

Abstract
The ionic mechanism underlying the acetylcholine-induced depolarization of layer V pyramidal neurons of rat prefrontal cortex was examined using whole-cell recording in in vitro rat brain slices. Consistent with previous results, pressure application of acetylcholine to layer V pyramidal neurons elicited a strong depolarization. Pharmacological analysis of this response indicated that it was mediated by the stimulation of muscarinic receptors as it was mimicked by muscarinic agonists, but not by nicotine, and was blocked by atropine.
The inward current responsible for the depolarization resulted from the activation of a voltage-dependent, cation nonselective current. Thus, the amplitude of the current was critically dependent on extracellular sodium concentration but not on extracellular potassium or chloride concentration. Examination of the I–Vrelationship for the muscarinic current using voltage clamp revealed that the current reversed near −15 mV and exhibited a strong voltage dependence, turning off rapidly in the subthreshold range. The voltage dependence of the current led to the appearance of a current associated with a conductance decrease when examined using steady-state voltage- or current-clamp measurements. This might have led to earlier misidentification of this response as mediated by a decrease in potassium conductance.
These results question the traditional interpretation that muscarinic depolarization in cortex is mediated by a decrease in potassium conductance. They indicate that the fundamental mechanism responsible for muscarinic depolarization in prefrontal cortex involves the activation of a voltage-dependent, cation nonselective current. This current might represent a previously unsuspected mechanism capable of mediating slow depolarization in the central nervous system.
- acetylcholine
- muscarinic receptors
- depolarization
- cation nonselective current
- pyramidal neurons
- cerebral cortex
The cerebral cortex receives a dense cholinergic innervation that is thought to regulate neuronal excitability and participate in the regulation of memory and learning (McKinney and Coyle, 1991). Previous studies aimed at elucidating the cellular and molecular mechanisms by which acetylcholine regulates neuronal excitability have focused predominantly on sensorimotor cortices (Krnjevic et al., 1971; McCormick and Prince, 1985, 1986; Schwindt et al., 1988; Constanti and Bagetta, 1991) and the hippocampus (Benardo and Prince, 1982; Halliwell and Adams, 1982; Madison et al., 1987;Benson et al., 1988). In these regions acetylcholine has been suggested to act on muscarinic–cholinergic receptors to increase membrane excitability by inhibiting potassium currents. Three main potassium currents have been postulated to participate in this response: a calcium-activated potassium current responsible for the afterhyperpolarization seen in these cells under control conditions; a voltage-independent “leak” potassium current; and the M-current, a voltage-dependent potassium current. The inhibition of the last of these has been suggested to account for the surprising observation that acetylcholine excites cortical neurons much more effectively near threshold than at their resting potential (ffrench-Mullen et al., 1983;McCormick and Prince, 1986; McCormick and Williamson, 1989).
We have previously reported that cholinergic stimulation of rat prefrontal cortex results in a large increase in membrane excitability (Andrade, 1991). An important component of this response is a membrane depolarization similar to that seen in the hippocampus (Madison et al., 1987) or other regions of cerebral cortex (Krnjevic et al., 1971;McCormick and Prince, 1985). However, our attempts, conducted over a period of two years, to prove conclusively that this depolarization was mediated by a decrease in potassium conductance proved unsuccessful. This led us to reexamine the view that the muscarinic depolarization in cerebral cortex is mediated via a decrease in potassium conductance. The results of these experiments are presented here. A preliminary account of this work has been published previously (Haj-Dahmane and Andrade, 1994).
MATERIALS AND METHODS
Preparation of the brain slices. Brain slices containing the medial prefrontal cortex (Krettek and Price, 1977) were prepared as previously described (Andrade, 1991). Briefly, adult male albino rats (200–250 gm) were anesthetized with halothane and killed by decapitation. The brain was quickly removed and cooled in ice-cold Ringer of standard composition (in mm: NaCl 119, KCl 2.5, MgSO4 1.3, CaCl22.5, NaH2PO4 1.0, NaHCO3 26.2, and glucose 11) continuously bubbled with 95% O2/5% CO2. The anterior forebrain containing the medial prefrontal cortex was then isolated, affixed to a stage with cyanoacrylate glue, and sectioned into 400 μm (nominal thickness) slices using a vibratome (Lancer series 1000, Ted Pella, Irvine, CA). The resulting slices were then transferred to an interface-type recovery chamber filled with a moist atmosphere of 95% O2/5% CO2, where they were allowed to recover for at least 1 hr at room temperature. After this period of recovery, slices were transferred one at a time to a recording chamber (Nicoll and Alger, 1981). In this chamber, the slices were submerged between two nylon nets and continuously perfused (3–4 ml/min) with normal Ringer saturated with 95% O2/5% CO2 at 30 ± 1°C.
Electrophysiological recordings. Whole-cell recordings were obtained from 225 layer V pyramidal neurons of the prelimbic and anterior cingulate subdivisions of the medial prefrontal cortex using the blind tight-seal recordings technique (Blanton et al., 1989). The recording pipettes were pulled from 1.2 mm (outer diameter) borosilicate glass (Glass Company of America, Bargaintown, NJ) using a Flaming–Brown-type horizontal puller (model PC80/PC, Sutter Instruments, Novato, CA). For the initial experiments aimed at characterizing the carbachol-induced current, pipettes were filled with a solution containing (in mm): KMeSO4 125, NaCl 5, MgCl21, HEPES 10, EGTA 0.02, ATP 2, and GTP 0.5. Because the carbachol-induced inward current proved insensitive to the buffering of intracellular calcium (see below), subsequent experiments were conducted using 10 mm EGTA/1 mm CaCl2 in the pipette. The free-calcium concentration under these conditions was estimated to be 10 nm. In some experiments, 10 mm BAPTA was substituted for the EGTA, as described in the text. The pH of these solutions was adjusted to 7.3–7.4 with KOH. This brought the intracellular potassium concentration to ∼135 mm. When filled with these intracellular solutions, electrode resistance ranged from 4 to 7 MΩ. Access resistances measured using the bridge compensation circuit of the amplifier were considerably higher and generally ranged from 8 to 25 MΩ. The osmolarity of the internal solutions was adjusted to ∼5 mOsm/l lower than the osmolarity of standard Ringer, using the appropriate salts. Perforated-patch recordings were obtained essentially as described by Korn and Horn (1989). Briefly, the tip of the electrode was filled with control solution and backfilled with the same solution containing 100 μg/ml nystatin or amphotericin B. After seal formation, the antibiotic perforated the membrane, allowing for recording the cell-membrane potential as well as limited current injection. In our hands, access resistances using this procedure generally exceeded 40 MΩ.
