

Abstract
Traumatic brain injury (TBI) modulates several cell signaling pathways in the hippocampus critical for memory formation. Previous studies have found that the cAMP-protein kinase A signaling pathway is downregulated after TBI and that treatment with a phosphodiesterase (PDE) 4 inhibitor rolipram rescues the decrease in cAMP. In the present study, we examined the effect of rolipram on TBI-induced cognitive impairments. At 2 weeks after moderate fluid-percussion brain injury or sham surgery, adult male Sprague Dawley rats received vehicle or rolipram (0.03 mg/kg) 30 min before water maze acquisition or cue and contextual fear conditioning. TBI animals treated with rolipram showed a significant improvement in water maze acquisition and retention of both cue and contextual fear conditioning compared with vehicle-treated TBI animals. Cue and contextual fear conditioning significantly increased phosphorylated CREB levels in the hippocampus of sham animals, but not in TBI animals. This deficit in CREB activation during learning was rescued in TBI animals treated with rolipram. Hippocampal long-term potentiation was reduced in TBI animals, and this was also rescued with rolipram treatment. These results indicate that the PDE4 inhibitor rolipram rescues cognitive impairments after TBI, and this may be mediated through increased CREB activation during learning.
Introduction
Traumatic brain injury (TBI) is a devastating injury that often results in lifelong cognitive deficits (Zaloshnja et al., 2008). Over 70% of people who sustain a TBI report memory deficits (Lew et al., 2006). The hippocampus, a region necessary for declarative memory formation, is highly vulnerable to brain trauma even when not directly damaged (Maxwell et al., 2003; Tomaiuolo et al., 2004). In experimental models of TBI, maintenance of hippocampal long-term potentiation (LTP) is significantly impaired (Reeves et al., 1995; Sick et al., 1998; Schwarzbach et al., 2006; Norris and Scheff, 2009). This suggests that the molecular mechanisms underlying LTP maintenance are impaired after TBI and may contribute to the cognitive deficits seen in TBI survivors.
The molecular basis for the impairments in hippocampal LTP caused by TBI are unknown, and understanding these biochemical mechanisms could direct the development of pharmacological therapies to improve cognition after TBI. We and several other laboratories have reported that TBI activates several protein kinases involved in LTP acutely but transiently, recovering to noninjured levels within hours to days after TBI (Yang et al., 1993; Dash et al., 2002; Mori et al., 2002; Atkins et al., 2006; Atkins et al., 2007; Folkerts et al., 2007). Upstream of these protein kinases are transient changes in AMPA- and NMDA-type glutamate receptors. The AMPA-type glutamate receptor subunit 1 is phosphorylated at a CaMKII site and dephosphorylated at a PKA site 1 h after TBI (Atkins et al., 2006). NMDA receptor levels and phosphorylation change biphasically, but like AMPA-type receptors, return to noninjured levels 2 weeks after injury (Kumar et al., 2002; Biegon et al., 2004; Bigford et al., 2009). However, nearly all of these experiments have addressed only acute and subacute changes, but not chronic time points; consequently, a gap in our knowledge is what are the biochemical mechanisms that underlie the chronic memory deficits observed in people living with TBI.
Rehabilitative strategies for restoring cognitive functioning at chronic times after injury have had some clinical success. Typical rehabilitative treatments for chronic TBI survivors have focused on delivering neurotransmitter receptor agonists, neurotransmitter reuptake inhibitors, or drugs that enhance neurotransmitter release, to boost dopaminergic, cholinergic, and/or adrenergic signaling (Arciniegas and Silver, 2006; Warden et al., 2006; Wheaton et al., 2011). Although these clinical studies demonstrate promise in developing a pharmacological treatment to improve cognition after TBI, the lack of an understanding of the underlying biochemical mechanisms that cause impairments in hippocampal synaptic plasticity and learning after TBI impedes substantial progress in the field. In a previous study, we found that, although basal phosphorylated levels of cAMP-regulated element binding protein (CREB) have returned to noninjured levels by 2 weeks after TBI, activation of this signaling molecule was impaired in hippocampal slices (Atkins et al., 2009). These results suggest that treatments to increase CREB activation during LTP induction and learning may rescue TBI-induced cognitive deficits. In this study, we investigated whether rolipram, a phosphodiesterase 4 (PDE4) inhibitor that prevents the degradation of cAMP, would rescue hippocampal LTP and learning deficits after TBI.
Materials and Methods
TBI.
All experimental procedures were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Miami Animal Care and Use Committee. Adult male Sprague Dawley rats (275–300 g, Charles River Laboratories) were anesthetized with 3% isoflurane, 70% N2O, and 30% O2. A 4.8 mm craniotomy (−3.8 mm bregma, 2.5 mm lateral) was made over the right parietal cortex and a beveled 18 gauge syringe hub was secured to the craniotomy. At 24 h after the craniotomy, the animals were reanesthetized, intubated, and mechanically ventilated (Stoelting) with 0.5–1% isoflurane, 70% N2O, and 30% O2. Pancuronium bromide (1 mg/kg) was administered through the tail artery for immobilization. A moderate (2.0 ± 0.2 atm) fluid-percussion pulse (14–16 ms duration) was delivered to the right parietal cortex. Sham-operated rats received all surgical manipulations. Rectal and temporalis muscle thermistors were used to maintain core and brain temperatures at 36.8–37.3°C. Blood gases (pO2 and pCO2), blood pH, and mean arterial pressure were maintained within normal physiological ranges (Tables 1 and Table 2). Animals were randomized into the surgery groups. Criteria for exclusion from the study were as follows: mortality, loss of >15% of body weight, nonresolving infection at the surgical incisions, inability to feed or drink, motor paralysis, listlessness, self-mutilation, excessive grooming leading to loss of dermal layers, spontaneous vocalization when touched, or poor grooming habits. Animals were monitored for health issues weekly after surgery. Five animals were excluded because of mortality from the TBI surgery. Attrition and removal from the study for sham surgery was 0% and for TBI animals was 9% (5 animals); all of which died within 1 h of the brain trauma. Investigators were blind to the animal surgery for all behavior and histology experiments. Before beginning the surgeries, the primary endpoint was prospectively selected and no interim data analyses were conducted.
