

Abstract
Submembrane [Ca2+]ichanges were examined in rat chromaffin cells by monitoring the activity of an endogenous Ca2+-dependent protein: the large conductance Ca2+- and voltage-activated K+ channel (also known as the BK channel). The Ca2+ and voltage dependence of BK current inactivation and conductance were calibrated first by using defined [Ca2+]i salines. This information was used to examine submembrane [Ca2+]i elevations arising out of Ca2+ influx and muscarine-mediated release of Ca2+ from intracellular stores. During Ca2+ influx, some BK channels are exposed to [Ca2+]i of at least 60 μm. However, the distribution of this [Ca2+]i elevation is highly nonuniform so that the average [Ca2+]i detected when all BK channels are activated is only ∼10 μm. Intracellular dialysis with 1 mm or higher EGTA spares only the BK channels activated by the highest [Ca2+]i during influx, whereas dialysis with 1 mm or higher BAPTA blocks activation of all BK channels. Submembrane [Ca2+]i elevations fall rapidly after termination of short (5 msec) Ca2+influx steps but persist above 1 μm for several hundred milliseconds after termination of long (200 msec) influx steps. In contrast to influx, the submembrane [Ca2+]i elevations produced by release of intracellular Ca2+ by muscarinic actetylcholine receptor (mAChR) activation are much more uniform and reach peak levels of 3–5 μm. Our results suggest that during normal action potential activity only 10–20% of BK channels in each chromaffin cell see sufficient [Ca2+]i to be activated.
- BK channels
- calcium
- calcium channels
- calcium stores
- chromaffin cells
- catecholamine secretion
- K+channel inactivation
The free cytosolic concentration of Ca2+([Ca2+]i) is an important regulator of numerous cellular functions, including neurotransmitter release, activation of ion channels, and cell death. Because elevations of [Ca2+]i have such a wide range of possible consequences, it is to be expected that cells would have developed methods to trigger the various Ca2+-dependent events independently of one another. However, exactly how this is accomplished is not entirely clear. What is known is that the spread of free Ca2+ away from Ca2+ sources is limited to short distances (Allbritton et al., 1992) and that the Ca2+ sensitivities of intracellular Ca2+-binding target proteins vary widely (Kasai, 1993). These observations have led to suggestions that localized Ca2+ elevations in the immediate vicinity of the various Ca2+-dependent proteins must be of central importance (Chad and Eckert, 1984; Simon and Llinas, 1985;Augustine and Neher, 1992a). If so, appreciation of the specific consequences of Ca2+ elevations requires the elucidation of both the features of the [Ca2+]i elevation, i.e., the magnitude, time course, and the spatial spread of Ca2+, and a knowledge of the precise localization and the Ca2+ sensitivity of the Ca2+-dependent target proteins. The present study examines the relationship between elevations of the submembrane [Ca2+]i in chromaffin cells and one such Ca2+-dependent target protein, the BK channel.
Amplitudes of [Ca2+]ielevations traditionally have been estimated using exogenous fluorescent Ca2+ buffers (Grynkiewicz et al., 1985). Although these buffers can monitor changes in bulk [Ca2+]i that follow physiological stimuli, limitations in the methods used to detect fluorescent signals make it difficult to follow the localized Ca2+ gradients predicted to occur near individual Ca2+ channels. Furthermore, introduction of these exogenous buffers alters many features of the [Ca2+]i elevations (Sala and Hernandez-Cruz, 1990; Zhou and Neher, 1993). Therefore, there is increasing interest in using intrinsic Ca2+-dependent proteins or processes to define the amplitudes and time course of [Ca2+]i elevations. In the squid giant axon (Adler et al., 1991) and frog hair cells (Roberts et al., 1990), this approach has supported the theoretical expectation that [Ca2+]i elevations during Ca2+ influx are large and rapid. Furthermore, these studies have suggested that particular Ca2+-dependent processes are coupled closely to sites of Ca2+ influx.
BK channels sense rapid changes in both membrane voltage and submembrane [Ca2+]i and contribute to action-potential termination in a number of cell types (Lancaster and Adams, 1986; Lang and Ritchie, 1987), including rat chromaffin cells (Solaro et al., 1995). In the present study, BK channels were used to examine submembrane [Ca2+]i elevations in rat chromaffin cells. The Ca2+ sensitivity of two features of BK current, the rate of inactivation and the fractional activation, was defined by direct introduction of defined [Ca2+]i into cells. Then this information was used to determine the amplitude, spatial distribution, and time course of submembrane [Ca2+]i elevations during depolarization-induced Ca2+ influx and muscarine-mediated elevation of [Ca2+]i. Our results indicate that, whereas some BK channels are located close to Ca2+ channels and are activated rapidly by the high [Ca2+]i that occurs around open Ca2+ channels during influx, most BK channels are located at a distance and are activated slowly by the bulk [Ca2+]i.
MATERIALS AND METHODS
Chromaffin cell culture. Methods of rat chromaffin cell isolation and maintenance of chromaffin cell cultures were as described in earlier reports (Neely and Lingle, 1992a; Herrington et al., 1995; Solaro et al., 1995). These were based on procedures described in several earlier studies (Fenwick et al., 1978; Role and Perlman, 1980; Livet, 1984).
Electrophysiological methods. Whole-cell and perforated-patch recordings were performed on cells 2–14 d after plating the cells, as previously described (Neely and Lingle, 1992a,Herrington et al., 1995; Solaro et al., 1995). In experiments in which an intracellular solution of defined [Ca2+]i was used, the standard whole-cell recording procedure was used (Hamill et al., 1981). Whole-cell voltage clamp was controlled with the Clampex program in the pClamp software package (Axon Instruments, Foster City, CA). Analysis of whole-cell current traces was done with our own software. Fitting of current waveforms or extracted data was done using a Levenberg–Marquardt algorithm for minimization of residuals after adjustment of function parameters.
In perforated patch-clamped experiments, uncompensated series resistances (Rs) were typically in the range of 8–15 MΩ, of which 80–90% was electronically compensated. Values for uncompensated series resistance and percentage of compensation are provided in the figure legends. Given the large size of the BK currents in most chromaffin cells, even 2 MΩ of uncompensated Rs can result in appreciable voltage errors. Therefore, analysis was limited to those cells in which voltage errors resulting from the residual uncompensatedRs were <20 mV. Series resistances with the standard whole-cell method were in the range of 4–8 MΩ, of which 80% was compensated.
Solutions. The standard extracellular solution contained (in mm): 140 NaCl; 5.4 KCl; 10 HEPES; 1.8 CaCl2, and 2.0 MgCl2titrated to pH 7.4 with N-methylglucamine (NMG). In experiments in which salines with defined [Ca2+]i were introduced into cells, CaCl2 was excluded in the external saline (0 [Ca2+]o). For perforated patch-clamp experiments, the pipette saline contained the following (in mm): 120 K-aspartate, 30 KCl, 10 HEPES(H+), and 2 MgCl2adjusted to pH 7.4 with NMG. Membrane permeabilization was accomplished with a mixture of amphotericin B (Rae et al., 1991) and pluronic acid, as described previously (Herrington et al., 1995). Osmolarity was measured by dew point (Wescor Osmometer, Wescor, Logan, UT) and adjusted between 290–310. Most experiments were done in the presence of 100 or 200 nm apamin to eliminate SK currents from the data traces. In experiments in which a defined [Ca2+]i was introduced into the cell, the pipette saline contained the following (in mm): 140 KCl, 20 KOH, 10 HEPES(H+), and HEDTA or EGTA with added CaCl2 to make the appropriate free Ca2+. 10 HEDTA was used for the 20 and 60 μm[Ca2+]i salines, 5 HEDTA for the 10 μm[Ca2+]i saline, and 5 EGTA for the 1 and 4 μm[Ca2+]i salines. In experiments in which cells were stepped to +111 mV after a step to +81 mV, the extracellular Na+ was replaced completely with NMG to prevent Na+ block of BK channels (Yellen, 1984) that results from intracellular accumulation of Na+. In the absence of NMG, steps from +81 mV to +111 mV often produce anomalous inward currents presumably reflecting the voltage-dependent block of BK channels by intracellular Na+. Removal of extracellular Na+ abolishes this effect. Extracellular solution changes and drug applications were accomplished via a multibarrel perfusion system, as described previously (Herrington et al., 1995). Voltages for perforated-patch whole-cell recordings have been corrected for a +9 mV liquid junction potential resulting from the use of aspartate-based pipette salines.
Estimates of the maximal available BK current. A modification of the Hodgkin–Huxley (H–H; 1952) model was used to estimate the maximal activatable current at +81 mV after any Ca2+ influx step or conditioning step. After a prepulse, peak current resulting from a step to +81 mV arises from two populations of channels: those that already are open at the time of the step and those that open after the step. Current through the first population simply decays in accordance with BKiinactivation properties. Thus,
 Equation 1Current resulting from the second population follows the usual H–H formalism and can be described by:
Equation 1Current resulting from the second population follows the usual H–H formalism and can be described by:
 Equation 2in which τh in both equations 1 and 2 represents the inactivation time constant of the channels, τm is the activation time constant,n is the cooperativity in activation,Iinst represents the instantaneous current amplitude, and Imax, the total activatable BK current in the cell. Together, these equations contain one additional free parameter, i.e., Iinst, over the usual H–H formalism, but this parameter is well defined by the current at t = 0. BK currents activated after steps to +81 mV were fit, therefore, with the following:
Equation 2in which τh in both equations 1 and 2 represents the inactivation time constant of the channels, τm is the activation time constant,n is the cooperativity in activation,Iinst represents the instantaneous current amplitude, and Imax, the total activatable BK current in the cell. Together, these equations contain one additional free parameter, i.e., Iinst, over the usual H–H formalism, but this parameter is well defined by the current at t = 0. BK currents activated after steps to +81 mV were fit, therefore, with the following:
 Equation 3Finally, an additional term was included to account for the small amount of steady-state voltage-dependent, but Ca2+-independent current active after inactivation of BK current.
