

Abstract
Cholinergic regulation of the activity of rabbit retinal ganglion cells and displaced amacrine cells was investigated using optical recording of changes in intracellular free calcium ([Ca2+]i). Labeling of neurons in the mature retina was achieved by injecting calcium green-1 dextran (CaGD) into the isolated retina. Nicotine increased ganglion cell [Ca2+]i, affecting every loaded cell in some preparations; the pharmacology of nicotine was consistent with an action at neuronal nicotinic receptors, and specifically it was κ-(neuronal-)bungarotoxin-sensitive but α-bungarotoxin-insensitive. Muscarine also raised [Ca2+]i, but it was less potent than nicotine, affecting only a subpopulation of ganglion cells, with an M1-like muscarinic receptor pharmacology. Neither the nicotine- nor muscarine-induced increases of ganglion cell [Ca2+]i were blocked by the glutamate receptor antagonists 6,7-dinitroquinoxaline-2,3-dione and aminophosphonopentanoic acid. Therefore, the effects of cholinergic agonists on ganglion cell [Ca2+]i were not attributable to an indirect effect mediated by glutamatergic bipolar cells. The effects of nicotine and muscarine were abolished in calcium-free solution, indicating that the responses depend on calcium influx.
Displaced (Cb) cholinergic amacrine cells were also loaded with CaGD and were identified by selective labeling with the nuclear dye 4′,6-diamidino-2-phenyl-indole. Cb amacrine cells did not respond to either nicotine or muscarine, but responded vigorously to the glutamate receptor agonist kainic acid. There is anatomical evidence indicating that cholinergic amacrine cells make synaptic contact with each other, but the present results do not support the hypothesis that communication between these cells is cholinergic.
Acetylcholine (ACh) is an important neurotransmitter in the mammalian retina, where it is localized primarily in two mirror symmetric populations of amacrine cells (for review, see Masland, 1986; Vaney, 1990). One population (Ca) is located in the inner nuclear layer (INL) with dendrites stratifying in sublamina a of the inner plexiform layer (IPL); the other population (Cb) is displaced to the ganglion cell layer (GCL) with dendrites stratifying in sublamina b of the IPL. The principal synaptic targets of cholinergic amacrine cells are ganglion cell dendrites (Famiglietti, 1983, 1991; Brandon, 1987); correspondingly, electrophysiological recordings have shown that many retinal ganglion cells are excited by ACh and nicotine (Straschill, 1968; Masland and Ames, 1976; Ariel and Daw, 1982; Ikeda and Sheardown, 1982; Schmidt et al., 1987).
It is not clear, however, whether the cholinergic input affects all ganglion cells (Ariel and Daw, 1982; Lipton et al., 1987) or only a subpopulation (Keyser et al., 1988; Hamassaki-Britto et al., 1991;Britto et al., 1994). Moreover, it is not known if the actions of ACh on ganglion cells are mediated solely by α-bungarotoxin-insensitive neuronal nicotinic receptors (Lipton et al., 1987; Aizenman et al., 1990) or whether some of the effects are mediated by either α-bungarotoxin-sensitive receptors (Vogel and Nirenberg, 1976;Swanson et al., 1987; Wada et al., 1989; Hoover and Goldman, 1992;Britto et al., 1994) or muscarinic receptors (Zarbin et al., 1986;Hutchins, 1994). Interpretation of pharmacological studies of ganglion cells is dependent on which retinal neurons, presynaptic to ganglion cells, are also cholinoceptive. Some studies have suggested that bipolar cells possess nicotinic receptors (Vogel and Nirenberg, 1976;Yazulla and Schmidt, 1976; Zucker and Yazulla, 1982; Hamassaki-Britto et al., 1994), raising the possibility that some of the cholinergic effects on ganglion cells might be indirect, mediated by glutamate release from bipolar cells. The cholinergic amacrine cells also appear to make reciprocal synapses with neighboring cholinergic cells (Millar and Morgan, 1987; Mariani and Herch, 1988;Famiglietti, 1991), but it is not known whether these synapses use ACh as their transmitter because the cholinergic cells also contain GABA and adenosine (Brecha et al., 1988; Vaney and Young, 1988;Blazynski, 1989).
A rapid way to assess the responses of retinal neurons would be to record from many cells simultaneously using optical recording. In this study, calcium-imaging techniques were used to simultaneously monitor the drug-evoked responses of many neurons in a small patch of retina. Although calcium-sensitive fluorescent dyes previously have been used as indicators of neuronal activity in intact or slice preparations of immature vertebrate nervous tissue (Yuste and Katz, 1991; O’Donovan et al., 1994; Wellis and Werblin, 1995; Wong, 1995), imaging mature nervous tissue has proven difficult because of the poor loading with membrane-permeable dyes. In this study on mature rabbit retina, the neurons were consistently labeled by injecting a dextran-conjugated calcium-sensitive dye into the retina. Using this method, the effects of cholinergic ligands on intracellular free calcium levels ([Ca2+]i) in ganglion cells and displaced cholinergic amacrine cells were explored.
Some of these results have been reported in preliminary form (Baldridge, 1995).
MATERIALS AND METHODS
Isolated rabbit retina preparation. Experiments were performed in accord with the Australian NHMRC code of practice for the care and use of animals for scientific purposes. Adult pigmented rabbits of either sex were killed with Nembutal (Boehringer Ingelheim, Artarmon, Australia) before enucleation. In some cases, rabbits were anesthetized with 4% Fluothane (Zeneca, Macclesfield Cheshire, UK) in air and given an intraocular injection of 0.4 μg 4′,6-diamidino-2-phenyl-indole (DAPI; Sigma, St. Louis, MO) in 20 ml of deionized water 1–2 d before they were killed. After enucleation, the anterior segment and vitreous were removed, and the posterior segment was immersed in Ames medium (Ames and Nesbett, 1981; Sigma) buffered to pH 7.4 with HEPES (Sigma) and bubbled continuously with oxygen at room temperature. The retina was dissected from the eyecup under fiber optic illumination, placed on a glass microscope slide, and cut with a scalpel blade into several pieces, typically ∼1 cm2. The pieces of retina were then mounted on black Millipore filters (HABP 045; Millipore, Bedford, MA) with the GCL uppermost.