Physiological parameters weight and atm
Physiological parameters
Hippocampal electrophysiology.
Hippocampal slices (400 μm) were prepared as previously described at 2 weeks after surgery (Atkins et al., 2009). These experiments were conducted over a period of 19 months and used animals from 16 separate vendor shipments. The ipsilateral hippocampi were dissected with a vibratome (Vibratome 3000) in sucrose-based artificial CSF (aCSF, in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 7 MgCl2, 0.5 CaCl2, and 5 d-glucose, equilibrated with 95% O2/5% CO2 at 4°C. Slices were transferred to a submerged recording chamber in aCSF (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgCl2, saturated with 95% O2/5% CO2.
Field potential recordings were obtained with borosilicate glass electrodes filled with 2 m NaCl (1–3 MΩ) placed in stratum radiatum of area CA1 at 31°C. A platinum-iridium cluster stimulating electrode (tip diameter 25 μm, FHC) was placed in the Schaffer collateral pathway. Input-output (I/O) curves were generated by measuring the fEPSP with increasing stimulus intensities. For LTP experiments, stimulus intensities were adjusted to give fEPSP slopes at 40–50% of the maximum fEPSP. Paired-pulse facilitation (PPF) was assessed with stimuli intervals between 50 and 200 ms. fEPSPs were recorded and filtered (1 kHz) with an Axopatch 200A amplifier (Molecular Devices) and digitized at 20 kHz with an A/D converter (PCI-6221, National Instruments). For LTP experiments, baseline recordings (0.033 Hz) were established for at least 20 min after completion of I/O curves. LTP was induced with high-frequency stimulation (HFS, 100 Hz, 1 s). The WinLTP program was used for data acquisition and analysis (Anderson and Collingridge, 2007). To detect a 20% difference in fEPSP slope after LTP induction between groups at 80% power and with a significance level of 0.05, the estimated sample size was 7 animals per group (Sick et al., 1998).
Drug administration.
Rolipram (Sigma-Aldrich) was dissolved in DMSO at 10 mm and 10 mg/ml for the hippocampal slice experiments and behavioral experiments, respectively. For hippocampal slice electrophysiology, rolipram was diluted in aCSF to 1 μm. Final DMSO concentration was 0.1%. For the behavioral experiments, rolipram (0.03 mg/kg) or vehicle (2 ml/kg, 5% DMSO in saline) was administered intraperitoneally 30 min before task acquisition. The dose of rolipram was chosen because of the known improvement at this dose on object recognition late phase consolidation in naive animals (Rutten et al., 2009). Animals were randomly allocated into the drug treatment groups, although pseudo-randomly so that both vehicle and drug were represented in each subgroup undergoing testing. Investigators were blind to the surgery and drug treatment during the behavior experiments and histology analysis.
Fear conditioning.
Contextual and cued fear conditioning was performed at 2 weeks after surgery, and retention was assessed at 24 h and 4 weeks after training. These experiments were conducted over a period of 4 months and used animals from 10 separate vendor shipments. The fear conditioning apparatus (30.5 × 24.1 × 21 cm, Coulbourn Instruments) had an electric grid floor (1.6 cm spacing, 4.8 mm diameter rods). On day 1, animals habituated to the box for 10 min (Schafe et al., 1999; Ploski et al., 2008). Preexposure to the context 24 h before training enhances contextual fear conditioning and makes the contextual fear conditioning more sensitive to anterograde hippocampal damage or inactivation (Rudy and Morledge, 1994; Rudy and O'Reilly, 1999; Matus-Amat et al., 2004; Brown et al., 2011). Anterograde hippocampal lesions do not always result in behavioral deficits with standard contextual fear conditioning (Phillips and LeDoux, 1994; Maren et al., 1997; Wiltgen et al., 2006). Thus, we reasoned that preexposure to the context would make the assay more sensitive to detect the hippocampal damage and synaptic plasticity deficits caused by the TBI. On day 2, animals received drug or vehicle 30 min before training. Animals were placed in the box for 120 s, and then a 30 s tone (75 dB, 2.8 kHz) was given. During the last 1 s of the tone, a 1 mA foot shock was delivered through the grid floor. Animals remained in the chamber for 60 s before they were returned to the home cage. Baseline freezing on the training day was assessed by averaging the freezing during the first 120 s before the tone. The box was cleaned with 70% ethanol between animals. (Levenson et al., 2004; Chwang et al., 2006). At 24 h and 4 weeks after training, contextual fear conditioning was assessed by placing the animals in the box and measuring freezing for 5 min. Cue fear conditioning was assessed 1 h after contextual fear conditioning by placing animals in a chamber with altered ambient light, white background noise, different textured chamber walls and floor, and a novel odorant. After 120 s, the cue (75 dB, 2.8 kHz) was presented for 60 s. Baseline freezing in the new context on the testing day was measured for 120 s before the cue. Cue fear conditioning was assessed by measuring the amount of time spent freezing for the 60 s duration of the tone. Freezing behavior was quantified using video-based analysis (FreezeFrame, Coulbourn Instruments). For assessment of CREB phosphorylation after fear conditioning, nontrained animals were exposed to the box, but not the foot shock. These animals were handled in parallel with the trained animals, and animals were randomly allocated into the training or nontraining groups as well as the drug treatment groups.
Water maze.
Water maze training was performed at 2 weeks after injury in a separate set of animals from the fear conditioned animals. The water maze and working memory experiments were conducted over a period of 2 months and used animals from 5 separate vendor shipments. The circular pool (122 cm diameter, 60 cm deep) was filled with opaque water at 24°C and surrounded by distinct, invariant extramaze cues. An escape platform, 9.3 cm in diameter, was submerged 1.5 cm below the water surface. At 30 min before acquisition, animals received rolipram or vehicle. Animals received four 60 s acquisition trials per day for 4 d with intertrial intervals of 4 min. If the rat failed to navigate to reach the platform within 60 s, it was guided to the platform and remained on the platform for 10 s. Path length to reach the platform, escape latency, and swim speed were analyzed with EthoVision software (Noldus Information Technology). After 4 training days, a probe trial (30 s duration) was given with the platform removed. To detect a 30% difference between groups at 80% power and with a significance level of 0.05, the estimated sample size was 8 animals per group (Bramlett et al., 1997a).