Equation 3Finally, an additional term was included to account for the small amount of steady-state voltage-dependent, but Ca2+-independent current active after inactivation of BK current.
RESULTS
Characteristics of Ca2+-dependent K+currents in rat chromaffin cells
Rat chromaffin cells display two major categories of Ca2+-dependent K+ current: one current results from voltage-dependent, large conductance BK channels (Neely and Lingle, 1992a), and the other results from voltage-independent, small conductance SK channels that are sensitive to apamin (Neely and Lingle, 1992a; Park, 1994). Activation of both types of current is observed with depolarizing steps that produce Ca2+ influx via voltage-dependent Ca2+ channels (Neely and Lingle, 1992a) and with the release of intracellular calcium by agents such as muscarine and caffeine (Neely and Lingle, 1992b; Herrington et al., 1995). Over the range of physiological potentials (−60 to +50 mV), chromaffin cell SK channels are more sensitive to Ca2+ than BK channels: maximal activation of SK channels occurs at [Ca2+]i of 2–4 μm (Park, 1994), whereas maximal activation of BK channels at 4 μm[Ca2+]i occurs only at membrane potentials of +90 mV and above (see Fig. 3). Thus, BK channels may be more suitable for monitoring submembrane [Ca2+]i elevations that exceed 4 μm. In the present study, we have used BK current to study submembrane [Ca2+]i elevations; SK current was blocked with 100 nm apamin.

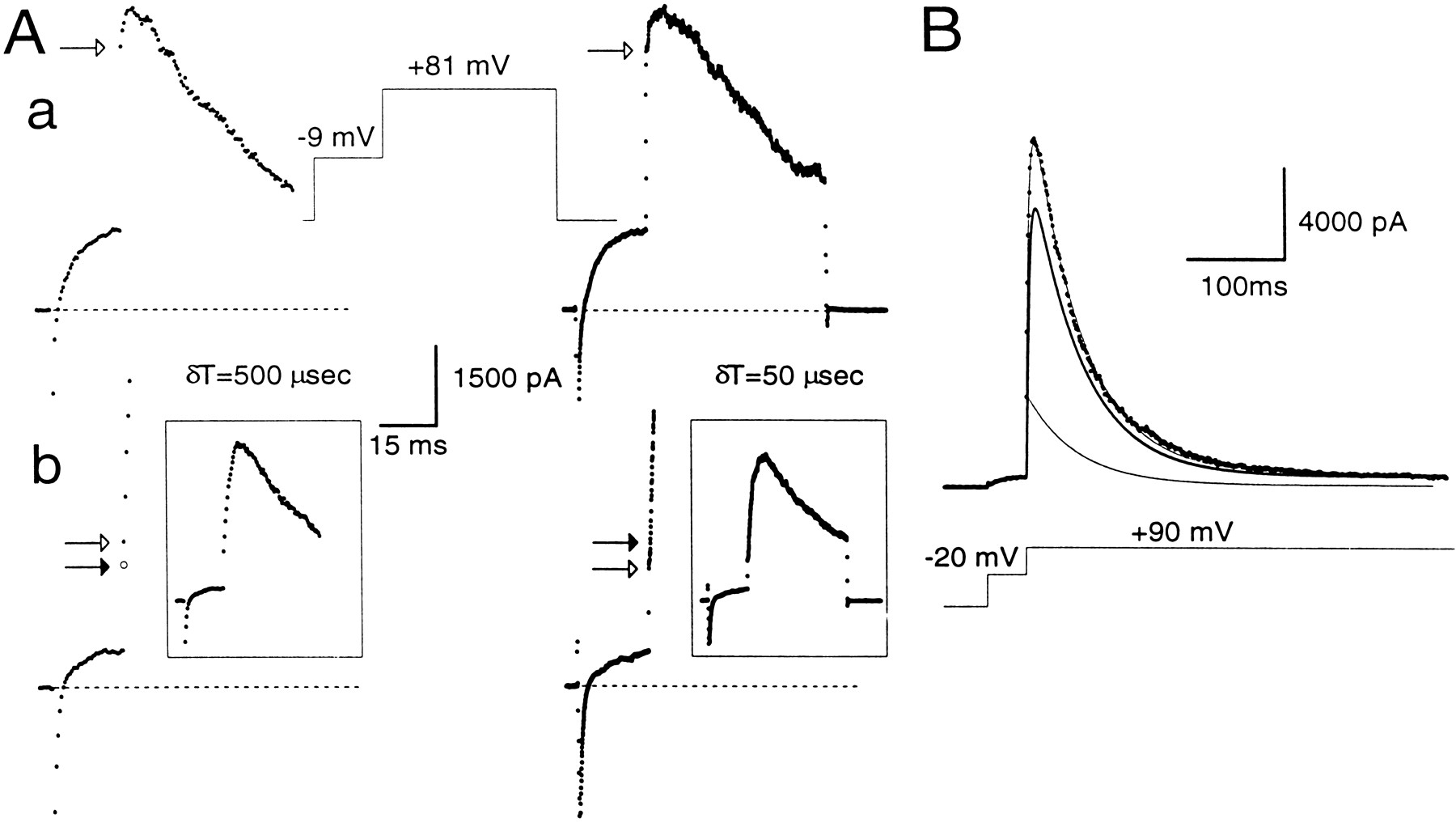
Estimation of instantaneous and peak currents.A, Cells were stepped for 20 msec to −9 mV and then stepped to +81 mV for 200 msec on the left and 50 msec on theright. A cell was clamped with the perforated-patch method in a, whereas in b the cell was clamped with the standard whole-cell method with a pipette containing 10 μm[Ca2+]i. On theleft, currents were digitized with a 500 μsec sampling interval and, on the right, with a 50 μsec sampling interval. In both cases, the step to −9 mV results in activation of outward current. However, with more rapid sampling, the step to +81 mV first reveals more rapid changes in current reflecting the finite voltage settling time and, subsequently, the more slowly developing current reflecting BK current activation. In a, as shown on the right, ∼300 μsec is required for the complete development of the “instantaneous” current. Because of the slower voltage-settling time of cells clamped with the perforated-patch method, current at 500 μsec provides a reasonable estimate of the ohmic component of BK current after the step to +81 mV. Openarrow in each case indicates the measured instantaneous current. In b, by contrast, only ∼100 μsec is required for the full development of the “instantaneous current.” For clarity, only the first few points after the voltage step to +81 mV are shown, and the complete current traces are shown in the insets.An open arrow in each case indicates the measured instantaneous current. The filled arrow on theright indicates the 500 μsec point. Sufficient BK current activation occurs between 100 and 500 μsec so that current at the 500 μsec point will overestimate the instantaneous current. As described in Materials and Methods, the change in current between 500 μsec and 1 msec was used to calculate the predicted current at 250 μsec, assuming a linear rate of current activation. This calculated value (filled arrow on left) provides a reasonable estimate of the instantaneous current observed with the faster sampling rate on the right. a,Rs, 12 MΩ;Cm, 5 pF; 80% compensated. b,Rs, 4 MΩ; Cm, 5 pF; 80% compensated. B, BK current activated at +90 mV by 10 μm[Ca2+]i included in the pipette was fit by a modified Hodgkin–Huxley model of current activation–inactivation. This model accounts for BK inactivation that might occur in the rising phase of the current. The peak current at +90 mV arises from a population of channels already open (the instantaneous current component), which decays in accordance with BKi inactivation properties (indicated by thethin line), and a second population, which follows the usual H–H formalism (indicated by the thick line). The true peak current is the sum of the peak current predicted by the usual H–H formalism and the instantaneous current. Standard whole-cell method. Rs, 5 MΩ;Cm, 6 pF; 80% compensated.
Two types of BK currents are found in rat chromaffin cells (Solaro et al., 1995). This is summarized in Figure1A. Ca2+ (4 μm) was introduced directly into each of two chromaffin cells using the standard whole-cell patch-clamp technique. In one cell, a depolarizing step to +90 mV resulted in a rapidly inactivating BK current (which we have termed BKi current, resulting from BKi channels). In the other cell, the step to +90 mV activated a sustained current (which we have termed BKs current, resulting from BKs channels). This sustained current is more reminiscent of BK currents generally found in most other cell types. BKi current is found in ∼75% of rat chromaffin cells; the remaining cells show either pure BKscurrent or a mix of BKi/BKscurrent (Solaro et al., 1995). In this paper, most results use cells with BKi current. In part, this reflects the greater likelihood of obtaining cells with BKicurrent, but also, as will be shown below, certain features of BKi current are particularly useful for answering some of the questions considered here. To provide assurance that our conclusions do not arise from some novel aspect of BKi current, the behavior of [Ca2+]i in cells with BKs current also is presented in some cases.

Properties of BK currents found in rat chromaffin cells. A, Examples of BKi and BKs current traces recorded with pipettes containing 4 μm are shown. Each cell was held at a holding potential of −60 mV and stepped to +90 mV for 400 msec. The voltage step evoked BKi current in the cell shown on the left and BKs current in the cell shown on the right. B, BKi currents were recorded with pipettes containing 1, 4, or 20 μm[Ca2+]i, respectively, at various voltages. Each cell was held at −60 mV and stepped from the holding potential to potentials ranging from −30 to +90 mV in increments of 10 mV. In all cases, the cell was bathed in an external saline containing 0 [Ca2+]o. C, The time constant of inactivation obtained by fitting current decay with a single exponential is plotted as a function of [Ca2+]i. Each point is the mean (± SEM) of data collected from 4–6 cells. D, The time constant of current inactivation is plotted against the membrane potential at each [Ca2+]i. Each point is the mean (± SEM) of data collected from 4–6 cells. The dottedlines in C and D indicate data from excised patches reported earlier (Solaro and Lingle, 1992). Error bars have not been indicated when smaller than symbol size.