In experiments aimed at examining the voltage sensitivity of the muscarine-induced inward current, it was necessary to isolate this current as much as possible within the limitation of the slice preparation. This was accomplished by using cesium gluconate-based intracellular solution of standard composition (in mm: Cs-gluconate 140, NaCl 5, MgCl2 2, CaCl2 1, HEPES 10, EGTA 10, ATP 2, and GTP 0.5, pH, 7.3–7.4). In addition, cesium (2 mm) and barium (100 μm) were also added to the bath to block inwardly rectifying currents, which are prominent in these cells. Finally, calcium-activated currents were further suppressed by the addition of cadmium (100 μm) to the bath.
Electrical signals were amplified with an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA). The membrane potential and electrode current were recorded on-line using a paper chart recorder (model 3200, Gould Instruments, Valley View, OH), whereas triggered events, such as ramps, were digitized using an Intel-based 80x86 computer equipped with a 12 bit A/D converter under the control of pClamp 5.5 software (Axon Instruments). Most voltage-clamp experiments were conducted using the single-electrode discontinuous voltage-clamp technique. The headstage output was continuously monitored on an oscilloscope, and the switching frequency (generally 3–6 kHz) and gain (generally 0.3–1.0 nA/mV) were adjusted to allow complete decay of the voltage transients between cycles. This procedure was preferred to the continuous single-electrode voltage-clamp mode because, although it was associated with higher noise, it allowed for better compensation (and continuous monitoring) of series resistance. All voltage-clamp experiments were conducted in solution containing tetrodotoxin (TTX; 1 μm). In a few experiments, 0.1–0.5% neurobiotin was included in the recording solution to allow for cell identification. At the end of the experiment, slices were fixed in 4% paraformaldehyde in 0.15% sodium phosphate buffer and processed using the avidin–biotin horseradish peroxidase histochemical technique essentially as implemented byFoehring et al. (1991), except that slices were prepared as whole mounts.
Quasi-steady-state current–voltage relationships were obtained by applying a slow depolarizing potential ramp at a speed of 5 mV/sec. In some experiments, the I–V curves were determined using 500-msec-long hyperpolarizing steps from −40 mV to between −120 and −40 mV. These experiments were conducted in continuous voltage-clamp mode to reduce noise. They were also conducted at room temperature to slow down the voltage-dependent inactivation of the current. Series resistance was compensated up to 70%, and the settling time of the current after a hyperpolarizing step (10 mV) was optimized generally to ≤5 msec. The data were plotted and analyzed using Origin software (Microcal Software Inc., Northampton, MA). The net carbachol-induced current was obtained by subtracting the controlI–V relationship from that obtained in the presence of carbachol. The values of membrane potential were corrected for the liquid junction potential that was present between the pipette solution and the bath solution (approximately +5 mV).
In most experiments, drugs were applied by superfusion dissolved in the Ringer at known concentrations. In some experiments, acetylcholine was applied to the vicinity of the cell by pressure ejection. In these experiments, a patch pipette containing 100 mmacetylcholine was placed immediately above the slice and the cell of interest, and acetylcholine was ejected by a brief pressure pulse (25–100 msec). To test the possible involvement of nicotinic receptors in the depolarizing response, nicotine was applied as a microdrop (3–7 μl, 10 mm) directly to the bath. This procedure has been shown to activate effectively nicotinic receptors in a comparable slice preparation (see below). The low sodium Ringer was prepared by substituting 119 mmN-methyl-d-glucamine for a comparable concentration of sodium chloride; the pH was adjusted to 7.4 with HCl. The low chloride Ringer was prepared by substituting sodium chloride with sodium isethionate. When high potassium solution was used, KCl was added to the normal extracellular solution. The osmolarity of these solutions was 285–290 mOsm/l.
Most drugs used in this study were obtained from Sigma (St. Louis, MO), except for TTX from Calbiochem (La Jolla, CA) and 6,7-dinitroquinoxaline-2,3-dione (DNQX) from Tocris Neuramin (Bristol, UK). Baclofen was a kind gift from Ciba-Geigy Corporation (Summit, NJ). Numerical data are presented as mean ± SEM. Statistical comparisons used Student’s paired or unpaired two-tailed t test or ANOVA; p < 0.05 was considered to be statistically significant.
RESULTS
Whole-cell recordings were obtained from 225 layer V pyramidal cells from the dorsal anterior cingulate and prelimbic subdivisions of the rat medial prefrontal cortex (Krettek and Price, 1977). In agreement with previous studies elsewhere in cortex (Connors et al., 1982; McCormick et al., 1985; Connors and Gutnick, 1990; Foehring et al., 1991), neurons in this region could be subdivided into broad classes according to their electrophysiological properties and firing pattern. The vast majority of cells encountered in this study could be classified on the basis of their electrophysiological properties as regular spiking cells (McCormick et al., 1985; Connors and Gutnick, 1990). No bursting neurons were encountered in these recordings, in accordance with previous studies in association cortex (Foehring et al., 1991). On a few occasions, recordings were obtained from fast-spiking presumed GABAergic (GABAA) interneurons (McCormick et al., 1985); these cells were not studied further.
Previous studies have established that regular-spiking cells correspond to morphologically identified pyramidal cells (Connors et al., 1982;McCormick et al., 1985; Connors and Gutnick, 1990; Foehring et al., 1991). To examine whether this correspondence also held for prefrontal cortex, cells were recorded using an intracellular solution containing neurobiotin and processed for HRP histochemistry. All of the cells with these electrophysiological properties recovered from layer V (n = 7) could be classified as pyramidal neurons. Therefore, in the remaining sections of this report, regular-spiking cells are referred to as pyramidal neurons.
Effects of acetylcholine on membrane potential
Pressure application of acetylcholine to layer V pyramidal neurons resulted in a strong depolarization of the resting membrane potential (Fig. 1A; n = 21). This effect was mimicked by bath application of muscarine (30 μm; n = 8; data not shown) or oxotremorine (30 μm, 5 of 7 cells tested; data not shown) and blocked by atropine (300 nm to 1 μm; n = 5; Fig. 1B). To determine whether nicotinic receptors could contribute to this depolarization, we examined the effects of fast bath applications of nicotine. No evidence for nicotinic receptor-mediated depolarization in pyramidal cells could be detected using this procedure (n = 6; data not shown), although the microdrop application technique was highly effective in activating neuronal nicotinic (n = 2) receptors in the chicken brain slice containing nucleus spiriforms lateralis, which expresses high levels of nicotinic receptors (Sorenson and Chiappinelli, 1990).

Acetylcholine depolarizes layer V pyramidal neurons of rat medial prefrontal cortex. A, Toptrace, Pressure application of acetylcholine depolarized this neuron and triggered a period of sustained spiking activity.Bottom trace, Two minutes after recovery, a second application of acetylcholine induced a comparable effect. B, After bath administration of 100 nm atropine, the acetylcholine response was almost completely inhibited. These recordings were obtained using potassium methylsulfate-based intracellular solution containing 20 μmEGTA.