Calcium indicator dye labeling. Using a 10 μl syringe fitted with a sharpened tapered 26 gauge needle (Hamilton, Reno, NV), a small amount (<1 μl) of 10% (w/v) calcium-green-1 conjugated to 10,000 MW dextran (Molecular Probes, Eugene, OR) and dissolved in deionized water was deposited within the retina. This was accomplished by ejecting the dye solution from the syringe, thus forming a drop at the needle tip, and then briefly inserting the needle into the retina. After application of dye, retinas were incubated in the dark for at least 2 hr at room temperature in Ames medium bubbled continuously with oxygen. Pieces of retina labeled with calcium-green-1 dextran (CaGD) were studied for 8–12 hr after retinal isolation.
Pharmacological treatment. Each retinal whole-mount was transferred to a chamber on an inverted microscope (Zeiss Axiovert 10), and the temperature was raised to 30–32°C. In the chamber, retinas were superfused continuously with Ames medium at 1 ml/min. In general, pharmacological agents were added to the superfusate or, in the case of agonists, pipetted directly into the chamber, which had a volume of 1 ml. Experiments involving κ-bungarotoxin were treated differently because only limited quantities of this drug were available: retinas were not superfused but were incubated in solutions of the drug, at room temperature, so that the solution could be reused and several retinas could be studied. This incubation was done in a chamber separate from the microscope and, therefore, images were not captured before and during the incubation. After transferring the retinas to the optical recording chamber on the microscope, it took 5–10 min before an agonist could be applied because of the time needed to both identify a suitable region for study and to reach a stable temperature. During this time, the retinas were superfused continuously with κ-bungarotoxin-free Ames medium.
Drugs. (−)-Nicotine di-d-tartrate, (+)-tubocurarine chloride, α-bungarotoxin, hexamethonium dichloride, and methyllycaconitine citrate were obtained from Research Biochemicals (Natick, MA); (+)-muscarine chloride, 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP), kainic acid, atropine sulfate, pirenzepine dihydrochloride, gallamine triethiodide, and EGTA were obtained from Sigma; NMDA, 6,7-dinitroquinoxaline (DNQX), andd,l-2-amino-5-phosphopentanoic acid (AP-5) were obtained from Tocris Cookson (Bristol, UK). κ-Bungarotoxin, also known as neuronal bungarotoxin, was obtained originally from Calbiochem (Lot 901673, La Jolla, CA) and was the generous gift of Professor D. J. Adams. In general, antagonists were used at a concentration that was found to completely block an agonist-induced effect and, at the same time, permit recovery of the agonist-induced effect after washout of the antagonist. Comparisons were not routinely made between agonist-induced responses before antagonist treatment and responses during antagonist treatment. This helped shorten the length of experiments to minimize the effect of photobleaching and allowed a greater number (4–6) of retinal pieces to be studied during the 8–12 hr period after retinal isolation.
Optical recording. The imaging system was identical to that described by Wong (1995). Using Image-1 (Version 4.0, Universal Imaging, West Chester, PA) and a computer-controlled filter wheel and shutter system (Lambda-10, Sutter Instruments, Novata, CA; Uniblitz D122, Vincent Associates, Rochester, NY), images were collected by a low-light silicon-intensified target (SIT) camera (Hamamatsu C2400, Hamamatsu City, Japan). CaGD fluorescence was produced by excitation from a 50 W mercury vapor lamp, filtered by −1.5 to −2.0 log neutral density filters and an appropriate filter set (485 nm excitation, 535 nm emission, 505 nm long-pass dichroic; XF23, Omega Optical, Brattleboro, VT). Labeled cells were always imaged with a Zeiss Achrostigmat 40× objective (1.30 numerical aperture), and each image was stored on optical disk (Sony LVR/LVS 5000) as an average of 32 frames. During agonist treatments, images were acquired as frequently as possible, about every 3 sec, but at other times acquisitions were made every 20–60 sec to minimize dye photobleaching. Storage on optical disk permitted subsequent quantitative analysis. DAPI-labeled nuclei were imaged using a fura-2 filter set (380 nm excitation, 510 nm emission, 400 nm long pass dichroic; XF04, Omega Optical) and also saved onto optical disk.
Quantitative analysis. The fluorescence intensity of labeled cell somata were measured using Image-1. Increases in the intensity of CaGD fluorescence indicate an increase of [Ca2+]i (Eberhard and Erne, 1991; Haughland, 1992). Changes in intensity were quantified from the raw images by determining the maximum increase in cell intensity (ΔF) during drug application and then normalizing the increase by dividing by the average intensity of the cell from the 10 images collected before drug application (ΔF/F). In the absence of any treatment, spontaneous changes in ΔF/F rarely exceeded 0.05 and never exceeded 0.10. However, responses were assessed conservatively in that a change in intensity was deemed to be significant only if ΔF/F was ≥0.10. Using Image-1, values of ΔF/F could also be converted into pseudocolor image representations (e.g., Fig. 1) allowing simultaneous presentation of cell location and cell response (ΔF/F). Changing values of ΔF/F, in response to drug treatments, are not linearly related to changes of [Ca2+]i and are intended only to provide a qualitative indication of changing [Ca2+]i.