For any membrane current to be a useful assay of submembrane [Ca2+]i, the dependence on [Ca2+]i of some parameter of channel function must be definable. Toward this goal, the [Ca2+]i dependence of two parameters of BK channel function, the conductance and the inactivation rate, are defined first. Then this information is used to address four questions: (1) What is the average [Ca2+]i detected by BK channels during depolarizing steps that produce Ca2+ influx via Ca2+channels? (2) Are there inhomogeneities in the [Ca2+]i detected by BK channels under such conditions? (3) What is the [Ca2+]i detected by BK channels during release of Ca2+ from internal stores by muscarine? (4) What is the duration of submembrane [Ca2+]i elevations with short and long Ca2+ influx steps?
Ca2+ and voltage dependence of BK current inactivation
The Ca2+ and voltage dependence of BKi inactivation previously has been defined for a limited set of [Ca2+]iand voltages by using ensembles of channel openings from excised patches (Solaro and Lingle, 1992). Because the present study relies on the behavior of BK channels in whole-cell recordings, the Ca2+ and voltage dependence of the inactivation time constant was determined with the standard whole-cell technique, using internal salines containing defined [Ca2+]i (Fig.1C,D). The previously published excised patch data are shown for comparison also (Solaro and Lingle, 1992). The whole-cell experiments extend the range of conditions ([Ca2+]i and voltage) over which the inactivation time constant is defined and indicate that the rate of inactivation becomes faster with increasing [Ca2+]i and membrane potential, as reported previously (Solaro and Lingle, 1992; Herrington et al., 1995). Furthermore, the time constants from the whole-cell experiments fall reasonably close to those determined from the patch experiments, suggesting that any discrepancy between the expected and the real [Ca2+]i at the membrane during whole-cell dialysis is minimal and much smaller than the changes that result from the various defined [Ca2+]i solutions. The usefulness of the inactivation time constant as an indicator of submembrane [Ca2+]i is, however, somewhat limited because it approaches its limiting value near only 4 μm[Ca2+]i at positive voltages.
Ca2+ and voltage dependence of the fractional activation of whole-cell BK current
Next, the Ca2+ and the voltage dependence of BK conductance was characterized by introducing a saline with a defined [Ca2+]i into the cell and measuring the fractional activation of BK current at various potentials in the absence of external Ca2+. Fractional activation of BK current was determined by comparing the tail of BK current elicited by a step to +90 mV after different activation potentials to the total amount of BK current activated at +90 mV. The +90 mV step was used because previous excised single-channel experiments indicated that complete activation of BK current occurs at this potential at [Ca2+]iof 4 μm or higher (Solaro, 1995). Thus, current at +90 mV provides a reasonable estimate of the maximum available current at [Ca2+]i of 4 μm and above.
A sample current record obtained with 10 μm[Ca2+]i and a conditioning step to 5 mV is shown in Figure2A. The trace shows that a large BK current is turned on at +90 mV, with some current activation occurring during the conditioning step. Careful examination of the trace also reveals that there is an immediate, instantaneous increase in the current when membrane potential is stepped from the conditioning potential to +90 mV. We will refer to this current as the “instantaneous current” (Fig. 2A). It arises because stepping the membrane potential to +90 mV increases the driving force on the K+ions moving through channels that are open at the conditioning potential, thereby producing an ohmic and immediate increase in the current. After the instantaneous increase in BK current, additional activation of BK current occurs in accordance with the open probability of the channels at +90 mV and the [Ca2+]i present in the cell. This produces a further increase in BK current, which subsequently peaks and then falls as inactivation gains predominance. We will refer to the maximum amplitude of the current at +90 mV as the “peak current.”

Peak and instantaneous currents in BKi cells. In A, 10 μm[Ca2+]i was introduced into the cell. First the cell was stepped from a holding potential of −60 to −5 mV for 20 msec and then stepped directly to +90 mV for 400 msec. After the step to 90 mV, there was a large, ohmic increase in current (the instantaneous current), which was followed by some additional transient activation of current. In B, currents were recorded using pipettes containing 4, 10, 20, or 60 μm free [Ca2+]i. The cells were stepped from the holding potential (−60 mV for cells with 4 and 10 μm[Ca2+]i; −80 mV for cells with 20 and 60 μm[Ca2+]i) to either −65 (left traces) or to −5 mV (righttraces) for 20 msec and then stepped to +90 mV. The instantaneous current at +90 mV became larger at the more positive conditioning steps and increased further as the [Ca2+]i was increased. In all cases, cells were bathed in an external saline containing 0 [Ca2+]o.Arrows on each trace denote the instantaneous (openarrows) and peak (closed arrows) current. Sampling period, 500 μsec.
Because the open probability of BK channels at +80 mV is near maximal at [Ca2+]i of 4 μm and above (Solaro, 1995), the peak current at +90 mV is a reasonably good estimate of the maximum available BK current at the time of the voltage step to +90 mV. Thus, the fractional activation of BK current at the different conditioning potentials can be determined simply by computing the ratio of the instantaneous to the peak current (see Fig. 4). This ratio defines the fraction of channels open at the conditioning potential compared with the total number of channels that can be opened at +90 mV.
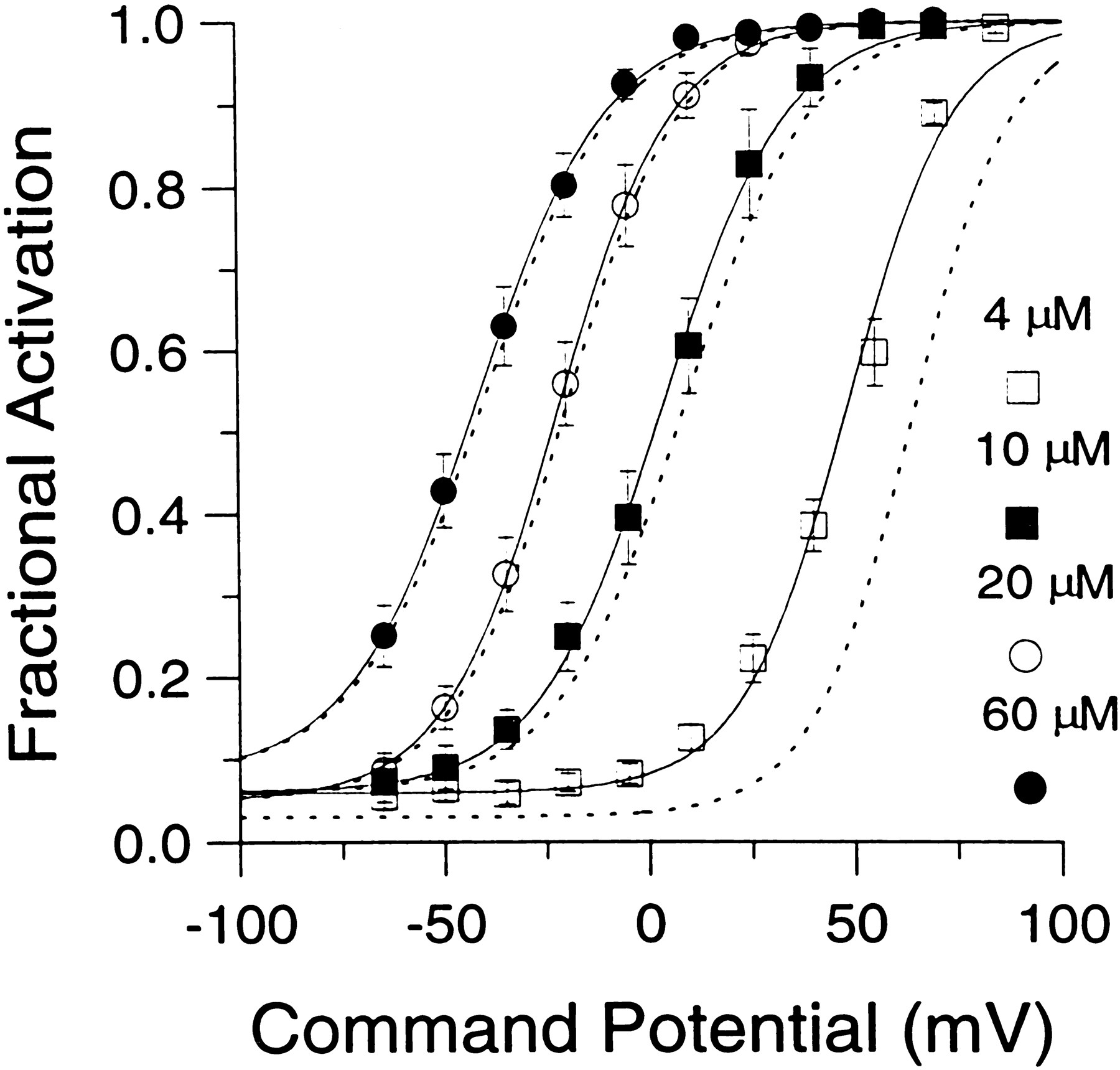
Fractional activation of BKicurrent at various [Ca2+]i and voltages. The instantaneous current was normalized to the peak current activated at +90 mV. This ratio represents the fractional activation of BK current at various conditioning potentials. Each point represents the mean (± SEM) of 6–7 cells. Procedures for estimating the instantaneous current and factors that may affect estimates of both peak and instantaneous current are described in Results. Fractional activation values obtained at each [Ca2+]i were fit with a single Boltzmann function that included a voltage-independent nonzero term. The voltage-independent term for each fit represents the amount of non-BK current activated by the voltage step to +90 mV during the first sampling interval. Fits were constrained toGmax of 1.0. The voltages of half-activation (V50) and slope factors at the various [Ca2+]i were as follows: 4 μm[Ca2+]i, 48.2 mV with a slope factor of 13.1 mV; 10 μm[Ca2+]i, 3.4 mV with a slope factor of 15.3 mV; 20 μm[Ca2+]i. −22.0 mV with a slope factor of 14.1 mV; and 60 μm[Ca2+]i, −41.6 mV with a slope factor of 15.8 mV. The dotted lines indicate the fractional activation data obtained by fitting each current trace with a modified Hodgkin–Huxley function and estimating the peak current from the fit, as described in Materials and Methods.