Because acetylcholine is rapidly degraded when applied to the slice, it is not practical to use it for routine bath applications. Therefore, for the rest of this study, we used its stable analog, carbachol. As illustrated in Figure 2, like acetylcholine, carbachol elicited a depolarization. This depolarization was concentration-dependent with threshold responses in the low micromolar range. Carbachol concentrations in the 30–300 μm range produced robust depolarizing responses that generally, but not always, reached spike threshold and initiated spiking activity (Fig. 2). The sensitivity to carbachol was the same whether measured using sharp microelectrode (Andrade, 1991), whole-cell, or perforated-patch recordings (Fig. 2). As previously seen for acetylcholine, atropine (100 nm to 1 μm) completely blocked the carbachol-induced depolarization (n = 5), and this antagonism could be surmounted by increasing the carbachol concentration.

Carbachol elicits a concentration-dependent depolarization of resting membrane potential of the cell. Current-clamp recording of the pyramidal neuron was obtained using the nystatin perforated-path recording technique. Bath administration of carbachol resulted in a concentration-dependent depolarization. In this and the subsequent figures, the bar above the traceindicates the period of the carbachol application.
Cholinergic stimulation could depolarize these cells through a direct effect or indirectly through the release of other neurotransmitters. The ability of carbachol to elicit a depolarization was preserved in the presence of TTX (n > 100; Fig.3A), indicating that the depolarization was not mediated by an action potential-dependent release of neurotransmitters. It was also preserved in the presence of the calcium channel blocker cadmium (100 μm; n = 6), which blocks synaptic transmission. Finally, it was not blocked by the ionotropic glutamate receptor antagonist DNQX at concentrations that blocked excitatory synaptic responses (n = 3). These results argued for a direct effect of acetylcholine on layer V pyramidal neurons.

The carbachol-induced depolarization behaves as if mediated by a decrease in potassium conductance when it is examined using a variety of traditional electrophysiological tests.A, Recording from a pyramidal neuron in current-clamp mode. Hyperpolarizing current pulses (140 ms, 0.2 nA) were applied periodically to monitor the input resistance. Administration of carbachol (30 μm) in the presence of TTX (1 μm) to this cell elicited a membrane depolarization associated with an apparent increase in membrane resistance. B1, Carbachol administration (30 μm) induced an inward current in a different pyramidal neuron voltage-clamped at −65 mV in the presence of TTX (1 μm). B2, Current–voltage (I–V) relationship obtained using a voltage ramp from −120 to −30 mV taken before (○) and during the superfusion with carbachol (•) from the cell illustrated in B1(holding current = 0.16 nA). C, Carbachol-induced current in a different neuron voltage-clamped at −65 mV (holding current = 0.11 nA). Superfusion with barium (2 mm) induced an inward shift of the holding current and a reduction of the amplitude of the carbachol-induced current.
Muscarinic receptor activation in central neurons has been reported to produce large increases in intracellular calcium (Reynolds and Miller, 1989; Yuste and Katz, 1991). Therefore, we examined whether changes in intracellular calcium concentration were required for the carbachol-induced depolarization. First, recordings were obtained by using an intracellular solution with low calcium-buffering capacity (20 μm EGTA). Under this condition, bath administration of carbachol (30 μm in the presence of TTX) resulted in a depolarization averaging 6.8 ± 2.5 mV (n = 11). Increasing the calcium-buffering capacity of the intracellular solution resulted in a significant reduction of the calcium-activated after hyperpolarization potential (from 2.3 ± 0.5 to 0.8 ± 0.3 mV; p < 0.05; n = 6), indicating effective buffering of calcium transients. However, no change was observed on the carbachol-induced depolarization (p > 0.5, one-way ANOVA). The depolarization elicited by 30 μm carbachol averaged 6.8 ± 2.5 mV (n = 11) when 20 μm EGTA was introduced into the recording solution and 6.8 ± 1.0 mV (n = 6) when 10 mm EGTA was used. Because these results indicated that a rise in intracellular calcium was not required to elicit the carbachol depolarization, most subsequent experiments were conducted using 10 mm EGTA in the intracellular solution to minimize the impact of calcium-activated currents on the analysis of the carbachol response.
Under voltage-clamp conditions, superfusion with carbachol (3–100 μm) elicited an inward current in the vast majority (>90%) of cells studied (n = 117). The mean current elicited by 30 μm carbachol applied to cells clamped at −65 mV was −119 ± 6.2 pA. As previously observed for current-clamp responses, the onset of the current was slow, taking close to 1 min to peak, and reversed slowly over 10–15 min after removal of the carbachol from the bath (Fig. 3B1). No consistent evidence for rundown or desensitization of the current was observed after repeated applications of carbachol (10–30 μm) in the time frame tested (up to 1 hr after whole-cell access).
Properties of the carbachol-induced inward current
Previous studies in a variety of preparations (Krnjevic et al., 1971; Madison et al., 1987; Benson et al., 1988), including cortical neurons in primary culture (Jones and Baughman, 1992), have reported that muscarinic receptors can elicit a membrane depolarization by inhibiting a voltage-independent “leak” potassium conductance. This suggested that a similar mechanism could be responsible for the muscarinic depolarization observed in prefrontal cortex. A variety of traditional manipulations gave results consistent with this possibility. For example, the depolarization was associated with an apparent increase in input resistance (Fig. 3A; n = 7), an effect often interpreted as a decrease in resting K+ conductance. Moreover, the carbachol-induced current could be seen to depend on the membrane potential of the cell, declining in amplitude as the voltage was shifted towardEK (Fig. 3B2; n = 88). Finally, barium (Fig. 3C; n = 14) and tetraethylammonium (TEA; n = 5), two often-used potassium channel blockers, increased the input resistance and significantly reduced the carbachol-induced current. These observations seemed to suggest that the depolarization induced by carbachol was related to a reduction of a potassium conductance.
To characterize the mechanism underlying this inward current,I–V curves were constructed for the carbachol-induced current. As illustrated in Figure 4,A and B, the current induced by carbachol depended on the membrane potential, increasing sharply at potentials more positive than −65 mV (Fig. 4). Below this potential, the carbachol-induced current diminished approximately linearly toward the predicted equilibrium potential for potassium (−104 mV). Surprisingly, in most cells studied (66 of 88 cells tested), the carbachol-induced current did not reverse at EK but persisted as an inward current even at potentials 10–20 mV negative to the predicted equilibrium potential for potassium (Fig. 4A;n = 66). In a smaller proportion of cells (22 of the 88 cells tested), the carbachol-induced current did reverse polarity at −101 ± 2.9 mV, a value close to the equilibrium potential for K+ (Fig. 4B). However, this reversal of the current at hyperpolarized potentials could only be observed after the first application of carbachol. Subsequent applications produced currents that remained inward even belowEK. Despite this, the inward current recorded near the resting membrane potential was not significantly changed from the first to the second application, suggesting that most of the effect of carbachol is mediated by an inward current that does not reverse at EK.

