

Abstract
Dendritic spines are postsynaptic structures that receive excitatory synaptic input from presynaptic terminals. Actin and its regulatory proteins play a central role in morphogenesis of dendritic spines. In addition, recent studies have revealed that microtubules are indispensable for the maintenance of mature dendritic spine morphology by stochastically invading dendritic spines and regulating dendritic localization of p140Cap, which is required for actin reorganization. However, the regulatory mechanisms of microtubule dynamics remain poorly understood. Partitioning-defective 1b (PAR1b), a cell polarity-regulating serine/threonine protein kinase, is thought to regulate microtubule dynamics by inhibiting microtubule binding of microtubule-associated proteins. Results from the present study demonstrated that PAR1b participates in the maintenance of mature dendritic spine morphology in mouse hippocampal neurons. Immunofluorescent analysis revealed PAR1b localization in the dendrites, which was concentrated in dendritic spines of mature neurons. PAR1b knock-down cells exhibited decreased mushroom-like dendritic spines, as well as increased filopodia-like dendritic protrusions, with no effect on the number of protrusions. Live imaging of microtubule plus-end tracking proteins directly revealed decreases in distance and duration of microtubule growth following PAR1b knockdown in a neuroblastoma cell line and in dendrites of hippocampal neurons. In addition, reduced accumulation of GFP-p140Cap in dendritic protrusions was confirmed in PAR1b knock-down neurons. In conclusion, the present results suggested a novel function for PAR1b in the maintenance of mature dendritic spine morphology by regulating microtubule growth and the accumulation of p140Cap in dendritic spines.
Introduction
Dendritic spines are postsynaptic protrusions that regulate synaptic transmission by receiving excitatory synaptic signals from presynaptic terminals. Dendritic spine morphology has been shown to strongly correlate with functions of neural circuits (Bourne and Harris, 2008; Bhatt et al., 2009; Holtmaat and Svoboda, 2009). The shapes and sizes of dendritic spines dynamically change in mature neurons and are accompanied by synaptic plasticity (Trachtenberg et al., 2002; Matsuzaki et al., 2004; Yuste and Bonhoeffer, 2004). In addition, abnormal morphology of filopodia-like protrusions occurs in several mental disorders, such as fragile X syndrome, an inherited form of mental retardation (van Spronsen and Hoogenraad, 2010).
Accumulating evidence has demonstrated that actin and its regulatory proteins are responsible for the regulation of dendritic spine morphology (Hering and Sheng, 2003; Li et al., 2004; Newey et al., 2005). However, the role of microtubules in dendritic spine morphology remains poorly explored, although recent studies have suggested that mature spine morphology is maintained by transient microtubule invasion into dendritic spines (Gu et al., 2008; Hu et al., 2008; Jaworski et al., 2009). In addition, inhibition of microtubule dynamics by a microtubule-disrupting agent or EB3 knockdown (KD) has been shown to transform dendritic spines into filopodia-like protrusions (Jaworski et al., 2009), which highlights the importance of microtubule dynamics in dendritic spine morphology. These results raised a new question about how microtubule dynamics are regulated in maintaining dendritic spines.
Partitioning-defective 1b (PAR1b)/MARK2 (microtubule affinity-regulating kinase 2), a serine/threonine protein kinase, was originally identified due to its ability to phosphorylate microtubule-associated proteins (MAPs) and to induce detachment from microtubules (Matenia and Mandelkow, 2009). PAR1b activity results in microtubule array breakdown when overexpressed in non-neuronal cultured cells, such as CHO cells (Drewes et al., 1997). PAR1b has been identified as an essential kinase for cell polarity, and microtubules are thought to mediate many of its effects on cell polarity (Matenia and Mandelkow, 2009). In neurons, PAR1b regulates axon–dendrite specification (Chen et al., 2006; Yoshimura et al., 2010), neurite elongation (Chen et al., 2006; Terabayashi et al., 2007), and neuronal migration (Sapir et al., 2008). In addition, PAR1b knock-out mice exhibit impaired spatial learning and memory (Segu et al., 2008). These results suggest that PAR1b regulates microtubule dynamics, which is required for dendritic spine maturation.
Results from the present study demonstrated that PAR1b localizes to dendritic shafts and spines in hippocampal neurons, and plays an essential role for dendritic spine maintenance in mature neurons. In addition, microtubule dynamics were analyzed in hippocampal neurons and Neuro 2a cells, demonstrating that PAR1b knockdown reduced the distance and duration of microtubule growth, providing the first direct evidence for PAR1b activity in maintaining microtubule growing state. Dendritic spine accumulation of green fluorescent protein (GFP)-p140Cap, which binds to the plus-end of microtubules and requires the presence of dynamic microtubules, was also reduced in PAR1b-depleted neurons. In conclusion, these combined results suggested that PAR1b maintains mature dendritic spine morphology by regulating microtubule growth.
Materials and Methods
Antibodies and reagents.
The following antibodies were used in the present study: chicken anti-GFP (Aves Labs); rabbit anti-GFP (Santa Cruz Biotechnology); mouse anti-PSD95 (Millipore); rabbit anti-synapsin1 (Abcam); rabbit anti-tau (Dako); rabbit anti-tau [pS262] (Invitrogen); rabbit anti-tRFP (turbo RFP; Evrogen); mouse omni-probe (Santa Cruz Biotechnology); mouse anti-α-tubulin (Sigma); mouse anti-β-actin (Sigma); mouse anti-EB-1 (Cell Signaling Technology); rat anti-EB3 (Abcam); rabbit anti-SNIP/p140Cap (Cell Signaling Technology); rabbit anti-PAR1b (Suzuki et al., 2004); mouse anti-MAP2 (HM1, Sigma); and Alexa488-, Alexa568-, and Alexa638-conjugated secondary antibodies (Invitrogen). Mouse anti-PAR1b monoclonal antibody was generated using the C-terminal fragment of human PAR1b (C2: 484–787 aa) fused with GST.