What are the possible errors in this approach of computing the fractional activation of BK current? One error may arise from imprecise estimates of the instantaneous current. This could result from two factors: the finite current sampling rate and the finite settling time of the imposed voltage change. In the first case, the magnitude of instantaneous current may be overestimated because of BK current activation during the finite sampling period. In the second case, early current points after a voltage step may reflect charging of cell capacitance and slow changes in the command voltage resulting from series resistance limitations. Because many of the experiments described in this paper required voltage protocols with sampling rates as slow as 500 μsec, the impact of these factors on estimates of instantaneous current are summarized in Figure 3 for the two types of whole-cell recording conditions. Currents were digitized at either 50 or 500 μsec. At typical Rsand Cm for cells in this study, voltage settling times are ∼250–300 μsec in cells clamped with the perforated-patch method and ∼75–100 μsec for standard whole-cell recordings. Thus, in perforated patch-clamped cells, the new command potential is not imposed fully for ∼250–300 μsec, and, in general, the first sampled point 500 μsec after the initiation of the voltage step is essentially identical to the estimate of instantaneous current obtained by sampling at 50 μsec intervals (Fig. 3Aa). In contrast, in standard whole-cell recordings, current through open channels seems to sense fully the newly imposed voltage within ∼100 μsec (Fig. 3Ab). Once the new command voltage is imposed, relatively smaller changes in BK current occur during the subsequent 50 μsec sampling periods, and the ohmic component through BK channels is apparent as the first point after the termination of rapid change in current. Thus, for standard whole-cell recordings, the current measured at 500 μsec after the voltage step overestimates the true amplitude of the instantaneous current.
To apply a correction for this sampling error, the following procedure was used. The initial phase of BK current activation is described well by a simple exponential function (Solaro et al., 1995). Therefore, BK current was assumed to activate in a primarily linear manner during the first few milliseconds after a voltage step. The measured current values at 500 and 1 msec were used, therefore, to calculate the amount of current that would have been observed at 250 μsec, assuming this linear rate of current activation. The 250 μsec extrapolation point was used because the amplitude of the instantaneous current defined at this time point agreed well with that obtained by directly sampling at 50 μsec intervals. Thus, fractional activation in cells dialyzed with defined [Ca2+]i salines was obtained by taking the ratio of the corrected instantaneous current and the measured peak current.
A second problem arose from the fact that the peak current would be somewhat underestimated by inactivation that might occur between the voltage step and the time of peak current. We attempted to estimate the extent of this inactivation by fitting current traces with a modified Hodgkin–Huxley model (1952) of current activation–inactivation, as described in Materials and Methods (Fig. 3B). The peak current given by this fit may somewhat overestimate the true peak current, because this model assumes independent activation and inactivation gating, whereas inactivation gating in BKi channels is coupled partially to activation gating (Solaro and Lingle, 1992). The fitting procedure, therefore, places an upper limit on the estimate of the true peak current. The voltages of half-activation (V50s) obtained by using amplitude estimates derived from this fitting procedure were shifted to the right by ∼15 mV at 4 μm[Ca2+]i, 5 mV at 10 μm[Ca2+]i, and by smaller values (1–2 mV) at 20 and 60 μm[Ca2+]i (Fig.4). These shifts in V50 at any individual [Ca2+]i were small compared with the differences in V50 among different [Ca2+]i. Any errors resulting from incorrect estimates of the true peak current are therefore small. Thus, when comparing the calibration data to fractional activation data produced by physiological [Ca2+]i elevations, we used only the directly measured peak current values.
A final potential source of error arises from contamination of the measured currents with the voltage-dependent, Ca2+-independent K+current. The amplitude of this current in rat chromaffin cells is typically ∼1–1.5 nA at +90 mV so that contamination of the estimate of both the instantaneous and peak currents is ∼5–8%. An error of this magnitude is substantially less than the differences that result from the various defined [Ca2+]i solutions.
The above considerations were applied in determining the fractional activation of BK current at various [Ca2+]i and voltages. Resulting fractional activation curves are shown in Figure 4. The procedure yielded roughly similar conductance–voltage curves for both BKi and BKs currents (Solaro et al., 1995). The values for the voltages of half-activation (V50) are within the range of estimates ofV50 obtained from single BK channels in excised chromaffin cells patches (Solaro, 1995). Qualitatively, the results indicate that, at ∼0 mV, submembrane [Ca2+]i must exceed 60 μm to result in maximal BK current activation, whereas at 4 μm[Ca2+]i there is minimal activation of BK current. At voltages exceeding +80 mV, BK current is activated maximally at [Ca2+]i of 4 μm or above. At 1 μm[Ca2+]i, BK current is activated detectably, although only at very positive potentials (Fig.1B). Finally, at 500 nm[Ca2+]i, virtually no BK current activation can be seen even at +90 mV (data not shown).
Fractional activation of BK current during depolarization-elicited Ca2+ influx
To use the fractional activation data of Figure 4 to assay the [Ca2+]i detected by BK channels during Ca2+ influx, the maximal activatable BK current must be defined for each cell. Therefore, the instantaneous and peak currents elicited by Ca2+influx in cells clamped in the perforated patch-clamp configuration were characterized. Figure 5A shows traces from a cell in which currents activated during a voltage step to +81 mV are compared with and without a 200 msec conditioning step to +1 mV. Test pulses to +81 mV were used, because there is minimal Ca2+ influx at this potential and BK currents can be viewed with minimal contamination from other currents. Because of the Ca2+ influx that occurs during the loading step to +1 mV, BK current activated during the step to +81 mV greatly exceeds the purely voltage-dependent K+ current activated by stepping directly to +81 mV. The instantaneous current after the Ca2+ loading step indicates that a substantial number of BK channels are open at this time. Examination of the fractional activation curves in Figure 4 indicates that, at +1 mV, submembrane [Ca2+]i must be in excess of 4 μm to produce an instantaneous current of this magnitude. Now, if the maximum number of BK channels that can be activated by the step to +81 mV can be defined, it would provide a way of more precisely defining the average [Ca2+]i detected by BK channels during Ca2+ influx.

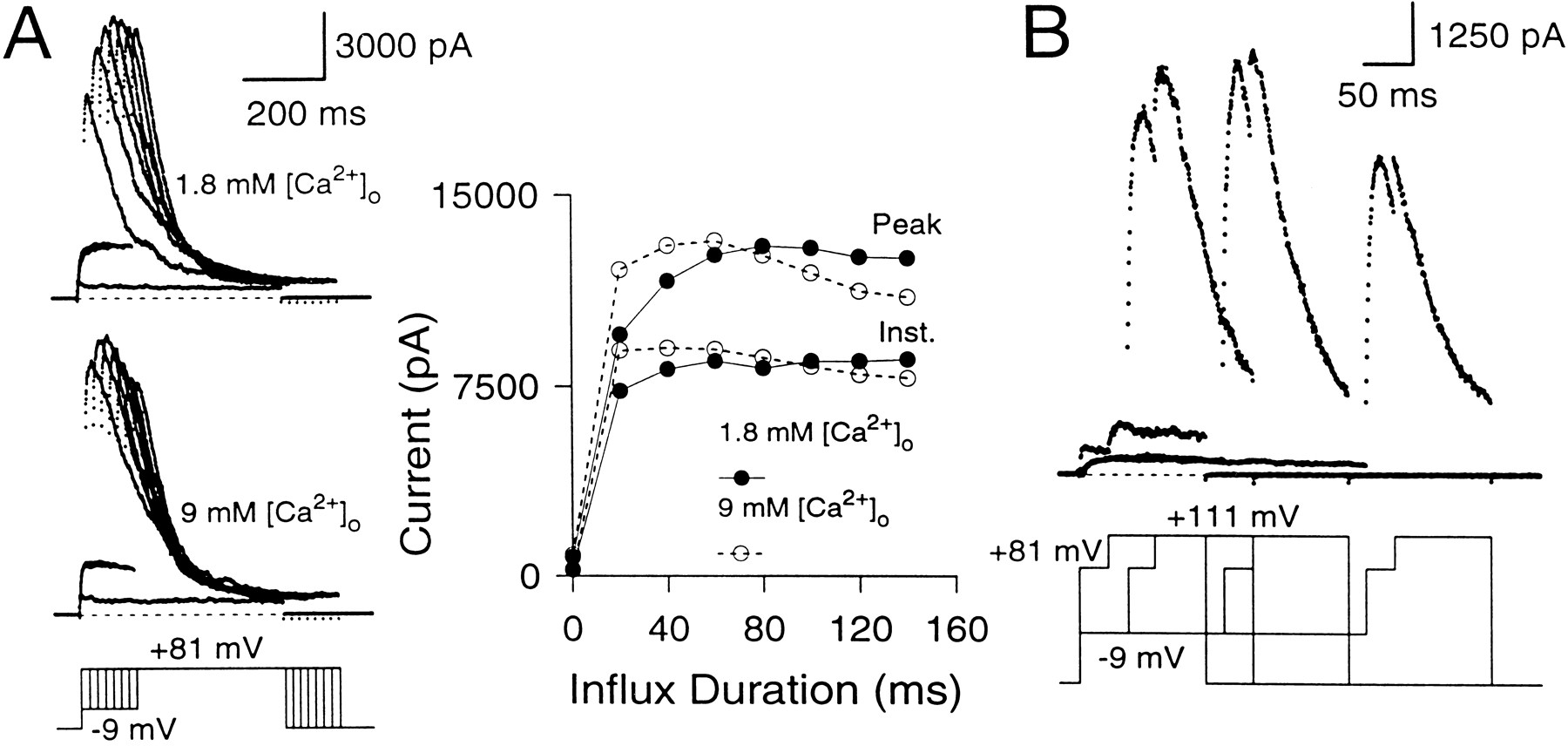
Effect of varying the duration of the Ca2+ influx step on instantaneous and peak currents. A, Outward current in a perforated patch-clamped cell is shown with and without Ca2+ influx. The cell was held at −69 mV and stepped to +81 mV with or without a conditioning step to +1 mV. On the left, the direct step to + 81 mV activates only Ca2+-independent, voltage-dependent K+ current. On theright, the step to +1 mV produces Ca2+influx and results in activation of BK current at +81 mV. Note the prominent instantaneous current at +81 mV. Closed andopen arrows indicate peak and instantaneous currents, respectively. B, The variation of peak and instantaneous currents with increasing influx is demonstrated in a cell with BKi current. The cell was held at −69 mV, stepped to −9 mV to produce Ca2+ influx, and then stepped to +81 mV. The duration of the influx step was varied from 0 to 140 msec in increments of 10 msec. The resulting current traces are shown on the left, and the instantaneous (squares) and peak (circles) current amplitudes at +81 mV for each Ca2+ influx step are plotted against the duration of the influx step on the right. Only every other current trace is shown for clarity. Perforated-patch method. Rs, 9.5 MΩ;Cm, 5.5 pF; 80% compensated. C, Same as B, but in a cell expressing BKs current. Perforated-patch method.Rs, 14 MΩ;Cm, 5 pF; 80% compensated. Sampling period, 500 μsec in all cases.