The inward current caused by carbachol fails to reverse polarity even at potentials more negative thanEK in the majority of cells studied.A, In most cells studied, the net carbachol current failed to reverse at EK. B, In a small proportion of cells, the net carbachol current reversed nearEK. The holding current for the cell illustrated in A was 0.15 nA and in B was 0.12 nA.
This failure to obtain a reversal of the carbachol current could simply reflect poor voltage control of the dendritic arbor, a possible site for the actions of carbachol. To minimize this problem,I–V curves were constructed for the inward current elicited by pressure application of acetylcholine localized to the immediate vicinity of the recording electrode. This procedure should result in an inward current spatially restricted to areas of the cell that were well clamped, presumably the soma and proximal dendrites. However, despite these precautions, no reversal of the acetylcholine inward current was found in four cells tested. This failure to obtain a consistent reversal of the muscarinic inward current contrasted with the unambiguous reversals obtained when the potassium current activated by baclofen was studied. In four cells tested, the outward current elicited by bath-applied baclofen (30 μm) consistently reversed at −104 ± 2.3 mV, a value not significantly different from the predicted equilibrium potential for potassium (data not shown).
Ionic mechanism
A possible explanation for the lack of reversal of the inward current would be that muscarinic receptors inhibit voltage-dependent potassium channels. To test this possibility, we determined the effect of ion substitutions on the amplitude of the inward current.
In an initial set of experiments, we examined the effects of raising extracellular potassium concentration on the amplitude of the carbachol-induced inward current. First, extracellular potassium was raised to 5 mm, a manipulation that should shiftEK by ∼18 mV and reduce the driving force for potassium almost by half. This manipulation resulted in only a slight reduction of the carbachol inward current (control = 100 ± 5.0 pA; 5 mm K+o = 80 ± 5.7 pA;Vh = −65 mV; n = 3;p > 0.05). Increasing the extracellular potassium concentration to 10 mm still failed to reduce significantly the carbachol-induced inward current (control = 136 ± 20 pA; 10 mm K+o = 110 ± 20 pA;n = 3; Vh = −65 mV;p > 0.05), although the driving force for potassium under these conditions was almost eliminated. Experiments using higher extracellular concentrations of potassium were not attempted because of the large increase in conductance observed.
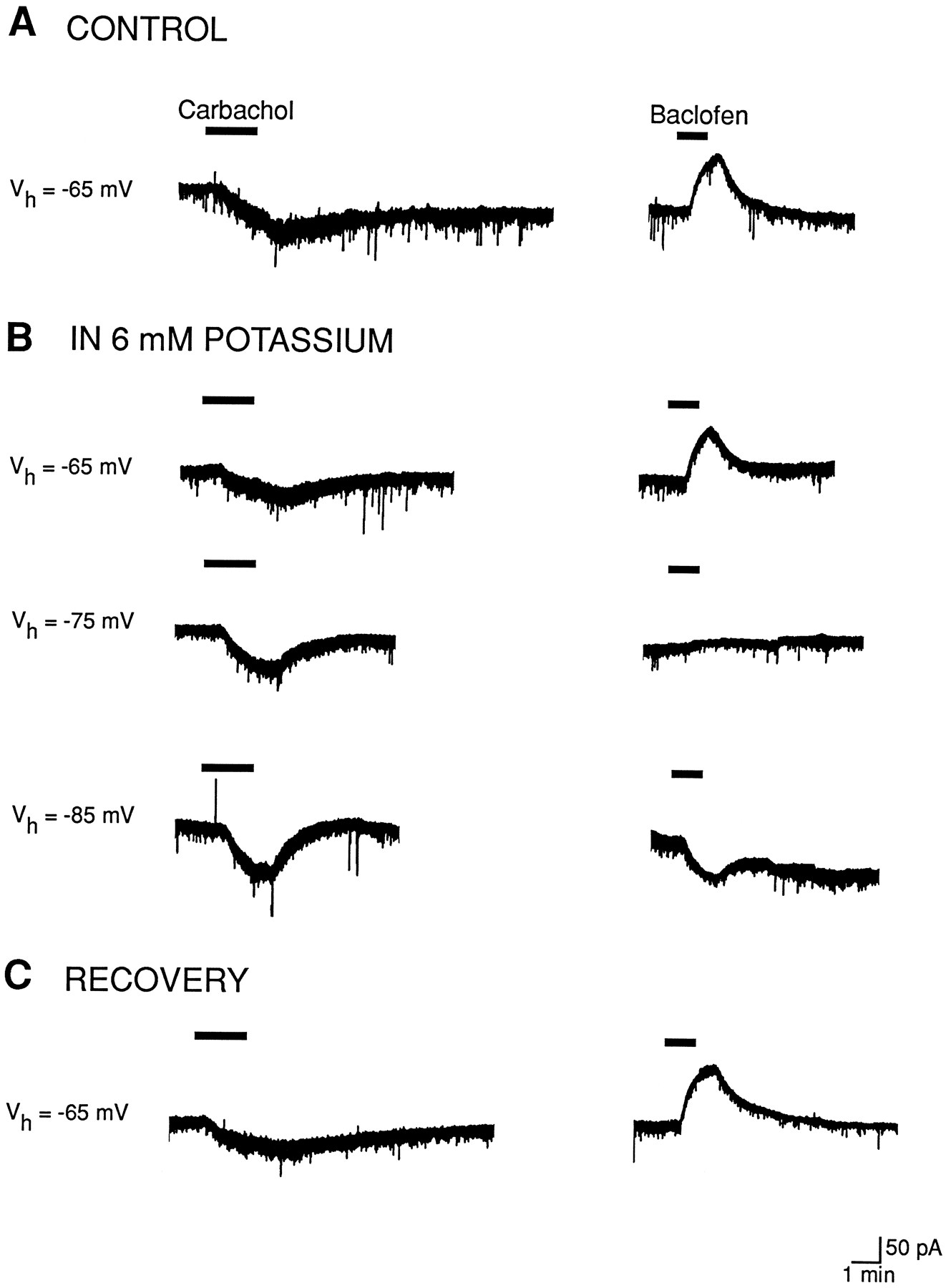
If carbachol elicited an inward current by reducing a potassium conductance, then carbachol should be without effect on cells that were voltage-clamped at EK. This prediction was tested by examining the effects of carbachol in 6 mm extracellular potassium, which shiftsEK into a voltage range where robust carbachol responses were seen under control conditions. As illustrated in Figure 5, carbachol still induced an inward current (n = 5) when cells were held atEK. Even more striking, carbachol was still able to elicit inward currents when tested at potentials negative toEK, at which point the baclofen-induced potassium current was reversed.