Duplex siRNAs were purchased from Sigma-Genosys, and nucleotide sequences were as follows: PAR1b knockdown Q3, sense, 5′-GAGGUAGCUGUGAAGAUCA-3′ and antisense; 5′-UGAUCUUCACAGCUACCUC-3′; and #1, sense, 5′-CUGAAUGAACCUGAAAGCAAA-3′ and antisense, 5′-TTTGCTTTCACCUUCAUUCAG-3′. For negative control, we used AllStars negative control siRNA (Qiagen).
Expression constructs.
For the generation of PAR1b small hairpin RNA (shRNA) constructs, pEB-Super-gfp (Masuda-Hirata et al., 2009) was used. In several of the experiments, pEB-Super-tRFP was used, which expresses turboRFP (Evrogen) simultaneously with an shRNA. The target sequences were 5′-GAGGTAGCTGTGAAGATCA-3′ (Q3) or 5′-GAATGAACCTGAAAGCAAA-3′ (#1). The 170 kDa cytoplasmic linker protein (CLIP170) cDNA, a kind gift from M. Iino (University of Tokyo, Tokyo, Japan), was cloned into the pEGFP-C vector (Clontech). The pGW1-GFP-p140Cap vector was used for GFP-p140Cap expression (Jaworski et al., 2009), and the GFP-MAP2b expression vector was generously provided by Gary A. Wayman (Washington State University, Pullman, WA). For ectopic expression of PAR1b, the human PAR1b, which is resistant to RNA interference (RNAi) sequences of Q3, was subcloned into the pEB6CAG vector (Suzuki et al., 2004) with IRES-GFP (Clontech). The PAR1b sequence was altered as follows: gaggtagctgtcaaaatta (italic nucleotides were substituted). The pEGFP-N2 vector was used for GFP expression (Clontech).
Cell culture and transfection.
Animal care and handling in experiments were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee at Yokohama City University, School of Medicine. Primary hippocampal neuronal cultures were established from ICR mouse embryonic hippocampal tissue (embryonic day 16) of either sex. Neurons were seeded on coverslips coated with poly-dl-ornithine (50 μg/ml, Sigma) at a density of 3 × 104 cells/cm2. Hippocampal cultures were plated in medium that contained DMEM (Invitrogen) with 10% fetal bovine serum (FBS), 4 mm glutamine (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin (Lonza). After 1 d, media were replaced with Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) and 2 mm glutamine. Neuro 2a cells were grown in Eagle's MEM supplemented with 10% FBS, 4 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. For differentiation, the medium was replaced with Eagle's MEM supplemented with 5% FBS, 2 μm all-trans-retinoic acid (Wako), 4 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. For live imaging, Neuro 2a cells were seeded on glass-based dishes (Iwaki).
Hippocampal neurons were transfected using Lipofecatamine 2000 (Invitrogen) for morphometric analyses. For biochemical assays, hippocampal neurons were transfected using amaxa Nucleofector (Lonza) according to manufacturer's instructions. For siRNA oligo transfection, Neuro 2a cells were transfected using siLentFect lipid reagent (Bio-Rad). At 24 h after incubation at 37°C in 5% CO2, the second transfection was performed. For biochemical assay and live imaging, cells were harvested at 2–4 d after transfection.
To establish GFP-CLIP170 stable transformants, Neuro 2a cells were transfected with the GFP-CLIP170 expression vector using Lipofectamine LTX according to manufacturer's instructions. Cells were cloned with a limiting dilution in 96 well plates in culture medium supplemented with 500 μg/ml G418 (Promega). After culturing for 2–4 weeks, wells containing a single colony were chosen for further study.
EB3-GFP expression in hippocampal neurons.
For EB3-GFP analysis, primary hippocampal neurons were transfected immediately after isolation with a nonsilencing control (NC) vector or PAR1b knock-down vector using amaxa Nucleofector. The cells were then seeded on glass-bottom culture dishes (MatTek) coated with poly-dl-ornithine. To express EB3-GFP in neurons, the Semliki Forest virus (SFV)-mediated gene delivery system was used as previously described (Jaworski et al., 2009). Cultured hippocampal neurons were infected at DIV8–DIV9 following addition of concentrated SFV infectious replicons to culture medium at 4 h before live cell imaging.
Image acquisition, processing, and morphometric analysis.
For live cell imaging, cells were placed into a closed, heat-controlled chamber (Leica Microsystems) with a CO2 supply system (Tokken) on an inverted microscope (DM IRE2, Leica Microsystems) equipped with an automated filter wheel exchanger (Ludl Electronic Products), a spinning-disc confocal system CSU10 (Yokogawa), a cooled CCD camera (ORCA-ER, Hamamatsu Photonics), Ar/Kr laser (Melles Griot), and an objective lens (HCX PL APO NA 1.40 100×, Leica Microsystems). Time-lapse image acquisitions were performed in the vicinity of glass to minimize the effect of comet movement along the z-axis. Images were acquired every 1 s using MetaMorph software (Molecular Devices). For quantification of GFP-CLIP170 and EB3-GFP movement, particles were manually tracked using the object tracking application of ImageJ software; results were exported to Excel software (Microsoft) and analyzed using StatView software (SAS Institute). GFP-CLIP170 and EB3-GFP particles were tracked in the cell body of Neuro 2a cells and in the dendrites of hippocampal neurons, respectively. More than 90 comets from more than nine cells were analyzed for each condition in each cell.