To define the maximal BK current that can be activated at +81 mV, Ca2+ influx was increased progressively by increasing the duration of a −9 mV Ca2+ loading step in an attempt to load the cell with Ca2+sufficiently so as to activate all BK channels (Fig. 5B). The resulting behavior of the instantaneous and peak currents shows several features worth noting. (1) Even the short duration Ca2+ influx steps produce a clear, instantaneous current. The peak current at +81 mV with such small influx steps is almost all instantaneous current. (2) Small increments in the duration of Ca2+ influx increase both the peak and the instantaneous currents. However, as the duration of the influx step is increased further (influx steps >30–40 msec), the instantaneous current plateaus, whereas subsequent increases in Ca2+ influx increase only the peak current. Eventually (typically after 100–300 msec of Ca2+influx), the peak current also reaches a maximal level (Fig.5C). (3) After long (typically 100 msec) Ca2+ loading steps, rates of inactivation of BKi current at +81 mV increase to values that suggest the residual submembrane [Ca2+]i is equal to or above 3–5 μm (Solaro et al., 1995). The fractional activation data of Figure 4 indicate that this [Ca2+]i should be sufficient to produce maximal BK conductance (gmax) at +81 mV. (4) With even longer Ca2+ loading steps, both the instantaneous and the peak current amplitude of BKi current begin to decrease. This decrease reflects inactivation of BKi channels at the potential of the Ca2+ loading step, as reported previously (Solaro et al., 1995).
Figure 5C shows the effect of varying the Ca2+-influx duration on the instantaneous and the peak current in a cell with BKs current. The basic behavior of the BKs current is similar to BKi current. However, in contrast to BKi current, the instantaneous current in BKs cells consistently shows a tendency to increase slowly with the loading-step duration. This point is discussed later.
Does the maximal peak current provide an estimate of the maximal available current at the time of the +81 mV voltage step? This was tested in two ways. First, Ca2+ influx through Ca2+ channels was increased by changing the extracellular [Ca2+] from 1.8 to 9 mm to test whether increases in Ca2+ influx result in the additional recruitment of any “silent” BK channels that are not active in the 1.8 mm[Ca2+]o saline (Fig.6A). In 9 mm[Ca2+]o, the instantaneous current amplitude elicited by short influx steps is increased relative to the current elicited in 1.8 mm[Ca2+]o. However, the maximal peak current amplitudes are similar in both cases, suggesting that all of the BK channels that are activated at +81 mV in the 9 mm[Ca2+]o saline also can be activated in the 1.8 mm[Ca2+]o saline, albeit at longer Ca2+ influx steps. Second, in three cells, the effect of an additional depolarization to +111 mV after the +81 mV step was tested. After Ca2+ influx steps of 200 msec or longer, this depolarization resulted in no additional increase in conductance over that observed at +81 mV (Fig. 6B), indicating that maximal conductance had been achieved. It is worth noting that because substantial inactivation of BK current occurs in many cells during the Ca2+ loading step, the maximal peak current at +81 mV will not represent all the BK current in a given cell but, rather, the activation of all available BK current.

The maximal peak current elicited at +81 mV with long Ca2+ influx steps defines the maximal available BK conductance. A, The cell was stepped from the holding potential (−69 mV) to −9 mV to produce Ca2+ influx and then stepped to +81 mV. Thetop traces show currents elicited in external saline containing 1.8 mm[Ca2+]o, whereas thebottom traces show currents in the same cell in external saline containing 9 mm[Ca2+]o. On theright, the instantaneous and peak currents are plotted against the duration of the influx step. In both salines, the maximal peak currents at +81 mV reach the same amplitude. Perforated-patch method. Rs, 15.5 MΩ;Cm, 7.8 pF; 80% compensated. B, The cell was stepped to −9 mV for varying durations to produce Ca2+ influx and then stepped to +81 mV for 50 msec to activate the BK current robustly. After 50 msec at +81 mV, the cell was stepped to +111 mV to detect the extent of BK current activation at +81 mV. External Na+ was replaced with N-methyl-d-glucamine (NMG) to circumvent the effects of intracellular Na+ block of BK channels at positive voltages (Yellen, 1984; see Materials and Methods). After short Ca2+ loading steps, there is some additional activation of BK current at +111 mV beyond what is seen at +81 mV. However, after Ca2+ influx steps of 150 msec and longer, additional activation at +111 mV does not occur, indicating that maximal activation of BK current has been achieved at +81 mV. Perforated-patch method.Rs, 11 MΩ;Cm, 4.5 pF; 80% compensated. Sampling period, 500 μsec.
The fractional activation of BK channels activated by Ca2+ influx steps long enough to produce maximal activation of BK current was determined by using the above approach in perforated patch-clamped cells at influx potentials in the −20 to +15 mV range (Fig. 7). Then these data were compared with the calibration data in Figure 4. The comparison showed that, after Ca2+ influx steps of long duration (100–400 msec), the average submembrane [Ca2+]i in different cells varies over a wide range, from somewhat under 10 μm in some cells to over 20 μm in others. However, in the majority of the cells, the average submembrane [Ca2+]i is ∼10 μm.

The fractional activation of BKi current elicited by large Ca2+ influx steps corresponds to an average submembrane [Ca2+]i of 10–20 μm. Ca2+ influx in perforated patch-clamped cells was elicited by loading steps between −20 and +11 mV, which were followed by steps to +81 mV. In each cell, the duration of the Ca2+ loading step was increased progressively by using the protocol shown in Figure 5. The fractional activation of BK current for a given Ca2+ influx potential was measured by calculating the ratio of the instantaneous to the peak current for the trace showing the maximal peak BK current. In some cases, traces for which the Ca2+ loading-step durations were longer than the duration that elicited the maximal peak BK current were used. The fractional activation data obtained at various Ca2+ influx potentials from 34 cells are shown here. Solid lines are the fractional activation calibration curves from Figure 4.
BK channels close to Ca2+ channels can be unmasked by EGTA
Although the above result indicates that the average [Ca2+]i detected by individual BK channels at the end of long Ca2+influx steps is ∼10 μm, it does not rule out large variations in the [Ca2+]i detected by individual BK channels arising from differences in the distances between BK and Ca2+ channels. Thus, BK channels located close to Ca2+ channels would experience high [Ca2+]i, and those located at a distance, low [Ca2+]i. If this is true, the exogenous Ca2+ buffers, EGTA and BAPTA, might differentiate between those BK channels close to Ca2+ channels and those more distant. The equilibrium Ca2+ affinities of these two buffers are comparable, but because the Ca2+ binding rate of EGTA is ∼100 times slower than BAPTA, EGTA should be unable to buffer [Ca2+]i as effectively as BAPTA in the immediate vicinity of Ca2+ channels (Neher, 1986; Stern, 1992). Thus, if BK channels are coupled closely to Ca2+channels, EGTA should be less effective than BAPTA in preventing their activation during influx. This approach has been used in past studies to demonstrate coupling between Ca2+ and BK channels in the frog hair cell (Roberts, 1993) and between Ca2+ channels and secretory vesicles in the terminals of the squid giant axon (Adler et al., 1991).