Carbachol elicits an inward current atEK as well as belowEK. A, The lefttrace illustrates the inward current induced by carbachol (30 μm), and the right traceshows the outward current elicited by baclofen (30 μm) in a cell held at −65 mV in the control condition (2.5 mm potassium) and in the presence of TTX (1 μm). B, Carbachol-induced currents (left traces) and baclofen-induced currents (right traces) recorded at different holding potentials (−65, −75, and −85 mV) in 6 mmpotassium and 1 μm TTX. Note that carbachol still induced an inward current even at potentials more negative thanEK. C, Recovery of carbachol and baclofen currents in 2.5 mm potassium. All of the traces represented in A, B, andC are from the same cell. The holding current at −65 mV in 2.5 mm potassium was 0.07 nA at the beginning of the experiment and 0.06 nA after recovery from 6 mm potassium. In 6 mmextracellular potassium, the holding current at −65 mV was 0.04 nA, at −75 mV was −0.17 nA, and at −85 mV was −0.27 nA.
In contrast to the results with extracellular potassium substitution, the carbachol-induced current was found to depend critically on the extracellular concentration of sodium (Fig. 6). Thus, replacement of 80% of the extracellular sodium withN-methyl-d-glucamine elicited a reversible reduction in the amplitude of the carbachol current (control = 138 ± 12 pA; low sodium = 36 ± 5.1 pA; p < 0.001;n = 5). No change in the cell membrane resistance was observed with the sodium substitution, suggesting that the reduction in the carbachol current was not secondary to the loss of voltage control of the dendritic arbor.

The carbachol current is dependent on the extracellular concentration of sodium. A, Effect of reducing extracellular sodium on the amplitude of the carbachol-induced current on a cell clamped at −65 mV (holding current = 0.17 nA) in the presence of TTX (1 μm). The lefttrace illustrates the carbachol response in control sodium (146 mm), the middle traceillustrates the response to carbachol observed in low sodium (26 mm), and the right traceillustrates the recovery. Dashed lines indicate the baseline current. B, I–V curves of the carbachol current recorded under control conditions (a), in 26 mm sodium (b), and recovery (c) from the same neuron illustrated in A.C, Summary plot of the amplitude of the carbachol-induced current recorded under control conditions (solid bar) and in 26 mm sodium (open bar). Error bars illustrate the SEM from five different determinations. **p < 0.01 versus control.
The above results suggested that the carbachol-induced inward current was mediated by an increase in sodium (or cation) conductance. This conclusion would appear at odds with the observation that the muscarinic depolarization is associated with an apparent increase in membrane resistance. These anomalous results are not peculiar to prefrontal cortex and have been observed in previous studies elsewhere (Benson et al., 1988; Shen and North, 1992a,b; Stevens et al., 1994), where they have been interpreted as resulting from a simultaneous increase in cation and decrease in potassium conductance. However, these results could also be explained if the inward current was mediated by the activation of voltage-dependent cation current. Therefore, the remaining experiments in this study were devoted to distinguishing among these possibilities.
We first examined the possible voltage dependence of the muscarine-induced inward current. To facilitate these studies, the inward current was isolated by recording with a cesium gluconate-based intracellular solution in the presence of cesium (2 mm), barium (100 μm), and cadmium 100 μm to block inwardly rectifying and calcium-activated currents, respectively. Under these conditions, carbachol was still capable of inducing a robust inward current while holding at −40 mV (Fig. 7A). Hyperpolarizing steps to between −120 and −60 mV revealed that the muscarinic inward current turned off within tens of milliseconds after hyperpolarization (Fig. 7B; n = 4), indicating a strong voltage dependence for the current. No fast-inactivating inward current was seen after recovery from carbachol. This indicated that the fast turn-off of the inward current observed at hyperpolarization was not simply an artifact of the substraction procedure used to isolate the net carbachol current. The effect of this voltage dependence on the appearance of the current is most vividly seen when the instantaneous and steady-state I–V relationships are compared. As illustrated in Figure 7C, the instantaneousI–V relationship is approximately linear, decreases with depolarization, and has an extrapolated reversal potential well-depolarized from rest, as expected for a current mediated by an increase in sodium (or cation) permeability. In contrast, the steady-state I–V curve shows a current that increases with depolarization and appears to be associated with a decrease in membrane conductance. From these results, it is clear that the apparent increase in membrane resistance associated with the muscarinic depolarization and inward current is the direct result of the voltage dependence of the underlying current.

Voltage dependence of muscarinic-activated inward current. A, Carbachol current recorded with cesium gluconate-based intracellular solution in the presence of cesium (2 mm), barium (100 μm), cadmium (100 μm), and TTX (1 μm) at −40 mV. B, Hyperpolarizing voltage step commands from −40 to −120 mV (protocol represented inupper right traces) were applied before and during carbachol application. B, Net carbachol current tracings were obtained by subtracting the currents obtained in control conditions from those obtained during carbachol application. The instantaneous muscarinic current increases in amplitude with hyperpolarization. However, the hyperpolarizing steps also resulted in a fast outward relaxation, reflecting the voltage-dependent turning-off of the muscarinic current. C, Comparison of the instantaneous and steady-state I–V curves for the carbachol-induced current obtained from the cell represented inA. The holding current at −40 mV was 0.05 nA.
If the carbachol-induced inward current is mediated by an increase in cation conductance, then it should reverse near 0 mV. We determined the reversal potential of the carbachol-induced current by using a slow linear voltage ramp from −80 to +10 mV. As illustrated in Figure8, the amplitude of the carbachol current exhibited characteristics consistent with its voltage dependence and reversed polarity at approximately −15 mV (Fig. 8; n = 4). The value of the reversal potential, as determined within the technical limitation of the brain slices technique, was more negative than would be expected for a pure sodium current. This suggested that potassium ions could play a role in the generation of the muscarinic inward current.

The carbachol-induced inward current reverses polarity near −15 mV. A, Current–voltage (I–V) plots were obtained using a linear voltage ramp from −80 to +10 mV before and during carbachol application in a neuron recorded with a cesium gluconate-based intracellular solution in the presence of cadmium (100 μm) and TTX (1 μm). B, Net carbachol current obtained by subtracting the I–V plots represented in A. The carbachol-induced current reversed polarity at a mean potential of −16 mV ± 2 mV (n = 4).