Immunofluorescence images were acquired using the disc confocal system as described above. Series stacks were collected from the bottom to the top covering all dendrites and protrusions, with a 0.5 μm step size in a visual field. The resulting images (6–12 images, depending on the sample) were then reconstructed according to z-stack projections of the maximum intensity. Microtubule images were acquired with a fluorescent microscope (Apotome, Carl Zeiss) equipped with a disc confocal system as described above, except a cooled CCD camera (ORCA-R2, Hamamatsu Photonics) and an objective lens (Plan-Apochromat NA 1.4 63×, Carl Zeiss) were also used. For dendritic spine morphometric analysis, hidden protrusions, which protruded toward the back or front of dendritic shafts, were excluded. Acquisition parameters were kept constant for all scans in the same experiment. In each experiment, >500 dendritic protrusions from randomly selected cells (more than seven neurons) were manually traced for each condition. The length and width of each protrusion were measured using MetaMorph software. The GFP-p140Cap localization index in Figure 6 was calculated as follows: an appropriate region of dendritic shaft (10 μm long) containing protrusions was selected, and the averaged GFP signal intensity of the protrusions was divided with intensity from the shaft region. To negate the effect of spine volume alteration, the above ratio was normalized to the ratio of the coexpressing tRFP signal intensity, which was similarly calculated. Regions were randomly selected (more than nine neurons/condition) from immunofluorescence images, and fluorescence was measured using MetaMorph software.
Immunofluorescence.
Cells were seeded onto coverslips and fixed with 3% paraformaldehyde in PBS for 10 min at room temperature, followed by two PBS wash steps and permeabilization with 0.1% Triton X-100 in PBS for 5 min at room temperature. The cells were then washed and soaked in blocking solution (PBS containing 10% calf serum) for 1 h at 4°C before incubation overnight at 4°C with appropriate primary antibodies diluted in 10 mm Tris-HCl, pH 7.5, containing 150 mm NaCl, 0.01% (v/v) Tween 20, and 0.1% (w/v) BSA. Microtubule staining was performed as previously described (Black et al., 1996). Cells were rinsed with PEM (80 mm PIPES, 5 mm EGTA, 1 mm MgCl2, pH 6.8) and then fixed and extracted with PEM containing 0.5% (w/v) NP-40 and 0.3% glutaraldehyde for 10 min, followed by two PBS wash steps and permeabilization with 0.5% Triton X-100 in PBS for 10 min at room temperature. To visualize mitochondria, 500 nm Mito Tracker Red CMX Ros (Invitrogen) was loaded into hippocampal neurons for 30 min at 37°C in 5% CO2 before immunofluorescence staining.
Results
PAR1b distributes in dendrites of mature neurons
Because endogenous PAR1b expression remains poorly understood in mature neurons, expression and subcellular localization in dissociated mouse hippocampal cultures was analyzed (Fig. 1). Expression of two PAR1b isoforms (∼80 and 90 kDa, respectively) was observed in the adult mouse brain and in hippocampal cultures (Fig. 1A). Expression levels increased with neuronal maturation; compared with developing neurons at DIV4 and DIV7, mature neurons at DIV14 and DIV21 expressed higher levels of PAR1b. Similar features were also verified by PAR1b monoclonal antibody, which recognized only the larger isoforms.

PAR1b is localized in dendritic shafts and dendritic spines in mature neurons. A, PAR1b expression in MDCK cells (lane 1), mouse adult whole brain (lane 2), and dissociated hippocampal cultures from DIV4 to DIV21 (lanes 3–6). Western blot analyses were performed using the indicated antibodies. EB1 and EB3 were used as expression markers for young and mature neurons, respectively. B, PAR1b knockdown (KD) in hippocampal young neurons. Mouse neurons were transfected with a NC or PAR1b KD vector, immediately following isolation and were subjected to Western blot analysis at DIV5. Analyses were performed with anti-PAR1b (polyclonal), anti-phospho Ser262 tau, anti-tau, and anti-actin antibodies. A stepwise dilution (11, 33, and 100%) of NC samples was loaded in lanes 1–3. Quantification of PAR1b knockdown efficiency (middle) and changes in tau Ser262 phosphorylation (right) are also shown. Values represent mean ± SEM (n = 4). C, Representative immunofluorescent image of mouse hippocampal neurons (DIV14) expressing PAR1b (white) and GFP (green). Neurons were transfected with the indicated knock-down vectors at DIV7. Transfected cells (asterisks) are identified by GFP signals simultaneously expressed from the knock-down vectors. D, High-magnification images of dendrite regions. Neurons are coimmunostained for PAR1b (white), GFP (green), and PSD95 (red). Arrows represent PSD95-positive areas, one of which is magnified in the inset. Scale bars, 10 μm.
When mouse neurons were electroporated with PAR1b knock-down vectors immediately following isolation, Western blot analysis of neurons at DIV5 revealed a 37% reduction in both PAR1b isoforms (Fig. 1B). Because the transfection efficiency estimated by fluorescent signals of coexpressing GFP was ∼60% (data not shown), these results suggested that PAR1b was knocked down efficiently in single neurons. Consistent with previous findings that PAR1b phosphorylates tau Ser262, and thereby inhibits its microtubule-binding activity (Biernat et al., 1993; Drewes et al., 1997), a decrease in Ser262 phosphorylation of tau was detected in PAR1b knock-down neurons, without any change in the total tau expression (Fig. 1B, right).