Video and pseudocolor images of the GCL from three pieces of rabbit retina (A–D, E–G, H–J) labeled with CaGD. A, Video image of CaGD-labeled ganglion cells imaged under 485 nm excitation. Some bundles of labeled ganglion cell axons are also visible. B, Pseudocolor representation of the response of the cells in A to 50 μmnicotine. Not every cell responded, but those that did showed responses of up to ΔF/F = 1.00, but more typically ΔF/F ∼ 0.50. The increase of CaGD fluorescence illustrated here and in all subsequent images indicates an increase of [Ca2+]i.C, Responses of cells from the same piece of retina to 50 μm muscarine. Compared with nicotine, fewer cells responded and the responses were weaker. D, Responses of cells from the same retina to 10 μmkainic acid. Virtually every CaGD-labeled cell responded, with some reaching ΔF/F ∼ 1.25. E, Video image of a piece of retina labeled with CaGD. F, Pseudocolor imaging illustrating the responses of the cells in E to 50 μm muscarine. Although several cells responded, the responses were weak. G, Response of the same cells to 50 μm nicotine. In this piece of retina, virtually every cell responded to nicotine with ΔF/F ∼ 0.50. H, Video image of a piece of retina stained with the nuclear stain DAPI and imaged under 380 nm excitation. Four nuclei were brightly labeled (red circles) and correspond to displaced (Cb) cholinergic amacrine cells. The other cells, with dimly stained nuclei, were characterized as presumptive ganglion cells (see Materials and Methods). These cells were also labeled with CaGD (not shown). I, Pseudocolor imaging of the same piece of retina as in H illustrating the response to 50 μm nicotine. Although several ganglion cells responded, the cholinergic amacrine cells did not.J, Both ganglion and amacrine cells (red circles) responded to 10 μm kainic acid. Scale bar in the last illustration of each set is 50 μm.
RESULTS
CaGD labels neurons in the adult rabbit retina
Several attempts were made to load neurons in the mature rabbit retina with the membrane-permeable dye Fura-2 AM using the same protocol that produces consistent labeling in the developing rabbit retina (Wong, 1995). Even after long incubation, virtually no neuronal labeling was evident except at the cut edges of the tissue. This labeling was restricted to cells immediately adjacent to the cut, and these cells did not show consistent responses to treatments, including raised extracellular potassium.
The first indication that dextran-conjugated dyes might label mature retinal neurons came from preliminary studies in which 1 mg of CaGD was injected into the vitreous 1–2 d before study. Although labeling was achieved on occasion, this method was not reliable and required large quantities of CaGD. Moreover, the best labeling occurred around the puncture site, suggesting that direct injection into the retina would produce optimal labeling. This turned out to be the case; injection of <1 μl of 10% CaGD into isolated retina labeled several classes of retinal neurons.
The labeling of neuronal somata by CaGD injection in the rabbit retina took two forms. First, there was labeling around the injection site: neurons were labeled in both the INL and the GCL but, in this study, only the labeled somata in the GCL were considered. As will be demonstrated later, both ganglion cells and displaced amacrine cells were labeled. Second, ganglion cell axons passing through the injection site were also labeled, giving rise to clusters of retrogradely labeled somata up to 800 μm from the injection site (away from the optic nerve head). An example of such retrograde labeling is illustrated in Figure 1A, in which labeled ganglion cell somata can be seen underneath the bundles of labeled axons. The labeling in Figure1E was also of this type, but the illustrated cluster of ganglion cells was offset from the bundles of labeled axons. The retrograde labeling provided the greatest number of labeled ganglion cells, particularly when injections were made near the visual streak, and was usually sought for experiments concerned specifically with ganglion cells. Labeling of displaced amacrine cells was not observed within the clusters of labeled somata distant from the injection site.
In retinas double-labeled with CaGD and the nuclear stain DAPI, it was evident that CaGD did not label every ganglion cell within a given region, with either the first or second type of labeling. However, labeling was not restricted to a particular type of ganglion cell as determined on the basis of soma size; labeled ganglion cell somata ranged in diameter from 9 to 32 μm, representing the full range of ganglion cells in the rabbit retina (Vaney, 1980; Peichl et al., 1987;Amthor et al., 1989a,b).
Cholinergic agonists increase [Ca2+]i in retinal ganglion cells
Nicotine increased the intensity of CaGD fluorescence in ganglion cells, indicating an increase of [Ca2+]i as shown for two pieces of retina in Figure 1, B and G. In some cases (Fig. 1G), 50 μm nicotine increased CaGD fluorescence significantly (ΔF/F ≥ 0.10) in virtually every CaGD-labeled ganglion cell (compare with Fig. 1E). In other cases (Fig. 1B), 50 μm nicotine produced a response in some, but not all, labeled ganglion cells (compare with Fig.1A). Ganglion cells that did not respond to nicotine did not appear to consist of a particular morphological type on the basis of soma size, which ranged in diameter from 9 to 31 μm and was similar to the size range of ganglion cell somata that did respond to nicotine (11–32 μm diameter). The average response to 50 μm nicotine of all ganglion cells tested (n = 217) was 0.50 ± 0.02 (mean ΔF/F ± SEM). Nicotine was effective at concentrations as low as 1 μm (Fig.2A,B, squares) and produced a maximum response at 10–100 μm (Fig. 2B, squares). The nicotinic agonist DMPP (Mulle and Changeux, 1990) also increased [Ca2+]i(Fig. 2B, triangles), but it was effective only at higher concentrations (≥10 μm) than nicotine.

Cholinergic agonists and kainic acid increase ganglion cell [Ca2+]i.A, Optical recordings from a ganglion cell illustrating the dose-dependent increase of CaGD fluorescence (ΔF/F), indicating an increase of [Ca2+]i in response to increasing concentrations of nicotine. B, Mean responses to increasing concentrations of nicotine (squares), the nicotinic agonist DMPP (triangles), and muscarine (circles) from all ganglion cells studied. C, Optical recording from a ganglion cell illustrating the dose-dependent response to increasing concentrations of kainic acid. D, Mean responses to increasing concentrations of kainic acid from all ganglion cells studied (squares). For comparison, the effect of the same dose of kainic acid on the firing rate of a ganglion cell has also been plotted (open circles). The ganglion cell firing rate data were taken from Massey and Miller (1988). In all cases, error bars are SEM.
Muscarine increased [Ca2+]i in a subset of nicotine-responsive ganglion cells (Fig. 1C,F): when both nicotine and muscarine were tested in sequence, about half the cells that responded to nicotine (n = 123) also responded to muscarine. Muscarine was less potent than nicotine, however, with 50 μm muscarine producing an average ΔF/F of only 0.20 ± 0.02. Although the threshold for a muscarine-induced response was similar to nicotine (near 1 μm), the greatest response to muscarine (100 μm) was still <0.30 (Fig. 2B, circles). No cells were observed that responded to muscarine but not nicotine.