A contribution by potassium could take either of two forms. The carbachol inward current might reflect the opening of cation nonselective channels through which potassium might flow in the outward direction. Alternatively, the carbachol inward current could be heterogeneous, involving a component mediated by a decrease in potassium conductance. Two sets of experiments were conducted to distinguish between these possibilities. If a decrease in potassium conductance made a significant contribution to the carbachol current, then intracellular cesium loading, which blocks a wide range of potassium channels, should reduce the effect of carbachol. This conjecture was tested by quantitatively comparing the effect of carbachol under control conditions and with a cesium-based intracellular solution. As illustrated in Figure 9, intracellular cesium loading failed to reduce the carbachol-induced current. Indeed, group comparisons indicated that the amplitude of the carbachol-induced current recorded using cesium as the predominant intracellular cation was essentially indistinguishable from that recorded using potassium (control = 113 ± 5 pA; cesium gluconate = 113 ± 12 pA; Fig. 9C; n = 6). In contrast, intracellular injection of cesium completely blocked the potassium conductance induced by baclofen (Fig. 9B,C;n = 3). These findings suggest that the carbachol-induced depolarization and inward current observed near rest are mediated predominantly, if not exclusively, via the activation of a voltage-dependent cation current.

The carbachol-induced current is not reduced by intracellular injection of cesium. A, Carbachol- and baclofen-induced currents were recorded using a potassium methylsulfate-based intracellular solution in the presence of TTX (1 μm). The left traceillustrates the inward current caused by carbachol (30 μm) at a holding potential of −65 mV. Theright trace illustrates the outward current elicited by baclofen (30 μm) in the same cell (holding current = 0.11 nA). B, Carbachol-induced (lefttrace) and baclofen-induced (right trace) currents were recorded using a cesium gluconate-based intracellular solution. Intracellular injection of cesium failed to reduce the carbachol current but suppressed the outward current elicited by baclofen. The holding current for this cell was −0.08 nA.C, Summary plot comparing the absolute amplitude of the carbachol- and baclofen-induced currents recorded using potassium methylsulfate-based (solid bar; n = 6) or cesium gluconate-based (open bar; n = 6) intracellular solutions. Error bars illustrate the SEM determined for each group. **p < 0.01 versus control.
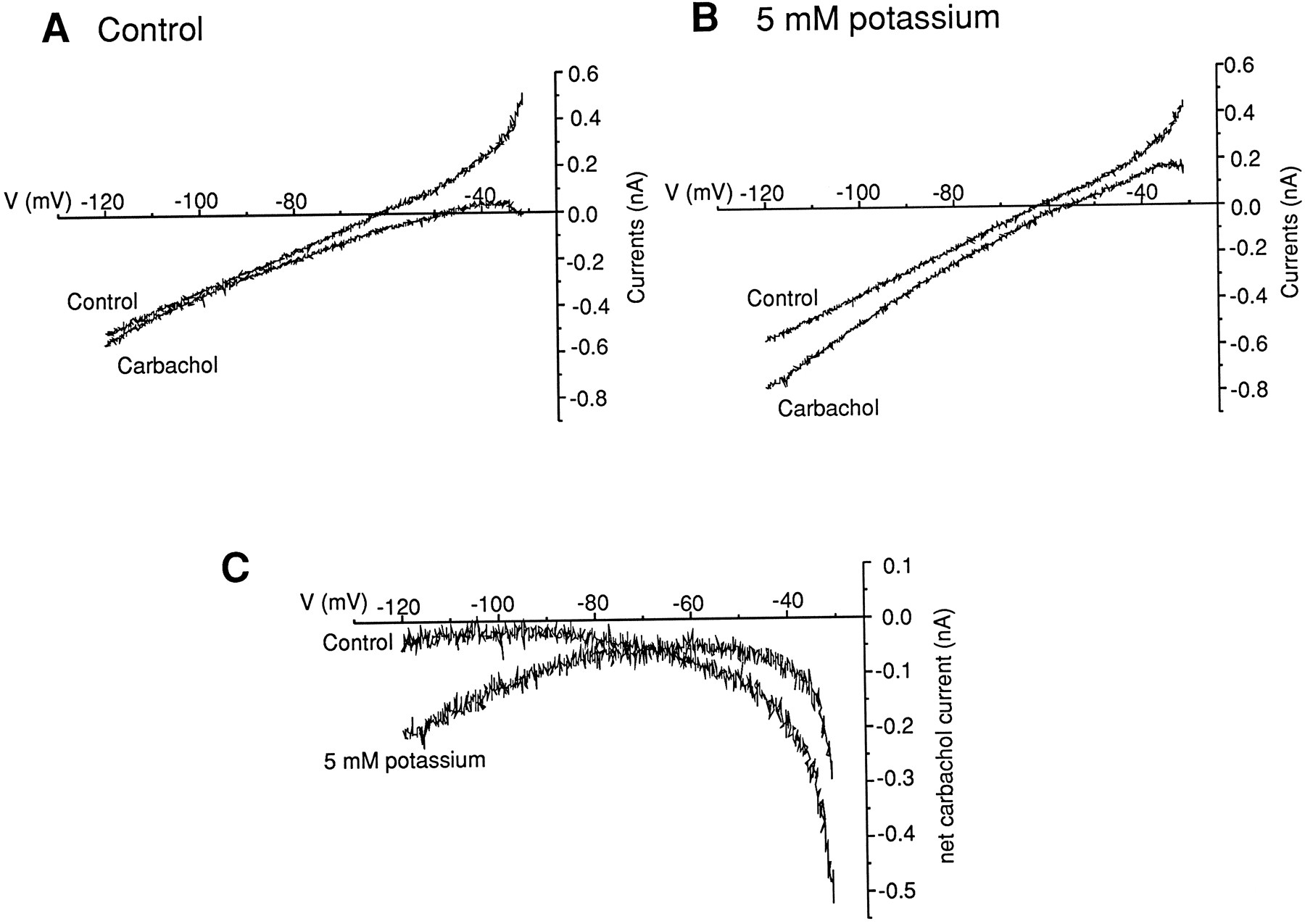
A current mediated by an increase in a cation nonselective current should be increased by elevating extracellular potassium, whereas a current that is mediated at least partly by a decrease in potassium conductance should be reduced by this manipulation. Thus, these two mechanisms make qualitatively different predictions regarding the behavior of the current in high extracellular potassium. As stated above, the increase in the extracellular potassium had little or no effect on the amplitude of the carbachol-induced current at potentials close to the rest (−65 mV). However, when we compared theI–V curves for the carbachol-induced inward current under control conditions (2.5 mmextracellular potassium) and in the presence of elevated (5 mm) extracellular potassium, we found that the increase of the extracellular potassium changed theI–V curves of the carbachol current (Fig.10; n = 5), with the carbachol current being enhanced at hyperpolarized potentials. This enhancement was also evident when carbachol was administered repeatedly while holding at progressively more hyperpolarized potentials (Fig. 5B). These results are consistent with the prediction for a cation nonselective current. In addition, the carbacholI–V curves were also found to cross between −60 and −70 mV, suggesting that the elevated potassium also decreased the carbachol current above this voltage (Fig. 10C; n = 5).