In Figure 1C, mouse hippocampal neurons were transfected with knock-down vectors at DIV7 and stained at DIV14 to analyze mature neurons (Fig. 1C,D). GFP coexpression allowed for the identification of transfected neurons and the visualization of cellular morphology. Immunofluorescent signal specificity was confirmed by significantly decreased PAR1b signals in cells expressing PAR1b shRNA (Fig. 1C, right, asterisks), but not in nonsilencing control (NC) shRNA cells (Fig. 1C, left panels, asterisks). PAR1b signals were observed as puncta within cell bodies and dendrites (DIV14) (Fig. 1C). Closer inspection revealed PAR1b distribution in dendritic shafts and concentration in dendritic spines, which contained the postsynaptic marker PSD95 (Fig. 1D, arrows). These subcellular PAR1b signals were also confirmed by PAR1b monoclonal antibody (data not shown). These results revealed PAR1b expression in dendritic shafts and spines.
PAR1b knockdown affects dendritic spine maintenance in hippocampal neurons
Compared with mature dendritic spines with mushroom-like morphology in control cells, DIV14 neurons subjected to PAR1b depletion for 7 d exhibited a dramatic decrease in mature dendritic spines (Fig. 1D). These results suggested that PAR1b was required for dendritic spine morphogenesis. To determine whether PAR1b is also required for the maintenance of normally developed dendritic spine morphology in mature neurons, PAR1b knock-down vectors were introduced into neurons at DIV17, when cells had formed numerous dendritic spines (Zhang and Benson, 2001; Schratt et al., 2006; Jaworski et al., 2009). At DIV21, dendritic protrusions in control cells mainly consisted of mushroom-shaped spines (Fig. 2A). In contrast, PAR1b knock-down neurons exhibited significantly reduced numbers of mushroom-shaped spines and increased filopodia-like or small head protrusions. The morphological differences of dendritic protrusions were clearly demonstrated by quantification of protrusion width and length (Fig. 2B,C). PAR1b-depleted neurons exhibited a decreased mean width of protrusions by 35%, from 0.52 ± 0.02 to 0.34 ± 0.01 μm, as well as an increased mean length of protrusions by 28%, from 1.38 ± 0.10 to 1.90 ± 0.12 μm. In contrast, the number of protrusions was not altered by PAR1b knockdown (6.05 ± 0.78 vs 6.07 ± 0.86 protrusions/10 μm for control vs PAR1b knockdown, respectively) (Fig. 2C, right graph). Since a significant percentage of protrusions in PAR1b knock-down cells remained positive for the presynaptic marker synapsin1 (Fig. 2A) (PAR1b KD: 72.4 ± 6.2%; vs NC: 78.9 ± 4.1%), degenerated protrusions in PAR1b knock-down neurons were considered to have formed functional synapses at least once before PAR1b knockdown.

PAR1b knockdown affects dendritic spine maintenance. A, Representative images of hippocampal neurons (DIV21) transfected with a NC vector (top panels) or PAR1b knock-down vector (PAR1b KD, bottom panels) at DIV17. Synapsin1 signals (red) represent presynaptic terminals. B, Distribution of dendritic protrusion width (left panels) and length (right panels) in NC cells (top panels) and PAR1b KD cells (bottom panels). More than 500 dendritic protrusions from randomly selected cells (more than seven neurons) were analyzed for each condition. C, Quantification of mean width, length, and density of dendritic protrusions in each cell. Bars represent mean ± SEM (n > 7). #p < 0.0001, *p < 0.01 by Student's t test. Scale bar, 10 μm.
The specificity of the effects of PAR1b knockdown was verified by an additional PAR1b knock-down vector (#1) (data not shown), as well as by results demonstrating that ectopic PAR1b expression restored mature dendritic spine morphology (Fig. 3). The PAR1b knock-down vector and a His-tagged, RNAi-resistant-PAR1b expression vector were coexpressed in mature, hippocampal neurons at DIV17; immunofluorescence was subsequently assessed at DIV21. tRFP and GFP were used as reporter proteins for the PAR1b knock-down vector and His-tagged-PAR1b expression vector, respectively (see Materials and Methods). Coexpression of RNAi-resistant PAR1b significantly restored the morphology of dendritic spines to the control level (Fig. 3). These results unequivocally revealed a novel and essential function of PAR1b in the maintenance of dendritic spine morphology in mature neurons.

PAR1b overexpression rescues the effect of PAR1b knockdown in dendritic spine maintenance. A, Representative images of hippocampal neurons (DIV21) cotransfected with PAR1b knock-down vector and GFP expression vector (top panels) or His-tagged-PAR1b expression vector (bottom panels) at DIV17. tRFP and GFP are simultaneously expressed from the PAR1b knock-down vector and His-tagged PAR1b expression vector, respectively. Nonsilencing control neurons were also examined as controls (data not shown; see below). B, Distribution of dendritic protrusion width (left panels) and length (right panels) in NC (top panels), PAR1b KD (middle panels), and PAR1b-rescued cells (bottom panels), which were analyzed similarly to those in Figure 2B. C, Quantification of mean width, length, and density of dendritic protrusions in each cell. Values represent mean ± SEM (n > 7). #p < 0.0001, *p < 0.01 by Student's t test. Scale bar, 10 μm.