Working memory.
On week 3 after surgery, the working memory task was performed using a delayed match-to-place task using the water maze described above (Hoskison et al., 2009). At 30 min before testing, animals received drug or vehicle. Four paired trials were given each day for 2 d with intertrial intervals of 4 min and delay of 5 s between trial 1 and 2. Trial duration was 60 s. The hidden platform remained invariant in location for each pair of trials. Upon reaching the platform, the animal remained on the platform for 10 s. After a 5 s delay, the animal was released into the water to again search for the hidden platform in the same location. Escape latency differences between the first location trial and subsequent match trial were measured. Data shown are from day 2.
Sensory threshold.
Animals received either drug or vehicle 30 min before sensory threshold testing for each task. At 24 h after retention assessment of the cue and contextual fear conditioning (6 weeks after surgery), shock threshold was assessed. Foot shock intensity started at 0.1 mA and increased by 0.02 mA every 30 s. The minimum shock intensity to induce flinching and jumping was recorded. At 7 weeks after surgery, the Von Frey test was used to test 8 specific calibrated filaments to deliver innocuous, mechanical stimuli in the range of 0.1–15 g of force. Rats were placed in a clear Plexiglas cage resting on an elevated wire mesh surface and acclimated for 30 min. Von Frey filaments (Stoelting) were pressed on the plantar paw region for 6 s. A brisk withdrawal from the filament was scored as a response. If a response was evoked, then the next lower force filament was tested. If there was no response, then the next higher force filament was tested. A total of six responses per paw were recorded to calculate withdrawal threshold (Dixon, 1980; Chaplan et al., 1994). At 24 h after Von Frey testing, the Hargreaves test was performed by assessing responses to a brief heat stimulus as previously described (Hargreaves et al., 1988). Rats were placed on an elevated glass surface and allowed to acclimate for 30 min. An infrared beam was positioned beneath a paw. Each paw was tested 4 times with an intertrial interval of 5 min, and the average of the last 3 trials was used to calculate withdrawal latency.
Hippocampal atrophy.
At 8 weeks after surgery, animals were anesthetized and perfused transcardially with 4% paraformaldehyde. Brains were serially sectioned coronally (50 μm thick). Serial sections (300 μm apart) were stained with hematoxylin and eosin plus Luxol fast blue. Atrophy was determined by tracing sections using Neurolucida 10.50 (MicroBrightField) and an Olympus BX51TRF microscope (Olympus America). Both the ipsilateral and contralateral hippocampi were contoured to calculate the difference in volume normalized to the contralateral volume.
Western blotting.
Animals received drug or vehicle 30 min before cue and contextual fear conditioning. Animals were fear conditioned with 3 tone-foot shock pairings during a 9 min trial (tone 1 min, 75 dB, 2.8 kHz, foot shock 1 mA, 1 s coterminating with tone, each tone-foot shock pairing separated by 2 min). At 20 min after fear conditioning, animals were anesthetized with 3% isoflurane and the ipsilateral hippocampus was dissected. Tissue was homogenized (Dounce homogenizer, 10 s, 4°C) in 750 μl of buffer (in mm): 15 Tris pH 7.6, 250 sucrose, 1 MgCl2, 1 EGTA, 1 DTT, 0.5 PMSF, 0.1 Na3VO4, 50 NaF, 2 Na4P2O7, 1.25 μg/ml pepstatin A, 10 μg/ml leupeptin, 25 μg/ml aprotinin, and 1× phosphatase inhibitor mixture set II (EMD Millipore). Homogenates were boiled with SDS sample buffer. Total protein was measured using Coomassie Plus (Thermo Fisher Scientific). Equal amounts of protein were electrophoresed, and blots were probed using antibodies against phospho-CREB Ser133 (1:1000, Cell Signaling Technology), total CREB (1:1000, Cell Signaling Technology), and β-actin (1:5000, Sigma-Aldrich). Epitopes were visualized with HRP-conjugated secondary antibodies (1:1000, Cell Signaling Technology) and enhanced chemiluminescence. Films were densitized using ImageJ 1.47c (National Institutes of Health). Changes in phosphorylated CREB were normalized to total CREB and β-actin levels to determine net phosphorylation changes in CREB. These behavior/biochemistry experiments were conducted over a period of 5 months and used animals from 6 separate vendor shipments. To detect a 30% difference in CREB phosphorylation between groups at 80% power and with a significance level of 0.05, the estimated sample size requirement was 6 animals per group (Atkins et al., 2009).
Statistical analysis.
Data represent mean ± SEM. Hippocampal electrophysiological recordings were analyzed with repeated-measures two-way ANOVA for I/O curves (animal group × current intensity), PPF (animal group × interstimulus interval), and LTP (animal group × time) with post hoc Bonferroni's tests. Hippocampal fEPSP slopes at 20 and 60 min after tetanus, cue and contextual fear conditioning, probe trial, swim speed, working memory on the match trial, sensory threshold tests, and hippocampal atrophy were analyzed with two-way ANOVA (surgery × drug; for probe trial factors were animal group × quadrant) and Bonferroni's post hoc tests. Hippocampal fEPSP slopes between 0 and 60 min after tetanus, water maze acquisition, working memory, and physiological parameters were analyzed with repeated-measures two-way ANOVA (animal group × time) followed by Bonferroni's post hoc comparisons. Water maze escape latency on the fourth acquisition day, percentage improvement in working memory, and Western blot data were analyzed with one-way ANOVA and post hoc Bonferroni's tests. Significance was set at p < 0.05.