Effect of elevated extracellular potassium on theI–V curves for the carbachol-induced inward current. A, Control and carbachol I–Vrelationships obtained in the presence of 2.5 mmextracellular potassium. B, Control and carbacholI–V relationships obtained in the presence of 5 mm extracellular potassium. C,I–V relationship for the net carbachol current observed in the presence of 2.5 and 5 mmextracellular potassium (holding current = 0.06 nA).
The above experiments did not address a possible contribution of chloride conductances to the carbachol-induced depolarization. To test for such a contribution, we examined the effect of changingECl on the carbachol-induced current. As illustrated in Figure 11A, reducing extracellular chloride from 126.5 to 7.5 mm(replacing NaCl with sodium isethionate) failed to induce changes in either the polarity or the amplitude of carbachol-mediated current (control = 98 ± 21 pA; low chloride = 100 ± 19 pA; n = 5). In contrast, lowering the extracellular concentration of chloride changed the polarity of chloride-mediated GABAAsynaptic potential (Fig. 11B), which confirmed that the manipulation was effective in altering the chloride gradient across the cell membrane.

The carbachol current is insensitive to changes in the chloride gradient. A, Carbachol-induced current recorded at a holding potential of −65 mV in the presence of TTX (1 μm), under control conditions (126.5 mm extracellular chloride, lefttrace), in low extracellular chloride (7.5 mm, middle trace), and during recovery (right trace). Lowering the extracellular concentration of chloride failed to induce changes in either the polarity or the amplitude of the carbachol current. B, Inhibitory postsynaptic potential evoked by stimulation of layer III (25–30 V for 500 μsec) in the presence of DNQX (10 μm) and APV (10 μm) in control conditions and in low chloride. Chloride substitution induced a change in the polarity of GABAA synaptic potential, indicating that this manipulation was effective in altering the chloride gradient across the cell membrane (holding current = 0.11 nA).
Effects of barium and TEA
Administration of barium or TEA was found to reduce strongly the amplitude of the carbachol-induced current. As illustrated in Figure3C, bath application of 2–5 mm barium elicited a large inward current and significantly reduced the amplitude of the carbachol inward current (n = 14). For example, 2 mm barium inhibited the inward current elicited by 30 μm carbachol by approximately half (control = 115 ± 15 pA; in barium = 55 ± 2.9 pA; n = 4;p < 0.05). The traditional interpretation of such results would be that barium, by blocking potassium channels, effectively occludes the muscarinic response. As such, this interpretation would suggest that the carbachol inward current is mediated by a decrease in potassium conductance. Such an interpretation, however, is at odds with the findings outlined above that indicate a predominant role for a cation nonselective current. One way out of this paradox would be if barium also blocked the cation nonselective current. This possibility was tested by assessing the effect of barium on the carbachol response near EK (at −55 mV in 10 mm extracellular potassium), a condition in which most, if not all, of the carbachol current would be carried through cation nonselective channels. Under this condition, barium administration elicited a much-reduced inward current yet inhibited the carbachol inward current as effectively as under control conditions (control = 143 ± 8.8 pA; in barium = 73 ± 13 pA; n = 3). Bath administration of TEA (2 mm) also inhibited the carbachol inward current (Fig. 12; n = 5) although, unlike barium, it did so without by itself eliciting an inward current.

TEA inhibits the carbachol current without by itself eliciting an inward current. Administration of carbachol (30 μm) in the presence of TTX (1 μm) elicited an inward current in a cell clamped at −65 mV. Bath administration of TEA (2 mm) after recovery from the carbachol failed to induce any inward current. However, a second application of carbachol in the presence of TEA elicited a greatly reduced inward current. Partial recovery of the carbachol current was observed after removal of the TEA from the bath (holding current = 0.17 nA).
DISCUSSION
In rat prefrontal cortex, acetylcholine administration to layer V pyramidal cells results in a membrane depolarization. This response appears to be signaled exclusively via muscarinic receptors, as mimicked by muscarine and oxotremorine but not by nicotine, and is blocked by low concentrations of atropine. Activation of muscarinic receptors has long been thought to depolarize cortical neurons by inhibiting potassium channels (Krnjevic et al., 1971; ffrench-Mullen et al., 1983; McCormick and Prince, 1985, 1986). Surprisingly, we find that the depolarization observed in this region is mediated predominantly, if not exclusively, by the activation of a voltage-dependent cation nonselective current.
Several lines of evidence support this conclusion. In the majority of cells tested, the muscarinic receptor-induced inward current decreased with hyperpolarization but did not reverse polarity even at potentials more negative than the theoretical estimateEK. This failure to reverse the carbachol current contrasted the unambiguous reversals of the baclofen-mediated potassium current at the predicted EK. Although it is easier to detect a clear inversion of a current associated with an increase in the membrane conductance, it is unlikely that the absence of an inversion in the case of the carbachol-induced current reflects poor voltage control. Indeed, when space-clamping artifacts were minimized by using localized applications of acetylcholine to the immediate vicinity of the recording site, the inward current still failed to reverse. Consistent with these findings, carbachol was able to elicit an inward current even when the cell membrane was held at EK in high potassium. Furthermore, intracellular injection of cesium did not affect either the amplitude or the voltage sensitivity of the carbachol-induced current. These observations indicate that the muscarinic current is unlikely to be mediated solely by a decrease in a leak or voltage-dependent potassium current.
The muscarinic inward current was greatly inhibited by lowering extracellular sodium. In contrast to sodium substitution, extracellular chloride replacement had no effect on the muscarinic receptor-induced inward current. From these results, the muscarine-induced inward current could have resulted from the activation of a sodium or a cation nonselective current. To distinguish between these possibilities, the reversal potential for carbachol-induced current was determined. Within the technical limitation imposed by the use of brain slices, the carbachol current was found to reverse polarity near −15 mV, a value more negative than the predicted equilibrium potential for sodium ions under our conditions (+53 mV). This suggests that the carbachol-induced current was most likely attributable to the activation of a cation nonselective rather than a pure sodium current. This was further confirmed by examining the effect of raising extracellular potassium on the amplitude of the carbachol current. Consistent with the activation of a cation nonselective current, the amplitude of the carbachol current was increased at potentials negative from rest. This effect, however, was somewhat larger than expected for a cation nonselective current. This suggests that other mechanisms might also contribute to the enhancement of the current in elevated extracellular potassium. Nevertheless, combined, these results argue for the activation of a cation nonselective current.