PAR1b knockdown leads to decreased microtubule growth in differentiated neuroblastoma cells
Recent studies have shown that dynamic microtubule properties are essential for dendritic spine maturation and maintenance (Gu et al., 2008; Hu et al., 2008; Jaworski et al., 2009). Since PAR1b controls cellular functions such as cell polarity via the regulation of microtubules (Cohen et al., 2004; Chen et al., 2006), the above-mentioned results suggested that PAR1b plays an essential role in mature dendritic spine maintenance by positively regulating microtubule dynamics. However, very little direct evidence exists with regard to PAR1b activity regulating microtubule dynamics. Therefore, the dynamic properties of microtubules in PAR1b knock-down cells were analyzed using live-imaging techniques.
Initially, microtubule dynamics were assessed in differentiated Neuro 2a cells, a mouse neuroblastoma cell line (Morrison et al., 2002), by monitoring movements of a CLIP170, which is a microtubule plus-end tracking protein, +TIP. Because +TIPs localize microtubule plus-ends only during the growing state (Akhmanova and Steinmetz, 2008), the growing ends of microtubules can be traced by monitoring GFP-CLIP170 movements. Because transient transfection resulted in significantly altered expression of GFP-CLIP170, which led to the difficulties in obtaining reproducible mobility results (Perez et al., 1999; Ligon et al., 2003), a Neuro 2a cell line was established that stably expressed GFP-CLIP170 at a low level. The cells were transfected with siRNA oligonucleotides for control (NC) or PAR1b knockdown (#1 and Q3). One day later, neural differentiation was induced by serum deprivation and retinoic acid application. Because immunoblotting analysis revealed >90% knockdown efficiency, almost all cells were assumed to be transfected with siRNA oligonucleotides, and PAR1b expression was effectively knocked down (Fig. 4A). When control cells were subjected to live imaging, dispersed comet-like GFP-CLIP170 signals, which were several micrometers in length, were observed as previously reported (Perez et al., 1999). The GFP-CLIP170 comets suddenly appeared, moved in a fixed direction for several micrometers, and subsequently disappeared (Fig. 4B). In the PAR1b knock-down cells, the distance of comet movement was significantly reduced. To quantify the effects of PAR1b on microtubule growth, the appearance and disappearance of each GFP-CLIP170 comet was tracked (Fig. 4C, red lines). In the control cells, the mean length of comet trajectory was 5.82 ± 0.86 μm. However, in PAR1b knock-down cells, the length decreased to 2.69 ± 0.26 μm (Fig. 4C,D, left). On the other hand, comet velocity remained unchanged between NC and PAR1b KD cells (NC: 0.31 ± 0.02 μm s−1; PAR1b KD (Q3): 0.30 ± 0.01 μm s−1) (Fig. 4D, right). The effects of PAR1b knockdown on microtubule growth distance and comet velocity were verified by an additional PAR1b siRNA oligo (#1) (2.93 ± 0.21 μm and 0.35 ± 0.01 μm s−1, respectively) (Fig. 4C,D). These results suggested that PAR1b knockdown severely impaired microtubule growth without significant effects on microtubule polymerization activity.

PAR1b knockdown causes defects in microtubule growth in Neuro 2a cells. A, Western blot analysis of total cell protein from Neuro 2a cells transfected with NC or PAR1b KD (#1 and Q3) siRNA oligonucleotides. A stepwise dilution (11, 33, and 100%) of NC protein was loaded into lanes 1–3. B, Representative time-lapse images of GFP-CLIP170 in Neuro 2a cells. Left, GFP-CLIP170 distribution at time 0. Scale bar, 10 μm. Right, Successive images every 2 s of the cropped region in the left panel (red box) are magnified. The arrow indicates CLIP170 comet movement, which appeared at 2 s and disappeared at 16 s. Scale bar, 1 μm. C, Representative images of CLIP170 comet trajectories (red lines) in differentiated Neuro 2a cells transfected with the indicated siRNA oligonucleotides. Scale bar, 10 μm. D, Quantification of mean lengths of GFP-CLIP170 comet trajectories (left) and mean velocities of GFP-CLIP170 comet movements (right). More than 90 comets from more than nine cells were analyzed for each condition. Mean ± SEM. *p < 0.01 by Student's t test.
PAR1b knockdown results in reduced microtubule growth in hippocampal neurons
The live-imaging analysis was subsequently extended to hippocampal neurons. For this purpose, an SFV-mediated gene delivery system was used, which monitored movement of EB3, a major member of +TIPs in hippocampal mature neurons (Jaworski and Hoogenraad, 2009). To obtain a high transfection efficiency of knock-down vectors in this assay, neurons were transfected with the vectors immediately after isolation, followed by infection with SFV encoding EB3-GFP at DIV8–DIV9. Neurons expressed EB3-GFP at 4 h after infection and exhibited comet-like movement of EB3-GFP particles along the dendritic shafts in distal or proximal directions. The length of EB3-GFP comet trajectories was shortened by PAR1b knockdown, as observed for GFP-CLIP170 in Neuro 2a cells (Fig. 5A). In control cells, the mean length of the EB3-GFP comet trajectory was 7.08 ± 0.66 μm, in contrast to 2.96 ± 0.33 μm in PAR1b knock-down cells (Fig. 5B, left). On the other hand, the mean comet velocity did not change significantly following PAR1b knockdown (NC: 0.20 ± 0.01 μm s−1; PAR1b KD: 0.18 ± 0.01 μm s−1) (Fig. 5B, right). These results demonstrated that, as we observed in Neuro 2a cells, PAR1b knockdown affected microtubule growth without reducing polymerization activity in hippocampal neuron dendrites. Immunofluorescent staining demonstrated that polymerized microtubule levels were not significantly affected in dendrites of PAR1b knock-down neurons (Fig. 5C) (relative signal intensity in dendrites: NC: 1 ± 0.12 vs PAR1b KD: 0.88 ± 0.10). This might indicate that tubulin polymerization in mature neurons is nearly at the steady state.