For comparison, the effect of the glutamate receptor agonist kainic acid on ganglion cell [Ca2+]i was also tested: virtually every CaGD-labeled ganglion cell showed a significant response during treatment with 10 μm kainic acid (Fig. 1D). In some cells, 10 μmkainic acid produced a ΔF/F of 1.25 or more (Fig. 1D) but, on average, ΔF/F = 0.80 ± 0.03 (n = 320). It is noteworthy that the threshold concentration for a kainic acid-induced effect (10 μm; Fig. 2D, square) was similar to that which increases ganglion cell firing rate (Fig. 2D, open circles; taken from Massey and Miller, 1988).
The effect of nicotine on ganglion cell [Ca2+]i is mediated by neuronal nicotinic receptors
The increase of ganglion cell [Ca2+]i produced by nicotine was blocked by (+)-tubocurarine, a nonspecific nicotinic receptor antagonist (Halvorsen and Berg, 1986; Vijayaraghavan et al., 1992). The effect of 50 μm nicotine was blocked completely by a 15 min treatment with 25 μm(+)-tubocurarine but was restored by a 30 min wash (Fig.3A). The effect of nicotine was also blocked by hexamethonium, which is believed to block the nicotine-gated channel (Ascher et al., 1979). After a 20 min treatment with 100 μm hexamethonium, the effect of 50 μm nicotine was blocked completely but recovered at least partially after a 40 min wash (Fig.3B).

The effect of nicotinic receptor antagonists on the nicotine-induced rise of ganglion cell [Ca2+]i. A, The effect of 50 μm nicotine (Nic) was blocked by 25 μm (+)-tubocurarine (Tbc) and 100 μm hexamethonium (Hex; B), and in both cases the response to nicotine recovered after washing. C, Neither 1 nor 50 μm nicotine were blocked by 2.5 μm α-bungarotoxin (α-Bgt).D, Ten micromolar methyllycaconitine (Mla) did not block 50 μm nicotine, and 1 μm methyllycaconitine failed to block the response to 1 μm nicotine. E, Summary of the results from all ganglion cell studies expressed as mean ΔF/F. Error bars are SEM.
The effect of nicotine on ganglion cell [Ca2+]i was not blocked by drugs selective for the α-bungarotoxin-sensitive nicotinic receptor (Ward et al., 1990; Vijayaraghavan et al., 1992). At 2.5 μm, α-bungarotoxin did not block the effect of 50 or 1 μm nicotine (Fig. 3C). The competitive antagonist methyllycaconitine, at 10 μm, did not block the increase of [Ca2+]i produced by 50 μm nicotine (Fig. 3D) but, in several retinas, did block the effect of 1 μmnicotine (not shown). Methyllycaconitine has nanomolar affinity for α-bungarotoxin-sensitive nicotinic receptors and micromolar affinity for α-bungarotoxin-insensitive nicotinic receptors (Ward et al., 1990; Vijayaraghavan et al., 1992) and, thus, the effect of 10 μm methyllycaconitine on the ganglion cell response to 1 μm nicotine could be attributable to an action at α-bungarotoxin-insensitive nicotinic receptors. Consistent with this possibility was the finding that lower concentrations of methyllycaconitine (1 μm), still well above the affinity of methyllycaconitine for α-bungarotoxin-sensitive receptors, failed to block the effect of 1 μm nicotine on ganglion cell [Ca2+]i (Fig.3D). Cells that did respond to 1 or 50 μm nicotine in the presence of either α-bungarotoxin or methyllycaconitine responded similarly, or more weakly, after washout. Cells that did not respond in the presence of these antagonists also failed to respond after a 30–60 min wash and, therefore, they were not considered.
The effects of these drugs on all ganglion cells studied are summarized in Figure 3E. On average, the response to 50 μm nicotine was ΔF/F = 0.50 ± 0.02 (n = 217), and the response to 1 μm nicotine was ΔF/F = 0.31 ± 0.03 (n = 43). In the presence of (+)-tubocurarine, there was no response to 50 μm nicotine (ΔF/F = 0.03 ± 0.01, n = 27) but after wash the response recovered (ΔF/F = 0.39 ± 0.06). Hexamethonium also blocked the 50 μm nicotine-induced rise of [Ca2+]i(ΔF/F = 0.04 ± 0.03, n = 12), an effect that was at least partially reversed by subsequent washing (ΔF/F = 0.24 ± 0.03). Neither α-bungarotoxin nor methyllycaconitine blocked the increase of [Ca2+]iproduced by 50 μm nicotine (ΔF/F = 0.49 ± 0.08, n = 9; 0.37 ± 0.04, n = 19, respectively); 2.5 μm α-bungarotoxin and 1 μm methyllycaconitine also failed to block the effect of 1 μm nicotine (ΔF/F = 0.33 ± 0.04, n = 26; 0.36 ± 0.03, n = 35, respectively).
The effect of 1 μm nicotine, but not 50 μm nicotine, on ganglion cell [Ca2+]i was blocked by previous incubation in 100 nm κ-bungarotoxin, a selective neuronal nicotinic receptor antagonist (Fig.4A). In a few cases, the response to 1 μm nicotine recovered after wash (Fig.4B) but, on average, the block was essentially irreversible (Fig. 4C). In control pieces of retina not treated with κ-bungarotoxin, 1 μm nicotine produced increases of [Ca2+]i that were typical or somewhat greater than usual (ΔF/F = 0.41 ± 0.08, n = 11). A higher concentration (1 μm) of κ-bungarotoxin blocked the response to 50 μm nicotine, but no recovery was achieved after washing for 1 hr. The poor and inconsistent recovery of ganglion cells from κ-bungarotoxin block has also been described by other investigators (Lipton et al., 1987; Aizenman et al., 1990).

The effect of nicotine on ganglion cell [Ca2+]i is blocked by κ-bungarotoxin. A, After a 40 min incubation in 100 nm κ-bungarotoxin (κ-Bgt), the effect of 1 μm nicotine (Nic), but not 50 μm nicotine, on ganglion cell [Ca2+]i was blocked.B, In some cells, the block of the response to 1 μm nicotine was reversed by washing for 1 hr.C, Mean ΔF/F of all the ganglion cells studied in response to 1 and 50 μmnicotine, shortly and 1 hr after incubation in 100 nm κ-bungarotoxin. Although some cells recovered after 1 hr wash as illustrated in B, on average the block produced by 100 nm κ-bungarotoxin was irreversible. Error bars are SEM.