BK currents were activated with intracellular EGTA in the range of 80 μm to 5 mm, using the standard whole-cell technique (Fig.8A). In 80 μmEGTA, the behavior of the instantaneous and peak current elicited by Ca2+ loading steps of different durations primarily approximates what is observed in cells studied with the perforated patch-clamped method. Instantaneous current reaches a plateau, whereas the peak current slowly increases with loading-step duration and then decreases as inactivation accumulates at −9 mV. However, at higher concentrations of EGTA, the results differ. At 400 μm EGTA, peak current exhibits an interesting biphasic behavior, decreasing at first but then rebounding as the influx step duration is increased further. The instantaneous current, however, shows only a steady decrement under these conditions. At 1 mm EGTA, almost all of the current activated at +81 mV at all Ca2+ loading steps is instantaneous current. This current becomes smaller as the influx duration is increased. At 5 mm EGTA, also, almost all of the peak current elicited with both the short and long influx steps is instantaneous current, which rapidly becomes attenuated as the influx step duration is increased. The average instantaneous current recorded with 5–20 msec voltage steps to −9 mV was 5253 ± 548 pA in perforated patch-clamped cells (n = 17), 5147 ± 642 pA with 80 μm EGTA (n = 10), 5041 ± 908 pA with 1 mm EGTA (n = 7), and 5833 ± 1293 pA with 5 mm EGTA (n = 5). Thus, about the same amount of BK current is activated by brief Ca2+ influx steps irrespective of the presence or absence of EGTA. In contrast, BAPTA at concentrations of 1 and 5 mm completely blocked activation of BK current, whereas 400 μm BAPTA eliminated most BK current (Fig. 8B).

EGTA eliminates the slowly developing increase in peak current but not the instantaneous current, whereas BAPTA eliminates all BK current. A, EGTA at concentrations ranging from 80 μm to 5 mm was introduced into cells, and BK current was elicited with Ca2+ influx. Cells were held at −69 mV, stepped to −9 mV to produce Ca2+ influx, and then stepped to +81 mV. The duration of the step to −9 mV was increased from 0 to 220 msec in increments of 20 msec. Shown below are the peak (circles) and the instantaneous (squares) currents plotted against the duration of the Ca2+ influx step. The first voltage step was ignored in this plot, because it does not result in any Ca2+ influx. The dotted linein each case indicates the amplitude of the steady-state Ca2+-independent, voltage-dependent current at +81 mV. As the EGTA concentration is increased, the slowly developing rise in peak current is eliminated. B, BAPTA at concentrations of 400 μm and 1 and 5 mm was introduced into each of three chromaffin cells. BAPTA even at 400 μm is quite effective in eliminating BK current. Note the different y-axis scales in A.
What do these results indicate? EGTA at concentrations of 1 mm or above abolishes the slowly developing increase in peak current seen in perforated patch-clamped cells (Fig.5) and in cells dialyzed with 80 μm EGTA (Fig.8A). However, it does not abolish the instantaneous current elicited with short influx steps, suggesting that the BK channels producing this current are located sufficiently close to Ca2+ channels so as to be unaffected by the buffering action of EGTA. The close proximity to Ca2+ channels probably leads to high [Ca2+]i around these BK channels, producing their rapid inactivation and leading to attenuation of the instantaneous current as the influx step duration is increased. In contrast, the gradual increase in the peak current seen in perforated patch-clamped cells (Fig. 5B) and with 80 μm EGTA (Fig. 8A) probably reflects the activation of BK channels at increasing distances from Ca2+ channels. The substantial time-dependent activation of this current at the long influx durations at +81 mV implies that the average open probability of these BK channels is low and suggests that they experience relatively low [Ca2+]i.
The current traces at 400 μm EGTA (Fig.9A) lend particular support to the idea of nonuniformity in the [Ca2+]i detected by different BK channels. This EGTA concentration seems optimal to produce a clear separation between the fall of the early instantaneous BK current and the slowly developing peak current. The rapid and severe inactivation of the early instantaneous current evident at the longer influx durations suggests that the BK channels producing this current are exposed to high [Ca2+]i. In contrast, the substantial time- and voltage-dependent activation of BK current at +81 mV over a period when the instantaneous current is abolished almost completely suggests that the BK channels contributing to the slowly developing current must be exposed to lower [Ca2+]i. The biphasic behavior of the peak current observed in cells with BKi current is not seen in cells with BKs current (Fig. 9B), emphasizing that it is a consequence of the inactivation behavior of BKi channels.

400 μm EGTA uncovers BK channels closely associated with Ca2+ channels from those at a distance. A, Two examples of BKi currents recorded in the presence of 400 μm EGTA in the cell are shown. The voltage protocol is the same as that in Figure 8. Amplitudes of the peak (circles) and instantaneous (squares) currents plotted against the duration of the influx step are shown on theright. The dotted line in each case indicates the amplitude of the steady-state Ca2+-independent, voltage-dependent current in each cell. In each case, the instantaneous current becomes progressively smaller as the duration of the influx step is increased. The peak current becomes smaller in the first few influx steps but then recovers as the influx step duration is still increased. B, The behavior of BKs current with 400 μm EGTA in the cell is shown. Instantaneous current is more or less steady, and peak current increases with increasing influx duration.
In sum, the above results indicate that some fraction of BK channels in chromaffin cells are sufficiently close to Ca2+channels to be unaffected by EGTA. Furthermore, when EGTA is used to unmask these BK channels, Ca2+ influx steps with durations >50 msec progressively and fully inactivate them. This rapid inactivation is consistent with the hypothesis that these BK channels are activated by relatively high [Ca2+]i. In contrast, in perforated patch-clamped cells, the instantaneous current always maintains a plateau, with only a small decrement after longer Ca2+ influx steps. The unmasking of the inactivation of some BK channels in the presence of high EGTA suggests that, in normal cells, the apparent plateau really reflects a balance of inactivation of BK channels closest to sites of Ca2+ influx and additional activation of channels just outside of these regions. Thus, the plateau may reflect a changing population of active BK channels. We envision the population of BK channels contributing to the instantaneous current in BKi cells as an annulus around points of Ca2+ influx that moves outward as the proximal channels become inactivated during the long influx steps. Furthermore, as the Ca2+ loading-step duration is increased, the instantaneous component would arise from a progressively larger population of BK channels sensing, on average, a progressively lower submembrane [Ca2+]i. In BKs cells, on the other hand, with longer Ca2+ influx steps the activation of more distant BK channels simply results in a gradual increase in the size of the instantaneous current, as noted earlier (Fig. 5C).
Submembrane [Ca2+]i elevations caused by intracellular Ca2+ release
Rat chromaffin cells display robust muscarinic acetylcholine receptor (mAChR)-mediated release of Ca2+ from intracellular stores, presumably via an IP3-mediated process (Neely and Lingle, 1992b;Herrington et al., 1995). The time course of the Ca2+ elevation can be monitored by repeated steps to +81 mV to activate BK current during the application of muscarine (Herrington et al., 1995). Using this protocol in cells with BKi current, large inactivating BK currents are seen during the muscarinic response, which typically lasts ∼15–20 sec (Fig. 10). At the peak of the muscarinic response, the inactivation time constants of BKi currents are typically 40–60 msec (Herrington et al., 1995; Solaro et al., 1995). Based on inactivation time constants from Figure 1, the implication is that the submembrane [Ca2+]i reaches 3–5 μm during the peak of the response. Analysis of the rates of inactivation of successive traces during the peak of the [Ca2+]i elevation also shows that [Ca2+]iremains elevated above 1 μm for several seconds (typically 4–6 sec with 15 sec 50 μm muscarine applications). Thus, during each of the 400 msec +81 mV voltage steps, it is unlikely that there are dramatic temporal variations in the [Ca2+]i. Instead, submembrane [Ca2+]i seems to remain fairly constant during these steps.

Ca2+ influx elevates the average submembrane [Ca2+]i to higher levels than does mAChR activation. A, A perforated patch-clamped cell was held at −69 mV and stepped repeatedly to +81 mV for 400 msec every 1.25 sec. After 5 sec, muscarine (50 μm) was applied for a period of 20 sec. Outward currents elicited before, during, and after the muscarine application are shown. For clarity, not all traces have been plotted. The peak current in each trace is plotted against time in the bottom plot. mAChR stimulation results in the release of Ca2+ from intracellular stores, which elicit large, inactivating BK currents here. Note that after the first surge of [Ca2+]i, BK currents are suppressed briefly before rebounding. This is attributable to Ca2+-induced inactivation of BK current and its subsequent recovery as [Ca2+]i levels fall (Herrington et al., 1995). Perforated-patch method.Rs, 13 MΩ;Cm, 6 pF; 80% compensated. B, The [Ca2+]i elevation resulting from mAChR activation is compared directly with that resulting from influx in the same cell. Top traceshows BK current elicited in normal external saline with a 25 msec influx step to −9 mV, followed by a 400 msec step to +81 mV.Bottom trace shows current elicited during a muscarine response from the same cell in a zero external [Ca2+] saline with the same voltage protocol applied every 1.25 sec. Note that the instantaneous current elicited with Ca2+ influx is significantly larger than that produced by mAChR stimulation, but the peak current is smaller and the rate of inactivation is slower. Closed and openarrows indicate the peak and instantaneous current, respectively. Perforated-patch method. Rs, 14.4 MΩ; Cm, 7 pF; 80% compensated. Sampling period, 500 μsec.