PAR1b knockdown affects microtubule growth in hippocampal neuronal dendrites. A, Representative images of EB3-GFP comet trajectories in DIV9 hippocampal neurons transfected with NC (top panels) or PAR1b KD vector (bottom panels). EB3-GFP comet trajectories in neuronal dendrites (white lines, left panels). Expression of EB3-GFP is superimposed with EB3-GFP comet trajectory images (EB3-GFP, middle panels). Transfected cells are identified by tRFP signals simultaneously expressed in knock-down vectors (red, right panels). Scale bar, 10 μm. B, Quantification of mean lengths of EB3-GFP comet trajectories (left) and mean velocities of EB3-GFP comet movements (right). More than 90 comets from more than nine cells were analyzed for each condition. Mean ± SEM. #p < 0.0001 by Student's t test. C, Representative images of polymerized tubulin in hippocampal neurons (DIV21) transfected with NC vector (top panels) or PAR1b KD vector (bottom panels) at DIV17. Neurons were coimmunostained for anti-α-tubulin antibody (MT) and anti-GFP antibody. Scale bars, 10 μm.
PAR1b knockdown impairs p140Cap localization in dendritic spines
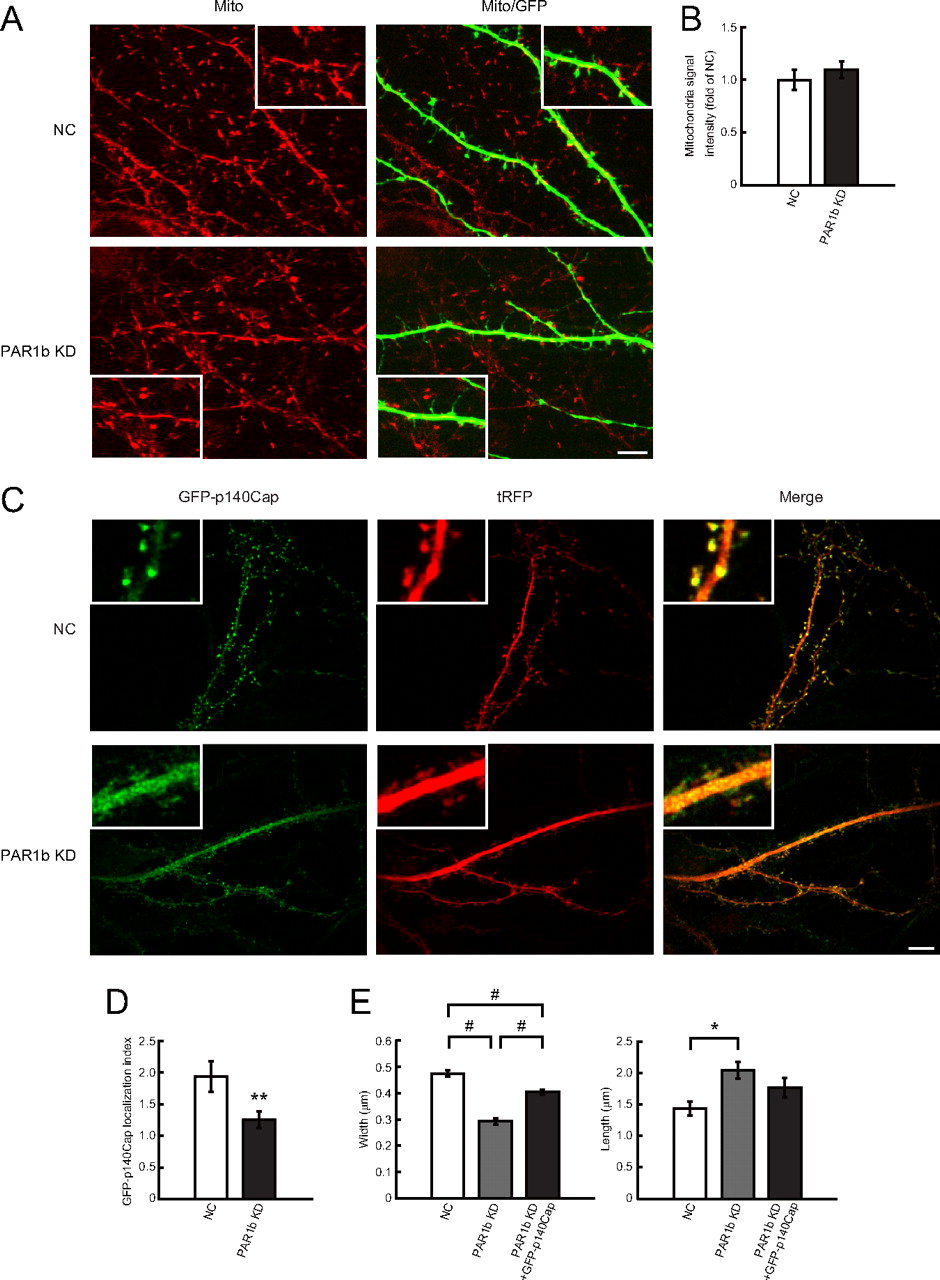
Thies and Mandelkow (2007) reported that tau overexpression in cultured hippocampal neurons induces mitochondrial mislocalization, thereby resulting in the disappearance of dendritic spines. They also demonstrated that co-overexpression of PAR1b can compensate for these effects. Figure 6, A and B, shows that PAR1b knock-down neurons did not alter mitochondrial localization or density in dendrites, which suggested that the spine degeneration phenotype due to PAR1b knockdown is the result of different mechanisms. In addition, because mitochondria are transported along microtubules, these results support the notion that PAR1b knockdown does not significantly affect steady-state microtubule organization in dendrites.