The nicotine-induced response is independent of glutamate receptors
The nicotine-induced rise of ganglion cell [Ca2+]i was not blocked by NMDA or non-NMDA glutamate receptor antagonists (Fig.5A). In the presence of the NMDA receptor antagonist AP-5 (100 μm; Davies et al., 1981) and the non-NMDA receptor antagonist DNQX (50 μm; Honoré et al., 1988), neither 10 μm kainic acid nor 10 μm NMDA produced a change in ganglion cell [Ca2+]i. However, subsequent treatment with 50 μm nicotine did increase [Ca2+]i. After washing for 30 min, the response to kainic acid and NMDA recovered. The results from all ganglion cells studied are shown in Figure5B. In the presence of 50 μm DNQX, alone or in combination with AP-5, there was no significant response to 10 μm kainic acid (ΔF/F = 0.08 ± 0.01, n = 70), but the response recovered after washing (ΔF/F = 0.56 ± 0.03). Similarly, in the presence of 100 μm AP-5, alone or in combination with DNQX, 10 μm NMDA had little effect on [Ca2+]i(ΔF/F = 0.01 ± 0.01, n = 88) but recovered after washing (ΔF/F = 0.45 ± 0.02). In the presence of both 50 μm DNQX and 100 μm AP-5, 50 μm nicotine produced an increase of [Ca2+]i(ΔF/F = 0.29 ± 0.03, n = 48).

The effect of nicotine on ganglion cell [Ca2+]i is not blocked by glutamate receptor antagonists. A, During incubation with a mixture of non-NMDA (50 μm DNQX) and NMDA (100 μm AP-5) receptor antagonists, the ganglion cell responses to 10 μm kainic acid (KA) and 10 μmNMDA were blocked. However, the effect of 50 μm nicotine (Nic) was not blocked by these antagonists. After wash, the response to both kainic acid and NMDA recovered. B, Summary of the results from all ganglion cell studies expressed as mean ΔF/F. Error bars are SEM.
The mechanism of the effect of muscarine on ganglion cell [Ca2+]i
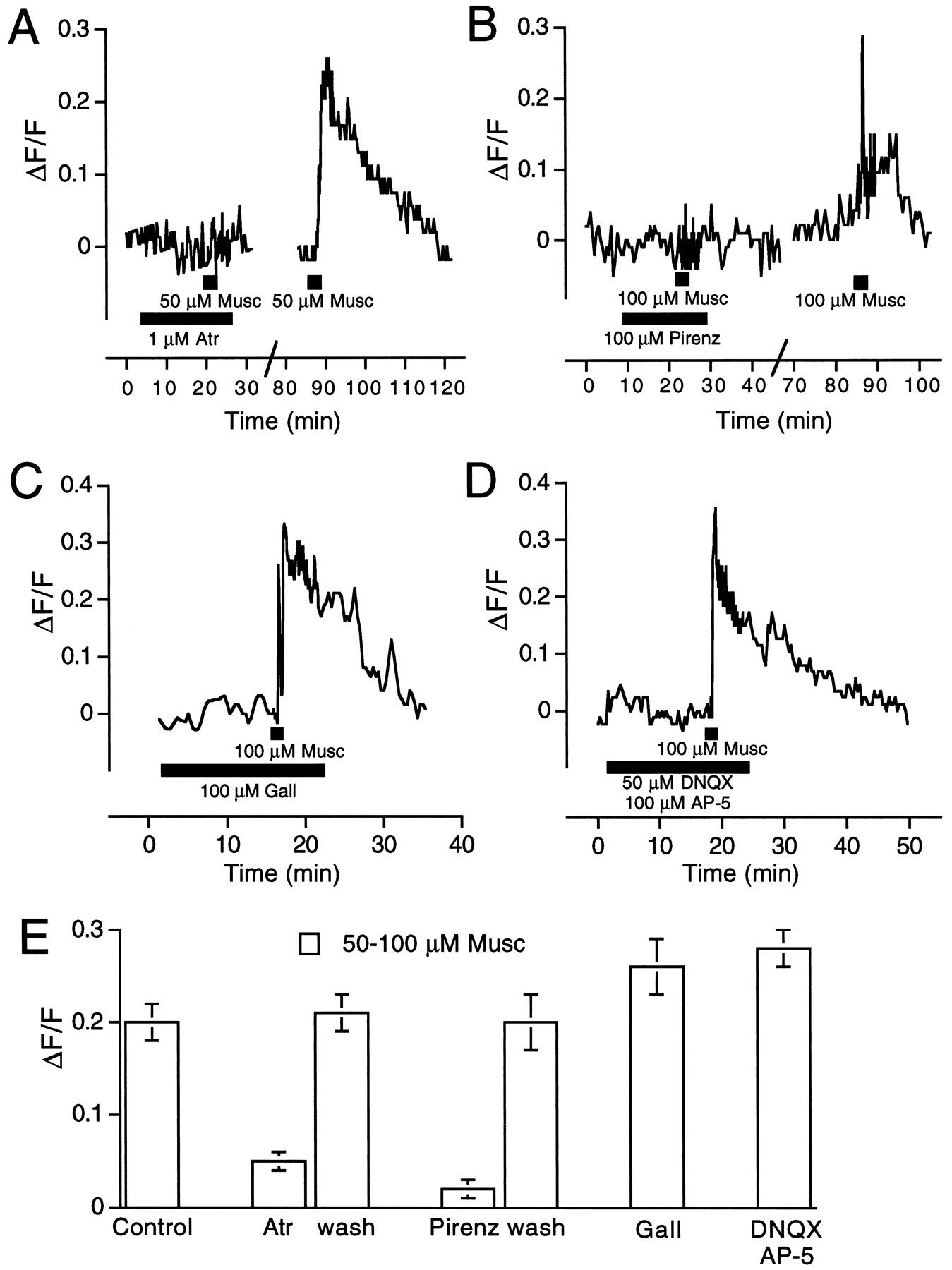
The increase of ganglion cell [Ca2+]i produced by 50 μm muscarine was blocked by atropine, a nonspecific muscarinic receptor antagonist. In the presence of 1 μm atropine, 50 μmmuscarine did not produce a significant increase of CaGD fluorescence but, after washing for 1 hr, the response to muscarine was restored (Fig. 6A). The effect of muscarine was also blocked reversibly by pirenzepine, an M1 muscarinic receptor antagonist (Burke, 1986), but at a rather high concentration (100 μm). In preliminary studies, 10 and 50 μm pirenzepine failed to block the response to 50–100 μm muscarine. In the presence of 100 μm pirenzepine, 100 μmmuscarine did not elicit a response but, after washing for 1 hr, the response to muscarine was restored (Fig. 6B). One hundred micromolar muscarine was not blocked by 100 μmgallamine, an M2 muscarinic receptor antagonist (Burke, 1986) (Fig.6C), or by the mixture of the NMDA and non-NMDA glutamate receptor antagonists AP-5 (100 μm) and DNQX (50 μm; Fig. 6D). The results from all ganglion cells studied are summarized in Figure 6E. The response to 50–100 μm muscarine was, on average, ΔF/F = 0.20 ± 0.01 (n = 36). In the presence of 1 μm atropine, there was no response to 50 μm muscarine (0.05 ± 0.01, n = 16), but the response to muscarine recovered after washing (ΔF/F = 0.21 ± 0.02). In the presence of 100 μm pirenzepine, 100 μm muscarine did not elicit a response (ΔF/F = 0.02 ± 0.01, n = 8), but after washing the response to muscarine recovered (ΔF/F = 0.20 ± 0.03). One hundred micromolar gallamine did not block the response to 100 μm muscarine (ΔF/F = 0.26 ± 0.03, n = 17), nor did the mixture of the glutamate antagonists AP-5 and DNQX (ΔF/F = 0.28 ± 0.02, n = 6).

