable that the disinhibitory effect of Taxol contributed to the improved regeneration seen in vivo.
Local Taxol application affects inhibitory scar formation
The damaged tissue around the epicenter of the lesion reacts to optic nerve injury with proliferation and activation of microglia and macrophages recruited from the bloodstream. In addition, astrocytes proliferate and together with the production of proteoglycans form a glial scar that limits axon regeneration (Silver and Miller, 2004). We found that Taxol affected, at least transiently, the astrocytic response to injury. The GFAP-free area appearing between the proximal and distal nerve stump was significantly increased in Taxol-treated animals 3 d after optic nerve crush compared with controls. This together with the finding that even low concentrations of Taxol significantly delayed the proliferation of primary astrocytes in culture suggests that Taxol may have influenced the initial proliferation of astrocytes or astrocytic hypertrophy in vivo. Most strikingly, the CSPG expression was significantly reduced around the lesion site of the optic nerve. This finding is consistent with a previous study that showed that reducing the proliferation of astrocytes by a cell cycle inhibitor decreased CSPG expression after spinal cord injury (Tian et al., 2006). Given that the inactivation of inhibitory CSPGs by chondroitinase ABC improves axon regeneration in the CNS, in particular when combined with an enhancement of the intrinsic growth capacity of neurons (Moon et al., 2001; Bradbury et al., 2002; Steinmetz et al., 2005), it is possible that the reduction in CSPG expression may have also contributed to the improved regeneration seen after Taxol treatment. Next to reactive astrocytes, macrophages at the injury site of the CNS act as a significant barrier for initial regeneration (Busch and Silver, 2007; Horn et al., 2008; Busch et al., 2009, 2010). Depletion of macrophages reportedly prevents axonal retraction after spinal cord injury (Horn et al., 2008). We also observed that Taxol treatment strongly, but only transiently, reduced the number of macrophages at the lesion site 1 and 3 d after crush. These data suggest that Taxol compromised the migration of cells, including macrophages, a finding that has also been shown by other studies (Ogasawara et al., 2001; Maranhão et al., 2008). The finding that macrophages may function as a barrier at the injury site opens the possibility that this reduction by Taxol may have also partially contributed to the improvement of axon regeneration.
In conclusion, the results of the current study demonstrate that the local application of Taxol enables RGCs to regenerate axons beyond the injury site of the optic nerve and that it markedly enhances the outcome of regeneration after additional stimulation by astrocyte-derived CNTF and LIF after lens injury. Because Taxol is an approved drug for treating humans, it is a promising candidate for the treatment of patients suffering, not only from optic nerve injury but potentially also from stroke or other traumatic CNS injuries. In fact, previously published data show that the systemic treatment of rats with Taxol was associated with an improved functional outcome after spinal cord injury, although it is not clear whether these effects were the result of improved axonal regeneration or caused by other mechanisms (Perez-Espejo et al., 1996). Optimization of modalities of drug application may further improve the beneficial outcome of Taxol on axon regeneration. Lens injury effects can be mimicked by intravitreal injections of Pam3Cys or the continuous release of CNTF (Leaver et al., 2006a,b; Hauk et al., 2010) and can be further enhanced by a coapplication of drugs elevating intracellular levels of cAMP (Cui et al., 2003; Park et al., 2004; Müller et al., 2007), raising hope that such combinatorial treatments with Taxol may promote even stronger regeneration in the optic nerve than has been achieved so far. Finally, in a modified form, these combinatorial treatments may be suitable as a treatment for other parts of the injured CNS as well, such as the injured spinal cord or brain.
Footnotes
This work was supported by the Deutsche Stiftung Querschnittlähmung. We thank Dr. Adrienne Müller for useful comments on this manuscript and Sabrina Winter for technical support.
- Correspondence should be addressed to Dr. Dietmar Fischer, Department of Neurology, Heinrich Heine University Düsseldorf, Life Science Center, Merowinger Platz 1a, 40225 Düsseldorf, Germany. dietmar.fischer{at}email.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}