PAR1b knockdown does not affect mitochondrial distribution, but does affect p140Cap localization, in mature neurons. A, Representative images of mitochondrial distribution in dendrites, as assessed by Mito Tracker Red (Mito) in DIV19 hippocampal neurons. Neurons were transfected with NC vector (top panels) or PAR1b KD vector (bottom panels) at DIV14. Scale bar, 10 μm. B, Mitochondrial localization of PAR1b knock-down neurons compared with NC neurons in dendrite regions. Values represent mean ± SEM (n = 9). C, Representative images of GFP-p140Cap localization in mature hippocampal neurons (DIV21). GFP-p140Cap-expressing vector was introduced in neurons in conjunction with NC vector or PAR1b KD vector at DIV17. Each knock-down vector simultaneously expressed tRFP. Immunofluorescence was assessed with anti-GFP antibody (green, left panels) and anti-tRFP antibody (tRFP, middle panels). D, Assessment of GFP-p140Cap accumulation in dendritic protrusions. Ratios of GFP-p140Cap intensities in dendritic protrusions versus dendritic shaft normalized with tRFP signals: 1.94 ± 0.24 in control cells and 1.26 ± 0.16 in PAR1b knock-down cells (see Materials and Methods for details). E, Quantification of mean width and length of dendritic protrusions in each cell. Values represent mean ± SEM (n = 9). #p < 0.0001, **p < 0.002, *p < 0.01 by Student's t test. Scale bar, 10 μm.
Jaworski et al. (2009) demonstrated that dynamic microtubules play an indispensable role in spine morphology by regulating spine accumulation of the EB3-binding protein p140Cap, which controls cortactin function and regulates actin organization in dendritic spines. Therefore, the localization of p140Cap in PAR1b knock-down neurons was analyzed by transiently expressing GFP-p140Cap in DIV17 mature neurons to assess the effects of PAR1b knockdown at DIV21. Similar to previous results, GFP-p140Cap expression was concentrated in dendritic spines in control cells (Fig. 6C) (Jaworski et al., 2009); a comparison of coexpressing tRFP signals suggested an approximately twofold accumulation of GFP-p140Cap in the spines (Fig. 6D). In contrast, GFP-p140Cap exhibited a decreased accumulation (∼1.3-fold) in spines of PAR1b-knock-down neurons (Fig. 6C,D). In these neurons, GFP-p140Cap expression was robustly distributed in dendritic shafts, which suggested that GFP-p140Cap accumulation in dendritic spines, rather than transport within dendritic shafts, was specifically impaired in PAR1b knock-down cells. Because p140Cap plays a critical role in dendritic spine maintenance (Jaworski et al., 2009), defects in p140Cap accumulation in dendritic spines could be responsible for the spine morphology defects observed in PAR1b knock-down neurons. This is supported by the results that GFP-p140Cap overexpression partially restored the mushroom-like morphology of dendritic spines in PAR1b knock-down neurons (Fig. 6E).
Discussion
Recent studies have established the critical importance of microtubule dynamics in mature dendritic spine morphology. However, further studies are needed to determine the regulation of microtubule dynamics for this activity. Results from the present study demonstrated that the cell polarity-regulating protein kinase PAR1b, which has been implicated in microtubule regulation, plays an essential role in mature dendritic spine morphology, at least in part by supporting the dynamic nature of microtubules. In mature neurons, PAR1b distributed in dendritic shafts and in dendritic spines. PAR1b knockdown in mature neurons resulted in a decreased number of mushroom-shaped dendritic spines and increased filopodia-like protrusions, as observed in EB3-depleted neurons or neurons treated with a microtubule-disrupting agent. PAR1b knockdown reduced the distance and duration of microtubule growth in hippocampal neurons. In addition, PAR1b knockdown resulted in delocalization of GFP-p140Cap from dendritic spines to dendritic shafts in neurons. This delocalization of p140Cap is one of the causes of the spine morphology defects in PAR1b knock-down neurons, since overexpression GFP-p140Cap partially rescued dendritic spine maturation. At present, it is not possible to exclude a possibility that PAR1b directly regulates p140Cap accumulation in spines. However, considering that dynamic microtubules are required for the spine localization of p140Cap (Jaworski et al., 2009), the present results are consistent with the notion that PAR1b indirectly affects spine accumulation of p140Cap via its activity regulating microtubule growth. Together, these results demonstrated that, in addition to regulation during the early stages of neuronal development, the microtubule-regulating activity of PAR1b played a critical role in the maintenance of mature dendritic spine morphology. Thies and Mandelkow (2007) have demonstrated that overexpression of PAR1b also induced filopodia-like dendritic protrusions as observed in PAR1b knock-down neurons. This may indicate that strictly regulated microtubule dynamics are essential for maintenance of mature spine morphology. The present study focused on the role of PAR1b in mature neurons. However, defects in dendritic spine morphology were also observed in DIV14 neurons transfected with the knock-down vector at DIV7 (Fig. 1D). These results suggested that PAR1b regulated not only dendritic spine maintenance, but also dendritic spine formation. Behavioral studies of knock-out mice have suggested that PAR1b is involved in learning and memory (Segu et al., 2008). Therefore, the present results might provide a molecular basis for these phenotypes, which could be due to impaired synaptic function resulting from morphological defects in dendritic spines.