In ∼25% of the cells examined, the carbachol-induced inward current reversed near EK. This suggests that in some cells a decrease in potassium conductance could contribute to the carbachol depolarization. Alternatively, other ionic mechanisms, such as a negative shift in the voltage dependence ofIh, could also account for the observed reversal. Unfortunately, the inability to obtain repeated reversals of the current under our experimental conditions did not allow us to address this issue. Thus, it remains possible that a small decrease in potassium conductance could contribute to the muscarinic depolarization seen in this region. Such a mixed ionic mechanism has been proposed previously to account for receptor-mediated inward current in other neuronal preparations (Shen and North, 1992a,b).
Receptor-mediated increases in cation nonselective currents have been reported in a variety of preparations (Benham, 1985; Freschi and Livengood, 1989; Inoue and Isenberg, 1990b; Shen and North, 1992a;Sims, 1992; Colino and Halliwell, 1993; Trimmer, 1994). In some preparations, these currents show clear voltage sensitivity (Benham, 1985; Freschi and Livengood, 1989; Inoue and Isenberg, 1990b; Inoue and Kuriyama, 1991), whereas in others they appear to be voltage-insensitive (Shen and North, 1992a,b; Colino and Halliwell, 1993). The cation nonselective current studied here is strongly voltage-dependent, increasing with depolarization and decreasing with hyperpolarization from rest. The technical limitations inherent to voltage-clamping neurons in brain slices made it impractical to attempt a quantitative characterization of the voltage dependence of this current. However, within these limitations, it was possible to show that carbachol caused an increase in chord conductance at the same time that it induced a decrease in slope conductance at or near the resting membrane potential. This explains the apparent increase in membrane resistance brought about by carbachol. To our knowledge, this is the first report of a muscarinic receptors-activated voltage-dependent cation current in the brain. However, a voltage-dependent cation current with properties remarkably similar to those observed here has been shown to mediate muscarinic response in guinea pig ileum (Inoue and Isenberg, 1990b).
Surprisingly, barium and TEA were found to inhibit the muscarinic inward current. Because both of these compounds are potassium channel blockers, on the surface this finding would seem to contradict the conclusion that the muscarinic inward current reflects predominantly the activation of a cation nonselective current. However, barium still inhibited the inward current when holding nearEK in elevated potassium. This suggests that barium reduced the muscarinic inward current by directly inhibiting the cation nonselective current. Similarly, TEA also inhibited the muscarinic inward current. However, in this case the inhibition occurred in the absence of a TEA-induced inward current. This observation indicates that TEA must block channels that open in response to muscarinic stimulation; if the inward current were mediated via the closure of potassium channels, TEA, by blocking the channels itself, would have to induce an inward current. The ability of TEA to block muscarinic receptors is unlikely to have contributed to the blockade of the current, because the TEA concentration used here was 10- to 100-fold lower than the Ki of TEA for muscarinic receptors (Caulfield, 1991). The present observation that TEA and barium can block a cation nonselective current is consistent with recent observations elsewhere in neuronal (Shen and North, 1992a,b) and non-neuronal cells (Inoue and Kuriyama, 1991).
Previous studies on receptor-activated cation nonselective currents have shown that some (Inoue and Isenberg, 1990a; Pacaud and Bolton, 1991; Sims, 1992), but not all (Shen and North, 1992a,b; Colino and Halliwell, 1993), of these currents are regulated by calcium. The cation nonselective current studied here clearly is not calcium-activated, as it can be seen in the presence of strong intracellular calcium buffering. However, in smooth muscle cells, the muscarinic-activated cation nonselective current, although not directly activated by calcium, is nevertheless modulated by the intracellular levels of this cation (Inoue and Isenberg, 1990a). It remains possible that a similar enhancement by calcium of the cation nonselective current might be present in cortical neurons.
The present conclusion that the muscarinic depolarization in these cells is not mediated by a decrease in potassium conductance but by the activation of a voltage-dependent cation nonselective current is consistent with previous studies on muscarinic responses in smooth muscle (Benham, 1985; Inoue and Isenberg, 1990b; Sims, 1992) and with the growing realization that cation nonselective currents also play an important role in mediating muscarinic responses in invertebrate (Freschi and Livengood, 1989; Trimmer, 1994) and vertebrate (Shen and North, 1992a; Colino and Halliwell, 1993) neurons. Interestingly, in the rat prefrontal cortex, a variety of receptor subtypes such as muscarinic cholinergic, α1-adrenergic, and serotonin type 2 induce an overlapping cluster of membrane responses, including the membrane depolarization studied here, a reduction of the afterhyperpolarization, and the appearance of a slow afterdepolarization (Andrade, 1991; Araneda and Andrade, 1991). We have previously proposed that this cluster of effects might reflect convergence of receptors coupled to the inositol–phospholipid signaling mechanism on a shared set of cellular mechanisms and ion channels (Araneda and Andrade, 1991). As such, the present results suggest that the cation nonselective current identified in this study might be the target for several other neurotransmitter receptors. Further studies will be required to test this prediction directly.
What is remarkable about the cation nonselective current studied here is its ability to mimic the behavior expected for a current resulting from a decrease in a resting potassium conductance when it is examined by using traditional electrophysiological tests. For example, its very steep voltage dependence around threshold secures that hyperpolarizing pulses delivered under current clamp would turn off the current. This causes a net outward current that adds to the hyperpolarizing pulse, in turn causing a larger voltage deflection and, thus, the appearance of an increase in membrane resistance. Moreover, the sensitivity to barium and TEA suggests the involvement of potassium channels. Finally, the voltage dependence of the current mimics the behavior expected for a potassium current at potentials positive toEk. The initial clue that potassium channels might not be involved, namely the failure of the current to reverse at EK, is easy to dismiss. Indeed, in this study, it was only the systematic exploration of the properties of the current that led us to question the interpretation that the current reflected a reduction in potassium conductance. It is noteworthy that neurotransmitter receptor-induced inward currents with voltage-dependent and pharmacological characteristics similar to those outlined above, but that generally fail to reverse atEk, have been reported frequently in the literature (Benson et al., 1988; Pan et al., 1994; Stevens et al., 1994). Such currents generally have been attributed to the closure of voltage-dependent potassium channels. It is possible that at least a proportion of these currents might be mediated by voltage-dependent cation nonselective current similar or identical to that reported here. Thus, it is also possible that the voltage-dependent cation nonselective current identified here may play an important role in mediating neurotransmitter actions in the brain.
Footnotes
This research was supported by MH49355 and the Alfred P. Sloan Foundation. We thank Dr. T. Egan for helpful suggestions and discussion and Dr. C. L. Arfken for reading this manuscript.
Correspondence should be addressed to Dr. Samir Haj-Dahmane, Department of Psychiatry and Behavioral Neuroscience, Wayne State University School of Medicine, 2309 Scott Hall, 540 East Canfield, Detroit, MI 48201.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}