Results
Hippocampal LTP deficits caused by TBI are restored with rolipram
Previous studies have demonstrated that TBI causes an impairment in expression of hippocampal LTP at the Schaffer collateral-CA1 synapse (Reeves et al., 1995; Sick et al., 1998; Schwarzbach et al., 2006; Norris and Scheff, 2009). To investigate potential pharmacological therapies that would restore hippocampal LTP and learning and memory deficits induced by TBI, we used the fluid-percussion brain injury model, a clinically relevant model of TBI that elicits reproducible deficits in hippocampal-dependent learning and memory (Dash et al., 1995; Bramlett et al., 1997a; Scheff et al., 1997; Pierce et al., 1998). In accordance with previous studies, we found that potentiation of the fEPSP slope at the Schaffer collateral-CA1 synapse was significantly decreased in hippocampal slices from TBI animals at 2 weeks after injury compared with sham animals (Fig. 1). Analysis of the fEPSP slope between 0 and 60 min after tetanus indicated a significant interaction of animal group × time (F(177,1947) = 1.70, p < 0.001). Rolipram, a phosphodiesterase inhibitor that boosts cAMP signaling, can convert a decaying form of LTP similar to what we observed with TBI into a robust, nondecaying form of LTP in hippocampal slices from aged animals or an Alzheimer's disease mouse model (Bach et al., 1999; Gong et al., 2004). Rolipram at1 μm had no significant effect on basal synaptic transmission in hippocampal slices from sham or TBI animals at 2 weeks after injury (Fig. 1A). When the average fEPSP response from 0 to 20 min after tetanization was analyzed by two-way ANOVA, no significant interaction of drug × surgery treatment was observed (F(1,33) = 0.12, p = 0.736), although a main effect of surgery was found (F(1,33) = 6.93, p = 0.013). An analysis of the average fEPSP response from 21 to 60 min after tetanization indicated a significant interaction of drug × surgery treatment (F(1,34) = 6.61, p = 0.015), with a significant difference in potentiation between TBI vehicle-treated versus TBI rolipram-treated slices (p = 0.008). These results indicate that rolipram rescued deficits in hippocampal LTP in slices from TBI animals from 21 to 60 min, but not at 0 to 20 min after tetanus (Fig. 1B,C).

Rolipram partially rescued TBI-induced deficits in hippocampal LTP. A, Normalized fEPSP slopes in hippocampal slices from sham + vehicle (n = 12 slices/8 animals), TBI + vehicle (n = 9 slices/8 animals), sham + rolipram (n = 8 slices/6 animals), and TBI + rolipram (n = 8 slices/7 animals) groups. There was a significant reduction in LTP in hippocampal slices from TBI animals at 2 weeks after injury. *p = 0.013, TBI + vehicle versus sham + vehicle (repeated-measures two-way ANOVA with Bonferroni's post hoc comparison). Rolipram (1 μm line) had no significant effects on baseline synaptic transmission or LTP in sham hippocampal slices but did rescue the deficits in hippocampal LTP from 21 to 60 min after tetanization in TBI hippocampal slices. Inset, Traces from representative recordings obtained before and 45 min after HFS. Average fEPSP slopes 0–20 min (B) and 21–60 min (C) after tetanus. The deficit in LTP was rescued with rolipram treatment between 21 and 60 min after HFS, but not between 10 and 20 min after HFS. ***p < 0.001 versus sham + vehicle (two-way ANOVA with Bonferroni's post hoc comparison). ##p = 0.008 versus TBI + rolipram (two-way ANOVA with Bonferroni's post hoc comparison).
TBI elicited a significant decrease in I/O curves at the Schaffer collateral-CA1 hippocampal synapse at 2 weeks after injury (Fig. 2A,B). To determine whether rolipram improved hippocampal LTP by rescuing this decrease in basal synaptic transmission, we analyzed I/O curves in TBI hippocampal slices treated with vehicle or rolipram by repeated-measures two-way ANOVA (Fig. 2A,B). A significant interaction of animal group × current intensity was found (F(24,328) = 5.09, p < 0.001). Although rolipram treatment (1 μm) caused a tendency to shift the I/O curve upward in TBI animals, this increase was not statistically significant at any current intensity compared with vehicle-treated TBI hippocampal slices. To assess presynaptic facilitation, we measured PPF with a range of interstimulus intervals. There were no significant differences in PPF with slices from TBI animals compared with sham animals (Fig. 2C). Rolipram treatment (1 μm) of the hippocampal slices did not significantly alter PPF in either sham or TBI hippocampal slices. Thus, presynaptic facilitation and basal synaptic transmission were not significantly altered by rolipram treatment at 2 weeks after TBI.

Effects of rolipram on basal synaptic transmission at the Schaffer collateral-CA1 synapse in the hippocampus. A, Superimposed representative traces of fEPSPs evoked by increasing current intensities. B, The I/O curve was shifted significantly downward in slices from TBI animals compared with slices from sham animals. Although rolipram (1 μm) caused the I/O curve to shift upward in TBI animals, this increase was not statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, TBI + vehicle versus sham + vehicle (repeated-measures two-way ANOVA with Bonferroni's post hoc comparison). ##p < 0.01, TBI + rolipram versus sham + vehicle (repeated-measures two-way ANOVA with Bonferroni's post hoc comparison). C, Effects of rolipram on PPF. Data represent the ratio of the second fEPSP slope to the first fEPSP slope. There were no significant differences between animal groups.