Previous studies have used microtubule immunostaining to indirectly address the effect of PAR1b on microtubule stability, and results from these studies revealed a cell type-dependent, complicated activity of PAR1b on microtubules. PAR1b overexpression has been shown to disrupt the microtubule array in cultured fibroblastic cells, such as CHO cells (Drewes et al., 1997); however, overexpression increases particular microtubule architectures (lateral microtubules) in epithelial cells (Cohen et al., 2004) and enhances the formation of microtubule-dependent neurites in neuronal cells (Biernat et al., 2002; Timm et al., 2003). The loss of PAR1b activity does not increase the amount of polymerized microtubules, but rather inhibits neurite extension (Timm et al., 2003) or disrupted microtubules in cells, such as epithelial cells (Cohen et al., 2004). Some of these results are thought to be the result of PAR1b activity-induced increases in microtubule dynamics (Matenia and Mandelkow, 2009), which could result in increased or decreased microtubule arrays, depending on the cell state (Erickson and O'Brien, 1992). However, very little direct evidence has been provided with regard to this PAR1b activity regulating microtubule dynamics. The present study analyzed the effect of PAR1b knockdown on microtubule dynamics in living cells, demonstrating that PAR1b depletion resulted in less continuous growing of the microtubule plus-end without affecting growing velocity (Fig. 3). This provides the first direct evidence that PAR1b positively regulates a dynamic property of microtubules in living cells. Previously, Sapir et al. (2008) demonstrated that PAR1b knockdown in cultured hippocampal neurons results in increased amounts of stabilized microtubules (detyrosinated α-tubulin) (Sapir et al., 2008), which was consistent with the notion that PAR1b is required for maintaining dynamic microtubules (Matenia and Mandelkow, 2009). However, the imaging data suggested that PAR1b knockdown results in longer EB3 durations on microtubule plus-ends, which was inconsistent with the present results and was unanticipated with the predicted function of PAR1b to increase microtubule dynamics. Since EB3 promotes microtubule growth by suppressing catastrophes (Komarova et al., 2005), relatively high expressions of EB3-GFP induced by transient transfection might perturb the effects of PAR1b knockdown (Sapir et al., 2008).
The present results from live imaging of PAR1b knock-down cells suggest that PAR1b suppresses the transition frequency of microtubules dynamics from the growing state to the pausing and/or shrinking state without affecting the polymerization activity itself. Interestingly, previous studies demonstrated that elevated MAP expression, such as MAP2 and tau, also decreases the distance of microtubule growth in cultured cells, where microtubule polymerization is at steady state with no net gain of microtubule mass (Dhamodharan and Wadsworth, 1995; Bunker et al., 2004). Therefore, the present results may be reasonably explained by the well known PAR1b activity inhibiting microtubule binding of MAPs. This is consistent with our results that overexpression of a somatodendritic MAP, MAP2b, in hippocampal neurons significantly decreased dendritic spine density and spine morphology (data not shown). Future studies are needed, however, to completely resolve the underlying molecular mechanisms for the novel PAR1b activity in mature neurons.
PAR1b belongs to a set of evolutionarily conserved proteins termed the PAR–aPKC (partitioning-defective–atypical PKC) system, which plays essential roles in cell polarity in various biological contexts (Suzuki and Ohno, 2006). Interestingly, previous studies have demonstrated that other components of the PAR–aPKC system, such as PAR6 and PAR3, are also concentrated in dendritic spines; PAR6/aPKC and PAR3 exhibit indispensable roles in dendritic spine morphology by regulating the actin cytoskeleton via Rac and Rho small GTPases, respectively (Zhang and Macara, 2006, 2008). Dendritic spines are highly polarized local structures maintained by intricate interactions between dynamic cytoskeleton reorganization and vesicle transport. Together, the results from the present study suggest that dendritic spines are one of the regulatory targets of the general cell polarity machinery, the PAR–aPKC system. An increasing amount of evidence suggests that the mode of dendritic spine regulation by the PAR–aPKC system is distinct from modes observed in other polarizing events (Iden and Collard, 2008). However, it is highly plausible that components of the PAR–aPKC system mutually interact and regulate dendritic spine morphology, although further studies are needed to confirm this.
Notes
Supplemental material for this article is available at http://www-user.yokohama-cu.ac.jp/∼ohnos/data.html. This material has not been peer reviewed.
Footnotes
This work was supported by KAKENHI (S.O., A.S., and K.H.) and the “Establishment of Research Center for Clinical Proteomics of Post-Translational Modifications” as part of the Special Coordination Fund for Promoting Science and Technology “Creation and Innovation Centers for Advanced Interdisciplinary” from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; and Personal Fellowship from the Netherlands Organization for Scientific Research-Earth and Life Sciences (NWO-ALW-VICI), TOP grants from the Netherlands Organization for Health Research and Development (ZonMW-TOP), and European Science Foundation-European Young Investigators award (ESFEURYI) (C.C.H.). We thank Gary A. Wayman for the GFP-MAP2b expression vector, Masamitsu Iino for CLIP170 cDNA, and Akio Yamashita for helpful advice.
- Correspondence should be addressed to Atsushi Suzuki, Department of Molecular Biology, Yokohama City University Graduate School of Medical Science, 3-9 Fuku-ura, Kanazawa-ku, Yokohama 236-0004, Japan. abell{at}med.yokohama-cu.ac.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}