The effect of muscarine on ganglion cell [Ca2+]i has an M1-like muscarinic receptor pharmacology. The effect of 50–100 μm muscarine (Musc) on ganglion cell [Ca2+]i was blocked by 1 μm atropine (Atr; A) and 100 μm pirenzepine (Pirenz;B), an antagonist with M1 > M2 muscarinic receptor affinity. The block produced by both antagonists was reversed by washing. C, The effect of muscarine was not blocked by 100 μm gallamine (Gall), an M2 muscarinic receptor antagonist, or the mixture of the glutamate antagonists DNQX (50 μm) and AP-5 (100 μm) (D). E, Summary of the results from all ganglion cell studies expressed as mean ΔF/F. Error bars are SEM.
The effect of nicotine and muscarine on ganglion cell [Ca2+]i depends on the presence of external calcium
The effect of both nicotine and muscarine depended on the presence of calcium in the superfusion solution. Ames medium was made nominally calcium-free by adding 5 mm EGTA, 2.5 mm MgCl2, and adjusting the pH to 7.4. During superfusion with this solution, [Ca2+]i was unaffected when challenged with either nicotine (Fig.7A,C; ΔF/F = 0.03 ± 0.01, n = 14) or muscarine (Fig.7B,C; ΔF/F = 0.03 ± 0.02, n = 11). After 30 min wash in the normal Ames medium, the response to both nicotine and muscarine recovered (ΔF/F = 0.34 ± 0.05 and 0.22 ± 0.05, respectively).

The effect of nicotine and muscarine on ganglion cell [Ca2+]i depends on extracellular calcium. When Ames solution was made nominally calcium-free by the addition of 5 mm EGTA, the response of ganglion cells to nicotine (Nic; A) and muscarine (Musc; B) was abolished. After subsequent washing, the response to both agonists recovered.C, The mean ΔF/F from all cells studied in response to nicotine and muscarine, during and after superfusion with calcium-free Ames medium. Error bars are SEM.
Cholinergic agonists do not increase [Ca2+]i in displaced cholinergic amacrine cells
In some experiments, retinal neurons were labeled with both CaGD and the nuclear stain DAPI. In the GCL of the rabbit retina, only the displaced cholinergic (Cb) amacrine cells are strongly labeled by DAPI at low concentrations (Masland et al., 1984; Tauchi and Masland, 1984; Vaney, 1984). Figure 1H shows four DAPI-labeled Cb amacrine cells (red circles) in the GCL; most of the other cells with weakly labeled nuclei are ganglion cells (Vaney et al., 1981; Hughes, 1985). When treated with 50 μm nicotine, the larger, weakly labeled ganglion cells showed a typical response, but the cholinergic amacrine cells did not respond (Fig. 1I). However, when subsequently treated with 10 μm kainic acid, both the ganglion cells and the displaced amacrine cells responded strongly (Fig. 1J).
The results from two other retinas are illustrated in Figure8A. In both cases, 50 μm nicotine failed to elicit a significant response in cholinergic amacrine cells, but a subsequent treatment with 10 μm kainic acid produced a response (Fig.8A, left). In presumptive ganglion cells from the same pieces of retina, nicotine and kainic acid both produced typical responses (Fig. 8A, right).

Nicotine does not increase [Ca2+]i in cholinergic amacrine cells. As illustrated in Figure 1H, displaced (Cb) cholinergic amacrine cells were identified in the GCL of the rabbit retina by their characteristic bright DAPI-labeled nucleus. Cells without this feature were identified as presumptive ganglion cells (see Materials and Methods).A, Optical recordings from two retinas illustrating the effects of nicotine (Nic) and kainic acid (KA) on these cells, and ganglion cells from the same pieces of retina. In both retinas, cholinergic amacrine cells failed to respond to 50 μm nicotine, but did respond to 10 μm kainic acid. In comparison, presumptive ganglion cells responded to both 50 μm nicotine and 10 μmkainic acid. B, The mean ΔF/Fof all cholinergic amacrine and ganglion cells studied in response to nicotine and kainic acid. Error bars are SEM.