TBI-induced learning impairments are reduced with rolipram
Given the improvements in hippocampal LTP with rolipram treatment, we tested whether learning and memory impairments after TBI could also be improved. Animals received sham surgery or moderate parasagittal fluid-percussion brain injury. At 2 weeks after surgery, animals received either vehicle or rolipram (0.03 mg/kg) 30 min before cue and contextual fear conditioning. Retention was assessed 24 h after conditioning, a time point that is susceptible to manipulations in CREB signaling (Bourtchuladze et al., 1994; Bourtchouladze et al., 1998). A significant interaction of surgery × drug treatment was observed for contextual (F(1,28) = 12.17, p = 0.002) and cue (F(1,28) = 6.38, p = 0.018) fear conditioning retention at 24 h after training. Both cue and contextual fear conditioning were significantly decreased in TBI animals treated with vehicle (cue, p = 0.013; context, p = 0.011) compared with sham animals treated with vehicle (Fig. 3A). In contrast, rolipram-treated TBI animals had significantly more freezing in both the context (p < 0.001) and in response to the cue (p = 0.022) compared with vehicle-treated TBI animals. At the dose used, rolipram had no significant effect on cue and contextual fear conditioning in sham animals compared with sham vehicle animals (context, p = 0.798; cue, p = 0.267). In TBI animals treated with rolipram, cue fear conditioning was indistinguishable from sham animals treated with rolipram (p = 0.374), and there was a nonsignificant trend for increased freezing in the context compared with sham animals treated with rolipram (p = 0.053). There were no significant differences in baseline freezing during the training day or the testing day before the cue presentation in the new context.

Cue and contextual conditioning at 2 weeks after TBI or sham surgery (n = 8/group). Animals received vehicle (5% DMSO in saline) or rolipram (0.03 mg/kg) 30 min before training. Fear conditioning was assessed at 24 h (A) or 4 weeks (B) after training without rolipram treatment. Both cue and contextual freezing were significantly increased in TBI animals treated with rolipram compared with vehicle-treated TBI animals. No significant differences between groups were observed in baseline freezing on the training day (Training) or baseline freezing on the testing day before the cue in the new context (Baseline). *p < 0.05, **p < 0.01, ***p < 0.001 (two-way ANOVA with Bonferroni's post hoc comparison).
To assess the persistence of these effects, animals were allowed to recover without treatment for 4 more weeks and then tested again for retention of cue and contextual fear conditioning in the absence of any drug treatment (Fig. 3B). A significant interaction of surgery × drug treatment was observed for cue (F(1,27) = 4.88, p = 0.036) and contextual (F(1,27) = 7.44, p = 0.011) fear conditioning at 4 weeks after training. Retention of cue and contextual fear conditioning was still observed in TBI animals that had been treated with rolipram, whereas TBI animals treated with vehicle were significantly impaired compared with TBI rolipram-treated animals (context, p = 0.004; cue, p = 0.002) or sham vehicle-treated animals (context, p = 0.011; cue, p = 0.017). No significant differences were observed between sham animals treated with vehicle versus rolipram (context, p = 0.485; cue, p = 0.778) or sham animals and TBI animals treated with rolipram (context, p = 0.276; cue, p = 0.594). These results demonstrate that a single treatment of rolipram at a dose that does not enhance learning in noninjured animals results in significant retention of both cue and contextual fear conditioning at 24 h and 4 weeks after learning.
To control for alternative interpretations, such as a change in shock threshold, we assessed foot shock threshold levels, and sensory threshold to mechanical stimuli (von Frey test) and heat (Hargreaves test). There were no significant decreases in foot shock threshold sensitivity, mechanical allodynia, or thermal hyperalgesia with rolipram treatment in sham animals or TBI animals (Fig. 4).

Hyperalgesia was not increased at 12 weeks after TBI or by rolipram (0.03 mg/kg i.p. 30 min before testing). A, Animals were tested for the first shock intensity to elicit a flinch or jump. B, The 50% withdrawal threshold for Von Frey filaments was assessed. C, Withdrawal latency to a 55°C stimulus was measured; n = 8/group.
Effects of rolipram on spatial learning after TBI
As an independent assessment of hippocampal-dependent learning, we also determined whether rolipram would rescue deficits in water maze acquisition and retention at 2 weeks after TBI. In a separate group of animals from the fear conditioned animals, water maze performance was assessed 2 weeks after sham surgery or moderate parasagittal fluid-percussion brain injury. Animals received vehicle or rolipram (0.03 mg/kg) 30 min before water maze training. During acquisition, TBI animals failed to display progressive learning (Fig. 5A,B). On the fourth day of task acquisition, analysis with a one-way ANOVA followed by Bonferroni's post hoc correction revealed that TBI animals treated with vehicle had significantly longer escape latencies (p = 0.010) and path lengths (p = 0.002) compared with sham animals treated with vehicle. In contrast, TBI animals treated with rolipram displayed a significant decrease in escape latency (p = 0.008) and path length (p = 0.009) to reach the hidden platform compared with TBI animals treated with vehicle on the fourth day of training, and these indices of learning were not significantly different from sham animals treated with vehicle or rolipram. At 24 h after the last acquisition trial, animals were tested for retention during a probe trial with the platform removed. A significant interaction of animal group × quadrant was detected in the probe trial (F(9,112) = 3.95, p < 0.001). TBI animals treated with vehicle spent less time in the target quadrant compared with rolipram-treated TBI animals or sham animals treated with vehicle or rolipram (Fig. 5C). There were no significant differences in swim speed in TBI animals or with rolipram treatment (Fig. 5D). Analysis of thigmotaxic behavior indicated no differences between groups for all acquisition days.

Rolipram reversed the TBI-induced cognitive deficits in the water maze task (n = 8/group). Escape latency (A) and path length (B) during acquisition. Animals were trained 2 weeks after sham or TBI surgery and then received vehicle (5% DMSO) or rolipram (0.03 mg/kg) 30 min before training. Escape latencies and path lengths of TBI + vehicle animals were significantly greater than TBI + rolipram animals or sham animals on the fourth day of training. **p < 0.01, TBI + vehicle versus other groups (one-way ANOVA with Bonferroni's post hoc comparison). ***p < 0.001, TBI + vehicle versus other groups (one-way ANOVA with Bonferroni's post hoc comparison). C, Time spent in each quadrant during the probe trial. All groups spent significantly more time in the target quadrant compared with the other quadrants, with the exception of the TBI + vehicle animals. **p < 0.01, target versus nontarget quadrants, ***p < 0.001, target versus nontarget quadrants (two-way ANOVA with Bonferroni's post hoc comparison). D, Swim speed was not significantly altered with TBI or rolipram.