We compared the submembrane [Ca2+]i elevation resulting from mAChR activation with the elevation caused by Ca2+ influx. The consequences of influx are demonstrated by the top trace in Figure 10B, in which the cell was stepped to −9 mV for 25 msec to elicit influx and then stepped to +81 mV. Ca2+ influx resulted in the typical activation of BK current, and, after a step to +81 mV, there was the usual instantaneous current with some additional slow current activation. Then external Ca2+ was removed, and 50 μm muscarine was applied to the cell, during which the previous voltage protocol was applied every 1.25 sec. At the peak of the muscarine-induced [Ca2+]i elevation, the step to −9 mV resulted in little detectable activation of BK current and little instantaneous current at +81 mV. However, a large, slowly activating current was observed that inactivates more rapidly (time constant = 48 msec) than the current resulting from Ca2+ influx (time constant = 69 msec). These inactivation rates indicate that, after termination of the 25 msec influx step, the average submembrane [Ca2+]i quickly falls below the [Ca2+]ipersisting during mAChR-induced submembrane [Ca2+]i elevation. However, the instantaneous current of the muscarine-induced response is significantly smaller than the instantaneous current resulting from influx. This indicates that, during the step to −9 mV, BK channels sense a higher average submembrane [Ca2+]i when the source of Ca2+ is influx through Ca2+ channels rather than release from intracellular stores.
These results argue that the mAChR-induced [Ca2+]i elevation is quite uniform over most of the response. Based on inactivation rates, release of intracellular Ca2+ by mAChR activation elevates the average submembrane [Ca2+] to ∼4 μm, yet little or no BK current activation occurs at −9 mV during the muscarine response. Because detectable instantaneous current after steps to +81 mV would be expected if even 10% of BK channels were exposed to 10 μm[Ca2+], the previous result suggests that few, if any, BK channels are exposed to [Ca2+] as high as 10 μm. Furthermore, at the peak of the response to muscarine, steps from +81 to +111 mV produce no additional time-dependent activation of BK current (data not shown), suggesting that there are very few BK channels exposed to [Ca2+] <4 μm. This implies that the mAChR-induced submembrane [Ca2+]i elevation may affect a significant portion of the membrane, with the minimal and maximal submembrane [Ca2+]i probably varying less than two- to threefold.
Short Ca2+ influx steps activate some BK channels with near-maximal open probabilities
Results obtained with high concentrations of exogenous buffers (Figs. 8, 9) suggest that the instantaneous current at +81 mV after short depolarizing steps to −9 mV arises from a fraction of BKi channels seeing a high [Ca2+]i, whereas the instantaneous current resulting from long influx steps arises from a population of channels seeing, on average, a lower [Ca2+]i. In the former case, if the BK channels underlying the instantaneous current see sufficient [Ca2+]i so as to be maximally or near-maximally activated at −9 mV, anadditional increment in cytosolic [Ca2+]i should have little or no effect on the amplitude of the instantaneous current. In contrast, the same additional increment in cytosolic [Ca2+]i should increase the instantaneous current resulting from long influx steps, in which the average [Ca2+]i is on the order of 10 μm (Fig. 7). This hypothesis was tested by using the elevation of [Ca2+]i by mAChR activation as a means to produce the additional [Ca2+]i increment. As argued earlier, mAChR activation produces a relatively uniform submembrane [Ca2+] of ∼3–5 μm. Furthermore, no BK channels detect [Ca2+]i sufficient to produce substantial activation at −9 mV.
BK current evoked by muscarine and a 5 msec Ca2+influx step were compared to the current evoked by the Ca2+ influx step alone. The 5 msec depolarization to −9 mV resulted in an instantaneous current of 4355 pA (Fig.11A). Subsequent mAChR activation immediately before the next depolarizing step elevated submembrane [Ca2+]i further but produced little change in the instantaneous current, yet the rate of BK current inactivation at +81 mV placed submembrane [Ca2+]i near 4 μm. This suggests that the BK channels activated during the short influx step already are sensing a [Ca2+]i of at least 60 μm. In contrast, after long influx steps, mAChR activation immediately before the influx step consistently enhances the instantaneous current. For instance, as shown in Figure 11B, after a 50 msec influx step, the instantaneous current increased from 4121 to 7119 pA. Assuming that the current elicited at +81 mV with mAChR activation is the maximal BK current available for activation, the fractional activation of BK channels at −9 mV increased from 0.31 to 0.54, indicating an increase in submembrane [Ca2+]i from 10 μm to ∼15 μm (from Fig. 4). This change in [Ca2+]i is consistent with the addition of the ∼4 μm[Ca2+]i that normally results from mAChR activation (Fig. 10). Essentially identical results were obtained in four cells with a 0.25% average increase in fractional activation after a short (5–10 msec) influx step and a 57% average increase after a long (50–100 msec) influx step. Together with the EGTA data (Fig. 8), these results suggest that the BK channels that contribute to the instantaneous current during short Ca2+ influx steps sense average submembrane Ca2+ concentrations of at least ∼60 μm, whereas BK channels that contribute to the instantaneous current with long influx steps sense average submembrane Ca2+ concentrations closer to 10 μm.

The average fractional activation of open BK channels after short depolarizations is higher than the average fractional activation of open BK channels after long depolarizations. A, BK current was elicited in a perforated patch-clamped cell with a step to −9 mV for 5 msec to produce Ca2+ influx and immediately followed by a step to +81 mV. The voltage protocol was repeated every 1.5 sec, and, immediately after the second episode, 50 μmmuscarine was applied. Shown here are the current traces immediately before and after the muscarine application. Instantaneous and peak current before muscarine application were 4355 and 5097 pA. Instantaneous and peak current after muscarine application were 4121 and 13232 pA. B, The Ca2+ influx step was increased to 50 msec in the same cell. As before, 50 μm muscarine was applied after the second episode. Shown here are the current traces immediately before and after muscarine application. Instantaneous and peak currents before muscarine application were 4414 and 7900 pA. Instantaneous and peak currents after muscarine application were 7119 and 12138 pA. Closedand open arrows indicate peak and instantaneous currents, respectively. Perforated-patch method.Rs, 14 MΩ;Cm, 6 pF; 80% compensated. Sampling period, 500 μsec.
What fraction of BK channels is sufficiently close to Ca2+ channels to be maximally activated by the short influx steps? To determine this, the instantaneous current at +81 mV activated by short Ca2+ influx steps was compared with the total BK current in the cell. Total BK current was determined roughly by steps to +81 mV during mAChR-induced [Ca2+]i elevation. Inactivation before the +81 mV step was minimized by holding at negative potentials before stepping to +81 mV, and the amount of inactivation during the rising phase of the current at +81 mV was accounted for by fitting the current with the modified Hodgkin–Huxley function described in Materials and Methods. Using this procedure, in 15 cells the instantaneous current activated at +81 mV after 5–10 msec depolarizations to −9 mV involved 17.7 ± 6.7% of the total population of BK channels. If the maximal BK current were really 50% higher, the fraction of BK channels exposed to [Ca2+] of 60 μm or higher would be 11.9 ± 4.5%.
Clearance of Ca2+ after influx
The persistence of submembrane [Ca2+]i after the termination of Ca2+ influx was studied by evoking Ca2+ influx with depolarizations of different durations. Then the amount of BK current activation that can occur after a variable recovery period after the termination of the influx step was determined (Fig. 12). This protocol showed that, with very small Ca2+ influx steps, submembrane [Ca2+] fell very rapidly; a step to +81 mV 5 msec after the termination of Ca2+influx resulted in no detectable BK current activation (Fig.12A). However, as the influx step was made longer, residual BK current activation was seen at progressively later time points after the termination of Ca2+ influx. With Ca2+ influx steps of 50 msec or longer, BK current activation often was seen hundreds of milliseconds after the termination of the influx step.

Submembrane [Ca2+]i drops rapidly after short influx steps but persists at significant levels for long durations after long influx steps. The cell was held at −69 mV, stepped to −9 mV to produce Ca2+ influx, and then stepped back to −80 mV for variable recovery periods before applying a step to +81 mV to activate BK current again. The recovery period duration at −80 mV ranged from no recovery (0 msec) to several seconds. In A, the influx step was 5 msec long; the recovery steps at −80 mV were incremented by 5 msec. In B, the influx step was 10 msec in duration; the recovery steps were incremented by 10 msec. In C, the influx step was 100 msec in duration; the recovery steps were incremented by 100 msec. InD, the influx step was 400 msec; the recovery steps were incremented by 200 msec. The inactivation time constants of BK currents elicited at +81 mV in D are shown in the inset on theright. Because channels recover from inactivation during the initial recovery steps, large increases in BK current elicited at +81 mV are observed as the recovery duration is increased. Perforated-patch method. Rs, 9 MΩ;Cm, 6 pF; 80% compensated.
In fact, when the Ca2+ loading steps are 200 msec or longer, rates of BK current inactivation at +81 mV often remain very fast even hundreds of milliseconds (200–400 msec) after the termination of the influx step. For instance, in the cell shown in Figure 12, the time constant of inactivation stays near its limiting value, ∼50 msec, for 200–300 msec after the termination of a 400 msec influx step. This indicates that the submembrane [Ca2+] remained ∼3–5 μm during this period. Longer periods of recovery after termination of influx result in the slowing of the inactivation rate, indicating a gradual fall in the submembrane [Ca2+]i. What is noteworthy is that even after long recovery times, e.g., 800 msec, the rates of inactivation still correspond to a submembrane [Ca2+]i above 1 μm. Note that the current elicited at +81 mV increases initially as the recovery step at −80 mV is increased (Fig.12D). This results from the recovery from inactivation of BK channels that have inactivated during the Ca2+loading step (Solaro et al., 1995).