All of the 41 cholinergic amacrine cells studied responded to 10 μm kainic acid (ΔF/F = 0.76 ± 0.06), but none produced a significant response to 50 μmnicotine (ΔF/F = 0.06 ± 0.01) (Fig.8B). In the adjacent ganglion cells, 50 μm nicotine increased [Ca2+]i(ΔF/F = 0.42 ± 0.03, n = 65) as did 10 μm kainic acid (ΔF/F = 0.81 ± 0.05). Fifty micromolar muscarine also failed to increase [Ca2+]i in cholinergic amacrine cells (ΔF/F = 0.03 ± 0.01, n = 17), but these cells did respond to 10 μm kainic acid (ΔF/F = 0.77 ± 0.06) (not illustrated).
DISCUSSION
Dextran-conjugated calcium indicator dye labels retinal neurons
Despite the potential utility of calcium imaging in mature nervous tissue, loading neurons in intact tissue with membrane-permeable calcium-sensitive dyes has proven difficult. It is unclear why these dyes fail to label mature nervous tissue, but perhaps the neuropil prevents adequate dye penetration (Regehr and Tank, 1991). In this study, neurons in the mature retina were labeled by injecting CaGD directly into the tissue. A similar technique has been used to label neurons in chick embryo spinal cord and ciliary ganglion (O’Donovan et al., 1993, 1994; Yawo and Chuhma, 1993). The labeling produced by dextran-conjugated dyes may be successful because dextrans diffuse well (Popov and Poo, 1992) and are actively transported (Glover et al., 1986; Nance and Burns, 1990; Manns and Fritzsch, 1991; Watanabe et al., 1993). In retina, labeling of neurons around the injection site probably results from loading at cut dendrites and diffusion to the soma. Ganglion cell labeling distant from the injection site is likely attributable to loading of cut ganglion cell axons followed by retrograde transport. The physiological properties of the neurons labeled by this technique could potentially be different from unlabeled cells, and this could have an influence on the apparent pharmacology. However, the pharmacological responses recorded using the optical method were consistent with the responses measured, when possible, using conventional electrode techniques (Massey and Miller, 1988; Zhou and Fain, 1995) adding to the confidence in the reliability of the optical technique.
Nicotine, acting at neuronal nicotinic receptors, increases [Ca2+]i in ganglion cells
Previous electrophysiological studies on rabbit and rat ganglion cells reported that ACh or nicotine affects all ganglion cells (Ariel and Daw, 1982; Lipton et al., 1987; Kaneda et al., 1995). However, studies of the distribution of both β4nicotinic receptor subunits in ground squirrel retina (Britto et al., 1994) and other subunits in chick retina (Keyser et al., 1988;Hamassaki-Britto et al., 1991; Britto et al., 1992) suggested that only a subpopulation of ganglion cells possesses nicotinic receptors. Nicotine produced dose-dependent increases of [Ca2+]i in rabbit retinal ganglion cells and, in several “responsive” preparations, affected every CaGD-labeled ganglion cell. It seems unlikely, therefore, that the nonresponsiveness of some ganglion cells in other preparations reflects the presence of distinct types of nicotine-insensitive ganglion cells in the rabbit retina: this would require that only the nicotine-sensitive ganglion cells were selectively or fortuitously labeled in the “responsive” preparations, which seems improbable given that the CaGD was injected in bulk into the tissue. Further evidence for a lack of selectivity comes from the finding that the soma size of nicotine-responsive and CaGD-labeled ganglion cells covers the full range of ganglion cell types (Vaney, 1980; Peichl et al., 1987;Amthor et al., 1989a,b). Although it is not clear why some ganglion cells did not respond to nicotine in some preparations, the demonstration in the “responsive” preparations that all CaGD-labeled ganglion cells were activated by nicotine supports the conclusion that all types of rabbit retinal ganglion cells are nicotine-sensitive.
α-Bungarotoxin binding has been demonstrated in nonmammalian retina (Vogel et al., 1977; Zucker and Yazulla, 1982) and mammalian retina (Vogel and Nirenberg, 1976), where both the IPL and GCL are labeled, consistent with a ganglion cell localization; moreover, in nonmammalian retinas, specific nicotinic receptor subunits that bind α-bungarotoxin (α7, α8; for review, see Deneris et al., 1991;Clarke, 1992) also have been detected (Britto et al., 1992; Keyser et al., 1993). In most vertebrates, however, the effect of ACh on retinal ganglion cells is mediated by α-bungarotoxin-insensitive “neuronal” nicotinic receptors, which are blocked by κ- (neuronal-) bungarotoxin (Lipton et al., 1987; Loring et al., 1989;Aizenman et al., 1990; but see Yazejian and Fain, 1993). The action of ACh at neuronal nicotinic receptors is consistent with the identification in chick retina (Hamassaki-Britto et al., 1991; Whiting et al., 1991; Britto et al., 1992; Keyser et al., 1993) of nicotinic receptor subunits sensitive to κ-bungarotoxin (α3, α4; for review, see Role, 1992; McGehee and Role, 1995).
In chick ciliary ganglion, α-bungarotoxin blocks nicotine-induced increases of [Ca2+]i (Vijayaraghavan et al., 1992), an effect associated with α7nicotinic receptor subunits (Vernallis et al., 1993). This subunit has been identified on chick retinal ganglion cells (Britto et al., 1992;Keyser et al., 1993), raising the possibility that α-bungarotoxin might block the effect of nicotine on ganglion cell [Ca2+]i. This hypothesis was not supported by the pharmacology of the effect of nicotine on rabbit retinal ganglion cell [Ca2+]i, which was consistent with an action at neuronal nicotinic receptors (Halvorsen and Berg, 1986; Lipton et al., 1987; Loring et al., 1989; Mulle and Changeux, 1990; Vijayaraghavan et al., 1992). Rabbit retinal ganglion cells may not possess α7 nicotinic receptor subunits or, as in the chick, they may have relatively few α7 subunits compared with another α-bungarotoxin-binding subunit, α8 (Britto et al., 1992; Keyser et al., 1993). In either case,the significance of α-bungarotoxin binding in retina remains uncertain.