One potential significant caveat is that rolipram may actually impair working memory. This has precedence in the literature because, in aged animals, working memory is worsened with treatments that increase cAMP and PKA activity, and infusion of Sp-cAMPS into the medial prefrontal cortex results in working memory deficits in young adult animals (Ramos et al., 2003; Runyan and Dash, 2005). To assess any potential detrimental effects of rolipram on working memory in TBI animals, we used a spatial working memory version of the water maze task and assessed the differences in escape latency between the first trial (location trial) and the second trial (match trial), which were separated by a 5 s delay (Boissard et al., 1992; Steele and Morris, 1999; Runyan and Dash, 2005). A significant interaction of surgery × drug treatment was observed (F(1,27) = 4.61, p = 0.041). No significant differences in escape latencies were detected between any groups on the location trial (Fig. 6). However, for all groups, there was a significant improvement in escape latency in the location trial compared with the match trial (Fig. 6A). Analysis of the match trial revealed a significant impairment in spatial working memory in TBI animals treated with vehicle compared with sham vehicle-treated animals (p = 0.013), and this deficit was rescued by rolipram treatment in TBI animals (p = 0.034). On the match trial, TBI animals treated with rolipram were indistinguishable from sham animals treated with vehicle or rolipram, and rolipram treatment had no significant effect on spatial working memory in sham animals. Analysis of the percentage improvement in escape latency revealed a significant difference between TBI vehicle-treated animals compared with sham vehicle-treated animals (p < 0.05) and TBI rolipram-treated animals (p < 0.05), whereas TBI rolipram-treated animals were not significantly different from sham vehicle- or rolipram-treated animals (Fig. 6B). No significant differences in thigmotaxic behavior were observed between groups for either the location or match trial.

Rolipram rescued TBI-induced deficits in working memory at 2 weeks after injury. A, All groups (n = 8/group) had a significant decrease in escape latency on the match trial, indicating working memory ability. **p < 0.01, ***p < 0.001, location versus match trial (repeated-measures two-way ANOVA with Bonferroni's post hoc comparison). However, TBI animals treated with vehicle were significantly worse compared with sham animals or rolipram-treated TBI animals on the match trial. #p < 0.01 (two-way ANOVA with Bonferroni's post hoc comparison). B, Percentage improvement on the match trial of the working memory task. *p < 0.05 versus sham + vehicle (one-way ANOVA with Bonferroni's post hoc comparison).
Next, to determine whether rolipram improved cognition by reducing the pathology caused by TBI, animals were assessed for hippocampal atrophy, a typical consequence observed after both experimental and clinical brain trauma (Bramlett et al., 1997b; Tate and Bigler, 2000). At 8 weeks after injury, the brains were serially sectioned and the ipsilateral and contralateral hippocampal volumes were determined. There was a main effect of surgery (F(1,28) = 25.43, p < 0.001), but no significant interaction of surgery × drug treatment (F(1,28) = 1.80, p = 0.191) for hippocampal atrophy. We observed significant hippocampal atrophy after TBI compared with sham animals. However, rolipram had no significant effect on hippocampal atrophy in TBI animals (Fig. 7).

A, Representative images of with hematoxylin and eosin plus Luxol fast blue-stained sections at −2.4 mm posterior from bregma. B, Quantification of ipsilateral hippocampal atrophy. n = 8/group. Scale bar, 1 mm.
To determine whether the improvement in hippocampal-dependent learning by rolipram was the result of effects on cAMP signaling, sham and TBI animals at 2 weeks after surgery were trained using cue and contextual fear conditioning. Before fear conditioning, animals received either rolipram (0.03 mg/kg) or vehicle. Nontrained animals that received exposure to the box, but no foot shock, were handled in parallel and used for comparison. At 20 min after conditioning, the hippocampi were dissected and analyzed by Western blotting for changes in phospho-CREB. We found that CREB phosphorylation increased in sham, but not TBI-vehicle treated, animals after fear conditioning. When TBI animals were treated with rolipram 30 min before training, the deficits in CREB activation after learning were rescued (Fig. 8).