Because detectable activation of BK current can occur at +81 mV with [Ca2+]i as little as 1 μm (Fig. 1), the complete absence of BK current after the shortest loading steps suggests that [Ca2+]i falls from at least 60 to <1 μm within 5 msec after the termination of Ca2+ influx. In contrast, after longer Ca2+ loading steps (>20 msec), [Ca2+]i falls at rates that depend on the magnitude of the Ca2+ load, although the high [Ca2+]i in the immediate vicinity of Ca2+ channels must still drop rapidly to the average cytosolic [Ca2+]i levels.
DISCUSSION
In this study, we determined the Ca2+sensitivity of BK channels in chromaffin cells and then examined the range of [Ca2+]i detected by BK channels after Ca2+ influx and the release of Ca2+ from internal stores. The key findings follow. (1) During Ca2+ influx, ∼10–20% of the BK channels in the cell detect submembrane [Ca2+]i probably as high as 60 μm. (2) Although these BK channels are associated closely with Ca2+ channels, most BK channels in chromaffin cells must be at some appreciable distance from Ca2+ channels. (3) The high concentrations of Ca2+ in the vicinity of Ca2+ channels fall rapidly after termination of Ca2+ influx. (4) Release of intracellular Ca2+ by mAChR stimulation produces a relatively uniform submembrane [Ca2+]i elevation of ∼3–5 μm.
We have assumed that the Ca2+ and voltage dependence of BK channel activation and inactivation are comparable in perforated-patch recordings and under the conditions of our calibration measurements involving standard whole-cell recordings. In view of reports of BK channel modulation (Reinhart et al., 1991;Twitchell and Rane, 1993; Bielefeldt and Jackson, 1994), this is an assumption that will have to be addressed further. However, at present, it is supported by the following observations. (1) After excision of BKi channels in hundreds of inside-out patches, we have never observed changes in inactivation properties or sensitivity to [Ca2+]. (2) Properties of BKi current in cells recorded with the perforated-patch method are identical immediately before and after activation of phospholipase C by mAChR activation (data not shown), implying that there are no persistent effects on BK channels of any mAChR-activated kinases. (3) The limiting BKiinactivation rates are similar both in perforated-patch and whole-cell recording, irrespective of the methods of [Ca2+] elevation (Neely and Lingle, 1992b;Herrington et al., 1995; Solaro et al., 1995). Although these are indirect arguments, there is no observation that requires us to reject this assumption.
Amplitudes of submembrane [Ca2+]ielevations detected by BK channels
The results argue that the submembrane [Ca2+]i elevations resulting from Ca2+ influx are highly nonuniform. Specifically, 5 msec after initiation of Ca2+influx, ∼10–20% of BK channels are exposed to [Ca2+]i high enough to produce maximal activation at −9 mV. This places the submembrane [Ca2+]i detected by these BK channels near 60 μm and possibly higher. Because these channels are activated by short Ca2+ influx steps rapidly and maximally, they are positioned ideally to play a role in rapid repolarization of the membrane potential during action-potential activity, a role previously demonstrated for BKi current in rat chromaffin cells (Solaro et al., 1995). The other 80% of BK channels are activated only by prolonged Ca2+ influx steps and detect lower [Ca2+]i. With long influx steps that activate all available channels, the average submembrane [Ca2+]i approaches 10 μm. Because the channels in this latter category are not exposed to [Ca2+]i sufficient to produce their rapid recruitment during single action potentials, it is unlikely that they participate in the process of action-potential repolarization. Instead, they might function under conditions that involve Ca2+ release from intracellular stores.
Spatial arrangement of BK and Ca2+ channels
BK channels in many cell types, including rat chromaffin cells, require [Ca2+]i in excess of 5 μm for activation at physiological potentials (McManus, 1991), indicating that they have to be positioned reasonably close to Ca2+ channels to see the required [Ca2+]i. However, this has been verified in only a few cell types. Among these, the frog saccule hair cells (Roberts et al., 1990) and the presynaptic terminals of the frog neuromuscular junction (Robitaille et al., 1993) are reported to possess BK channels tightly colocalized with Ca2+ channels. In the frog hair cell, this colocalization is reported to lead to [Ca2+]i in excess of 1 mm around the BK channels. Colocalization remains less certain in hair cells from other species (Art et al., 1995). In the neurons of Helix, only a subset of Ca2+-dependent K+ channels are coupled to Ca2+ channels (Gola and Crest, 1993), and, in bovine chromaffin cells, BAPTA is more effective than EGTA in suppressing Ca2+-activated K+ current (Marty and Neher, 1985), suggesting that at least some of the Ca2+-activated K+ channels are colocalized with Ca2+ channels.
Possible distances between BK channels and Ca2+channels can be calculated based on the published formalism for the diffusion profile of Ca2+ away from single Ca2+ channels (Neher, 1986; Stern, 1992). Assuming single channel Ca2+ currents in the range of 0.2–0.5 pA, these equations indicate that a BK channel sensing [Ca2+]i of 60 μm must be 13–30 nm from the nearest Ca2+ channel. Thus, the 10–20% of BK channels in rat chromaffin cells that see [Ca2+]i of 60 μm or higher are probably within this distance from Ca2+ channels. However, it is important to note that this estimate is critically dependent on the single channel Ca2+ current value, a parameter subject to errors in its estimation (Silberberg and Magleby, 1993). Despite this uncertainty, a key question that arises is whether the BK channels “close” to Ca2+ channels are coupled to Ca2+ channels by some specific mechanism or whether an apparent “coupling” can arise simply from random positioning of a large number of BK and Ca2+channels in the membrane.
Two facets of the results are consistent with the view that the BK channels exposed to the highest [Ca2+]i may constitute a distinct population coupled to Ca2+ channels by a specific mechanism. First, the relative insensitivity of the instantaneous current produced by the short Ca2+influx steps over a wide range of EGTA concentrations suggests that the BK channels underlying this current are not located at a continuum of distances from Ca2+ channels. This result is inconsistent with a scenario in which channels are distributed randomly. Second, in the presence of 400 μm EGTA, BK currents exhibit an interesting biphasic behavior (Figs. 8, 9), which may be explained most simply by the presence of two populations of BK channels: one population colocalized with Ca2+ channels that is activated rapidly during Ca2+ influx and a second population that is activated only with prolonged Ca2+ influx and, presumably, located further away from Ca2+ channels. In addition, simulations show that a random distribution of BK and Ca2+channels in the cell membrane is insufficient to account for 10–20% BK channels detecting [Ca2+]i of 60 μm or higher (Prakriya et al., 1996).
Clearance of Ca2+ after influx
The results show that the high [Ca2+]i in the vicinity of open Ca2+ channels must fall quite rapidly on termination of Ca2+ influx. After very short influx steps (5 msec) comparable to the widths of single-action potentials (Solaro et al., 1995), BK channels that are activated almost maximally during Ca2+ influx fail to be activated by steps to +81 mV just 5 msec after the termination of Ca2+ influx. This represents a [Ca2+]i drop from at least 60 to under 1 μm—almost two orders of magnitude. Diffusion of Ca2+ from points of Ca2+ entry can, in principle, account adequately for this rapid fall in submembrane [Ca2+]i. In contrast, after Ca2+ influx durations of 50 msec or longer, average submembrane [Ca2+]i relax to under 1 μm over hundreds of milliseconds and even seconds. The time course of this relaxation presumably reflects the nature of Ca2+ sequestration and removal agents such as mitochondria, Na+/Ca2+ exchangers, and Ca2+/ATPases. Mitochondria have been shown to sequester Ca2+ during Ca2+influx and gradually release it into the cytosol on termination of influx (Werth and Thayer, 1994). Furthermore, there is recent evidence that mitochondria are involved in shaping part of the [Ca2+]i fall after very long influx steps in rat chromaffin cells (Herrington et al., 1996;Park et al., 1996).
Implications for secretion
Our results indicate that influx of Ca2+raises the submembrane [Ca2+]i to at least 60 μm in some locations, whereas release of intracellular Ca2+ by mAChR activation raises submembrane [Ca2+]i to peak levels of only ∼4 μm. Although the absolute magnitude of the [Ca2+]i elevations produced by mAChR activation is less than that produced by influx, the elevation produced by mAChR activation persists much longer and seems to affect a significantly greater portion of the chromaffin cell membrane. Based on estimates of the Ca2+dependence of exocytosis from bovine chromaffin cells (Knight and Baker, 1982; Augustine and Neher, 1992b) indicating that secretion can be initiated at [Ca2+]ilower than 1 μm, our results are consistent with previous reports implicating mAChRs in eliciting secretion in rat chromaffin cells (Wakade and Wakade, 1983; Malhotra et al., 1988). This contrasts with bovine chromaffin cells, in which release of Ca2+ from intracellular stores by mAChR activation produces relatively weak elevations in [Ca2+]i (Cheek et al., 1989; O’Sullivan et al., 1989) and is ineffective in producing secretion (Fisher et al., 1981; Kim and Westhead, 1989).
In sum, our results indicate that during brief depolarizations, although most BK channels in the cell are not activated at all, 10–20% of the BK channels are driven to near-maximal open probabilities. The calibration data suggest that these BK channels are exposed to [Ca2+]i of at least 60 μm. Furthermore, they are insensitive to slow Ca2+ buffers, suggesting that they are located close to Ca2+ channels. Because rat chromaffin cells express multiple types of Ca2+channels (M. Prakriya and C. J. Lingle, unpublished observations), it remains to be determined whether an association exists between the BK channels exposed to the highest [Ca2+]i and a specific Ca2+ channel subtype.
Footnotes
This work was supported by Grant DK-46564 from National Institutes of Health. We thank Suzanne Swanson for preparation and maintenance of the cell cultures, and Drs. Jeanne Nerbonne, Zhuan Zhou, and Robert Wilkinson for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Christopher Lingle at the above address.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}