It is likely that the effect of nicotine on ganglion cell [Ca2+]i is primarily attributable to the activation of voltage-gated calcium channels (Karschin and Lipton, 1989). However, the response may also be partly attributable to either calcium influx through the nicotine receptor-gated channel (Adams and Nutter, 1992; Mulle et al., 1992;Vernino et al., 1992) or the release of calcium from intracellular stores induced by calcium influx (Lei et al., 1992) (for review, seeSimpson et al., 1995). In each case, however, the response depends on the presence of extracellular calcium, consistent with the demonstration, in this study, that the effect of nicotine was abolished when the superfusion solution was made calcium-free.
The effect of nicotine does not depend on glutamate receptor activation
Treatment with the glutamate receptor agonists kainic acid and NMDA produced robust increases in ganglion cell [Ca2+]i. This is consistent with the excitatory effect of glutamate on ganglion cells in the mammalian retina (Aizenman et al., 1988; Massey and Miller, 1988,1990). Although ganglion cells are affected directly by ACh (Lipton et al., 1987), a significant part of the nicotine-induced response might be attributable to an indirect action of nicotine at bipolar cells, particularly given the localization of nicotinic receptors in the outer plexiform layer of several species (Vogel and Nirenberg, 1976; Yazulla and Schmidt, 1976; Zucker and Yazulla, 1982; Hamassaki-Britto et al., 1994). However, direct evidence that the effect of nicotine on ganglion cell [Ca2+]i is not attributable to an indirect action at bipolar cells is provided by the demonstration that the effect was not blocked by the glutamate antagonists DNQX and AP-5.
Muscarine increases ganglion cell [Ca2+]i
Muscarinic receptors have been identified in the IPL and GCL of the mammalian retina (Zarbin et al., 1986; Hutchins, 1994), but their functional significance is not clear. In rabbit retina, muscarinic antagonists only weakly affect ganglion cell responses (Ames and Pollen, 1969; Masland and Ames, 1976), whereas in cat retina, muscarinic antagonists decrease both light-induced and ACh-induced effects on ganglion cells (Schmidt et al., 1987; Kaneda et al., 1995). However, muscarine-induced currents were not detected in isolated ganglion cells from cat and rat retinas, and the ACh-induced currents were unaffected by atropine in the rat cells (Lipton et al., 1987;Kaneda et al., 1995).
Muscarine increased the [Ca2+]i in some rabbit ganglion cells, but only weakly compared with nicotine. It is difficult to reconcile the muscarine-induced [Ca2+]i increase with the failure of muscarine to elicit currents in isolated ganglion cells. An obvious possibility is that the effect of muscarine on ganglion cell [Ca2+]i was indirect. The effect was not blocked by glutamate receptor antagonists, indicating that it was not attributable to indirect action at bipolar cells. Muscarine still might act through amacrine cells (Cunningham and Neal, 1983), but cholinergic amacrine cells seldom, if ever, make synapses with noncholinergic amacrine cells (Famiglietti, 1991).
If the effect of muscarine on ganglion cell [Ca2+]i was attributable solely to release of calcium from intracellular stores, a current may not necessarily result. The pharmacology of muscarine was consistent with an M1-like muscarinic receptor, which could lead to phosphoinositide hydrolysis (Cutliffe and Osborne, 1987; Ghazi and Osborne, 1988; Moroi-Fetters et al., 1988) and inositol (1,4,5)-trisphosphate-induced release of calcium from intracellular stores (for review, see Berridge, 1993). However, the muscarine-induced increase of ganglion cell [Ca2+]i is unlikely to be attributable solely to calcium release from internal stores because the effect of muscarine was abolished in calcium-free solution.
Nicotine does not increase [Ca2+]i in displaced cholinergic amacrine cells
In the rabbit retina, the principal outputs from the cholinergic amacrine cells are to the dendrites of ganglion cells (Famiglietti, 1983, 1991; Brandon, 1987), but synapses between cholinergic amacrine cells have also been identified (Millar and Morgan, 1987; Mariani and Herch, 1988; Famiglietti, 1991). Neither nicotine nor muscarine affected [Ca2+]i in displaced cholinergic amacrine cells, indicating that synapses between these cells are not cholinergic. This conclusion is consistent with a recent report that neither ACh nor carbachol elicited currents in cholinergic amacrine cells recorded in rabbit retinal slices (Zhou and Fain, 1995). A sparse population of ACh receptors, located distally on dendrites of cholinergic amacrine cells, may produce a signal that is too weak to affect somatic [Ca2+]i. Alternatively, any cholinergic input to cholinergic amacrine cells may be masked by tonic inhibition. Such inhibition could come from other amacrine cell types or perhaps from the cholinergic amacrine cells themselves, which contain several neuroactive substances in addition to ACh.
Cholinergic amacrine cells contain and release GABA (Brecha et al., 1988; Vaney and Young, 1988; O’Malley and Masland, 1989); moreover, they are directly modulated by GABA (Zhou and Fain, 1995) and possess GABAA receptors (Greferath et al., 1993, 1995). Cholinergic amacrine cells also show adenosine-like immunoreactivity (Blazynski, 1989), and both adenosine (Blazynski et al., 1992) and ATP (Neal and Cunningham, 1994) modulate ACh release in the retina. Although adenosine or ATP could be released at the synapses between cholinergic amacrine cells, the effect on ACh release could also be indirect, mediated by other types of amacrine cells (Neal and Cunningham, 1994).
Footnotes
This work was supported by grants to D. I. Vaney and R. O. L. Wong from the National Health and Medical Research Council of Australia and a fellowship to W.H.B. from the Natural Science and Engineering Research Council of Canada. I thank Professor D. J. Adams for generously supplying the κ-bungarotoxin, D. I. Vaney and R. O. L. Wong for helpful discussions and critical reading of this manuscript, and J. C. Nelson, D. K. Crook, D. M. Thomas, and C. Macqueen for technical help.
Correspondence should be addressed to Dr. William H. Baldridge, School of Optometry, Faculty of Science, University of Waterloo, Waterloo, Ontario, Canada N2L 3G1.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}