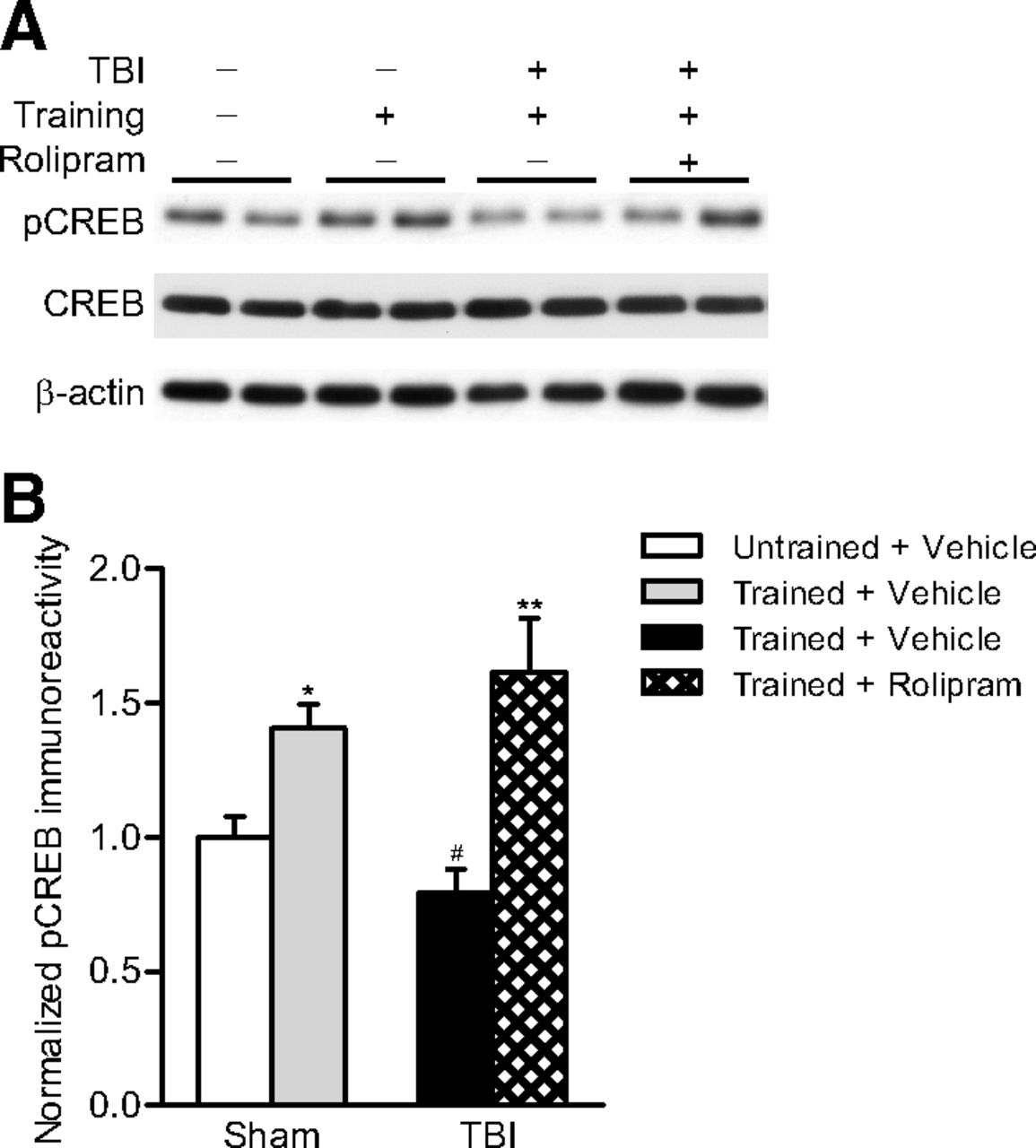
Phosphorylated CREB levels were assessed in the hippocampus at 20 min after cue and contextual fear conditioning. A, Representative Western blots. B, Densitometric analysis. CREB phosphorylation significantly increased in sham animals after training, but not in TBI animals treated with vehicle 30 min before training (n = 6/group). Rolipram rescued the TBI-induced deficits in CREB phosphorylation after learning. *p < 0.05 versus untrained sham + vehicle animals, **p < 0.01 versus untrained sham + vehicle animals, (one-way ANOVA with Bonferroni's post hoc comparison). #p < 0.01 for TBI + vehicle versus TBI + rolipram animals (one-way ANOVA with Bonferroni's post hoc comparison).
Discussion
In both clinical and experimental TBI, chronic learning deficits are common, but not well addressed with current pharmacological treatments (McAllister, 1992; Skelton et al., 2000; Arciniegas and Silver, 2006; Warden et al., 2006). Thus, it is important to identify new therapies that specifically modulate the molecular mechanisms that underlie chronic memory deficits after TBI. In this study, a low dose of rolipram improved acquisition and retention of several hippocampal-dependent learning tasks, as well as spatial working memory 2 weeks after TBI. Rolipram attenuated deficits in both hippocampal LTP and CREB activation during learning in TBI animals. These results suggest that rolipram may ameliorate deficits in the mechanisms of hippocampal synaptic plasticity impaired by TBI or augment activity in the remaining intact neurons and synapses with normal biochemical activity to compensate for the cellular and synaptic loss associated with TBI.
To ensure that the improvements in fear conditioning with rolipram were the result of effects on memory processing, foot shock threshold, mechanical allodynia and thermal hyperalgesia were evaluated (Crain and Shen, 2008). There were no sensory alterations with either rolipram or TBI. Baseline freezing was also unaltered, suggesting that rolipram did not result in generalization of the fear conditioned response. In addition, there were no significant differences in physiological measures after TBI of vehicle- and rolipram-treated animals. Hippocampal atrophy was similar between treatment groups, providing support for the model that rolipram improved learning and memory by altering cAMP signaling rather than the pathology caused by TBI.
In this study, parasagittal fluid-percussion brain injury resulted in cue fear conditioning deficits that were rescued with rolipram. The underlying mechanism(s) for rolipram's effect on cue fear conditioning after TBI is unclear. It is unknown whether this brain injury model produces pathology within the amygdala. Using a model with relatively comparable pathology with parasagittal fluid-percussion brain injury, lateral fluid-percussion brain injury did not produce neuronal loss in the amygdala but did alter levels of GAD67, NMDA receptors, immediate early gene expression, and astrocyte activation (Abrous et al., 1999; Reger et al., 2012; Rodgers et al., 2012). One study using the central fluid-percussion brain injury reported no overt cell loss in the amygdala but did observe alterations in a gustatory neophobia task indicative of damage to the amygdala and hippocampus (Hamm et al., 1995). Further studies are needed to determine the changes that occur in the amygdala after parasagittal fluid-percussion brain injury.
In animals with memory deficits resulting from normal aging, sleep deprivation, or Alzheimer's disease, rolipram improves hippocampal-dependent learning (Barad et al., 1998; Bach et al., 1999; Gong et al., 2004; Vecsey et al., 2009). Furthermore, rolipram reverses memory deficits caused by an ERK inhibitor and in transgenic mice expressing a truncated form of CREB-binding protein (Bourtchouladze et al., 2003; Zhang et al., 2004). Low doses of rolipram in the range used in this study do not typically alter cognition in uninjured animals, only in animals that are altered biochemically or behaviorally to induce cognitive impairments (Winsky and Harvey, 1987; Egawa et al., 1997; Imanishi et al., 1997; Barad et al., 1998; Bourtchouladze et al., 2003; Gong et al., 2004; Rutten et al., 2009; Vecsey et al., 2009). Perhaps PKA signaling is robust enough in normal animals that modest augmentation does not elicit a significant behavioral effect; however, if the system is downregulated, such as after TBI, rolipram may provide enough of a functional boost to restore activation of CREB signaling during learning.
The use of rolipram in the clinical setting is hampered by specificity considerations. Rolipram equally inhibits all PDE4 isoforms and causes side effects of emesis and nausea, precluding its clinical development (Robichaud et al., 2002; Bruno et al., 2009). The PDE4D isoform in particular is a critical modulator of long-term memory formation and emesis (Li et al., 2011) However, select knock-down of PDE4D within the hippocampus enhances learning without affecting emesis. The development of isoform-selective inhibitors that block the relevant PDE4 isoforms to enhance learning without side effects of nausea and emesis may be a more promising therapeutic route. We have previously found that rolipram given early after TBI increased hemorrhage in the vulnerable cortex at 3 d after injury (Atkins et al., 2012). These results suggest that, although acute rolipram after TBI worsens recovery, delayed treatment improves functional outcome without adversely affecting pathology.
A clinically relevant finding of our study is that the TBI animals showed persistent improvements in fear conditioning even 4 weeks after the single rolipram dose that was given before training. Four weeks is sufficient for hippocampal-dependent learning to become insensitive to hippocampal lesions (Maren et al., 1997). Given rolipram's short half-life, it is possible that rolipram contributed to stabilization of synaptic circuitry via altered gene expression mediated by CREB (Bourtchouladze et al., 2003). This interpretation has support from several studies demonstrating that boosting cAMP-PKA signaling during learning augments encoding of memories (Bourtchuladze et al., 1994; Abel et al., 1997; Barad et al., 1998; Pittenger et al., 2002; Gong et al., 2004; Navakkode et al., 2004). This robust, persistent effect of rolipram is clinically relevant because nonselective PDE4 inhibitors, such as rolipram, can be given only a few times before homeostatic mechanisms result in a rebound, decreasing cAMP levels below basal levels and inducing PDE4D protein expression (Giorgi et al., 2004; Dlaboga et al., 2006). Furthermore, chronic, high doses of rolipram impair hippocampal-dependent memory formation (Giralt et al., 2011). These results indicate the therapeutic utility of low doses of PDE4 inhibitors to augment memory formation during rehabilitation training, without the requirement to be onboard during recall, thus reducing potential homeostatic mechanisms that would eventually decrease efficacy.
In addition to altering CREB signaling within neurons, there are other alternative interpretations of how rolipram enhanced learning in TBI animals. Rolipram is a vasodilator and may have increased cerebral blood flow (Rutten et al., 2009). At the dose used in this study in naive animals, there is no significant change within the hippocampus in local cerebral blood flow and a modest decrease in local cerebral glucose utilization, resulting in hyperemia. Fluid-percussion brain injury chronically decreases cerebral blood flow and causes microvascular loss in the hippocampus (Dietrich et al., 1996; Park et al., 2009; Hayward et al., 2010). Vascular reactivity to rolipram may be altered after TBI, such that rolipram improved learning and memory performance by improving the chronic decrease in cerebral blood flow after TBI.
PDE4 selectively degrades cAMP compared with cGMP. There are also modest increases in cGMP with high concentrations of rolipram, suggesting that rolipram could work through cGMP, although this is unlikely (van Staveren et al., 2001). In addition, rolipram enhances neurotransmitter availability by activating cholinergic and noradrenergic systems (Schoffelmeer et al., 1985; Asanuma et al., 1993). Thus, beyond stimulating CREB activation, rolipram may have improved modulatory neurotransmitter signaling in the hippocampus during learning to improve TBI-induced memory impairments.
Not all cognitive domains are improved with rolipram or therapies that boost cAMP signaling. In aged animals, working memory deficits are exacerbated with treatments that increase cAMP signaling; and in young adult animals, infusion of drugs to increase phosphorylated CREB in the medial prefrontal cortex also worsens working memory (Ramos et al., 2003; Runyan and Dash, 2005). Working memory is significantly impaired after TBI, and CREB-mediated expression of the α1-adrenergic receptor within GABAergic neurons contributes to working memory deficits (McAllister et al., 2004; Kobori et al., 2011). Thus, any cognitive enhancer developed preclinically needs to evaluate this memory domain as well as reference memory. In these studies, we found that rolipram improved spatial working memory deficits induced by TBI. This particular behavioral paradigm is sensitive to hippocampal lesions, and the improvements observed in this study may have been the result of effects on the hippocampus (Morris et al., 1986; Boissard et al., 1992; Steele and Morris, 1999). Although the platform was moved between trial pairs with only a 5 s delay to reveal working memory deficits, the deficits observed may actually reflect spatial memory rather than spatial working memory (Clark et al., 2001). Further experiments are required to determine whether rolipram enhances reference and working memory at later time points after injury because the relevant therapeutic targets may change over time. For example, in the prefrontal cortex, the molecular mechanisms that underlie working memory deficits after controlled cortical impact are quite dynamic. At 14 d after injury, GABA receptor antagonists improve working memory, but not at 4 months after injury (Hoskison et al., 2009). Further work is required to determine the therapeutic time window and optimal dosing of PDE4 inhibitors as cognitive enhancers across multiple memory modalities for TBI.
Currently, PDE4 inhibitors are being tested in a clinical trial as cognitive enhancers for healthy adults (NCT01433666). Our data indicate that rolipram is highly effective in a preclinical model of brain injury to improve cognitive deficits, and this may be due, in part, to boosting signaling through the transcription factor CREB and enhancing hippocampal synaptic plasticity. These studies support the development of isoform-specific PDE4 inhibitors or alternative pharmacotherapies to stimulate CREB signaling as viable therapeutic routes to improve cognition in TBI survivors.
Footnotes
This work was supported by the National Institutes of Health Grants NS069721 and NS056072 and the Miami Project to Cure Paralysis. We thank Dr. W. Dalton Dietrich for critical reading of the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Coleen M. Atkins, Miami Project to Cure Paralysis, University of Miami Miller School of Medicine, 1095 NW 14 Terrace, Miami, FL 33136. catkins{at}med.miami.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}