

Abstract
A long-standing question in neurobiology is whether astrocytes respond to the neuronal release of neurotransmitters in vivo. To address this question, acutely isolated hippocampal slices were loaded with the calcium-sensitive dye Calcium Green-1 and the responses of the astrocytes to electrical stimulation of the Schaffer collaterals were monitored by confocal microscopy. To confirm that the responsive cells were astrocytes, the slices were immunostained for the astrocytic marker glial fibrillary acidic protein. Stimulation of the Schaffer collaterals (50 Hz, 2 sec) resulted in increases in the concentration of intracellular calcium ([Ca2+]i) in the astrocytes located in the stratum radiatum of CA1. The astrocytic responses were blocked by the sodium channel blocker tetrodotoxin, the voltage-dependent calcium channel blocker ω-conotoxin-MVIIC, and the selective metabotropic glutamate receptor antagonist α-methyl-4-carboxyphenylglycine (MCPG). These results suggest that the astrocytic responses were induced by stimulation of metabotropic glutamate receptors on the astrocytes by neuronally released glutamate. The astrocytic responses to neuronal stimulation were enhanced in the presence of the K+ channel antagonist 4-aminopyridine (4-AP). Inhibition of the astrocytic responses in the presence of 4-AP required the presence of both MCPG and the ionotropic glutamate receptor antagonist kynurenic acid. These results suggest that higher levels of neuronal activity result in stimulation of both metabotropic and ionotropic glutamate receptors on the astrocytes. Overall, the results indicate that hippocampal astrocytes in situ are able to respond to the neuronal release of the neurotransmitter glutamate with increases in [Ca2+]i.
- hippocampus
- astrocytes
- in situ
- intracellular calcium
- metabotropic glutamate receptors
- ionotropic glutamate receptors
- CA1
- stratum radiatum
- neural–glial interactions
A very fundamental question in neurobiology is whether astrocytes respond to the neuronal release of neurotransmittersin vivo. More than 25 years ago, Orkand et al. (1966)demonstrated that amphibian glial cells in the optic nerve of the mudpuppy depolarized in response to neuronal stimulation. Because they had also demonstrated that the membrane potential of glial cells accurately reflects changes in extracellular K+concentration ([K+]o) (Kuffler et al., 1966), they concluded that the glial responses to neuronal activity were the result of an increase in [K+]o. As the [K+]o increased, the glial cells depolarized. As a result of this work, glial cells were viewed as accurate K+ electrodes that depolarized in response to the increases in K+ associated with neuronal activity. During the ensuing years, many groups demonstrated that mammalian glia also depolarized in response to neuronal stimulation (Futamachi and Pedley, 1976; Miller et al., 1977;Roitbak and Fanardjian, 1981). They also concluded that the glial responses were attributable to increases in [K+]o (Futamachi and Pedley, 1976; Miller et al., 1977; Roitbak and Fanardjian, 1981).
Once the technology for making purified glial cultures was available (McCarthy and de Vellis, 1980), it was demonstrated that astroglia contain a wide variety of neurotransmitter receptors coupled to different second-messenger systems (for review, see Murphy and Pearce, 1987). Conceptually, the view of astrocytes as simple K+ sensors was replaced by the view that astrocytes in vivo respond to neurotransmitters released from activated neurons. Although experimentally there is no direct evidence that astrocytes in vivo respond to neuronally released neurotransmitters, there is evidence of immunoreactivity for receptors, including β-adrenergic receptors (Aoki et al., 1987; Aoki and Pickel, 1992) and AMPA receptors (Martin et al., 1993), on astrocytes in situ. Recent experiments have demonstrated that hippocampal astrocytes in situ contain glutamate (Jabs et al., 1994; Porter and McCarthy, 1995a), adenosine (Porter and McCarthy, 1995b), α-adrenergic (Duffy and MacVicar, 1995), and GABA receptors (Fraser et al., 1995) coupled to increases in the concentration of intracellular calcium ([Ca2+]i). Therefore, astrocytes in situ contain functional receptors. However, it is unknown whether these receptors are activated by neuronal activityin situ. It has been demonstrated that hippocampal astrocytes in situ respond to perfusion of NMDA and that these responses can be blocked by the sodium channel blocker tetrodotoxin (TTX) (Porter and McCarthy, 1995a). Because TTX blocks synaptic transmission, these results suggest that NMDA activates receptors on the neurons and that the neurons release a substance such as a neurotransmitter or K+ that causes the astrocytes to respond. The data suggest that astrocytes in situ can respond to neuronal activity, but they do not distinguish between astrocytic responses to released neurotransmitter versus an increase in [K+]o. A clear demonstration that gray matter astrocytes do respond to the release of neurotransmitters from active neurons is lacking in the literature.
Because astrocytes make up a very large percentage of the brain’s mass and are believed to be involved in physiological processes such as the maintenance of the brain microenvironment (Walz, 1989) and in pathologies such as cerebral edema (Kimelberg, 1995) and epilepsy (Heinemann et al., 1995), it is important to understand how they respond to changes in neuronal activity.
To address these issues, we used calcium-sensitive dyes, confocal microscopy, electrophysiology, and immunocytochemistry to determine whether gray matter astrocytes in acutely isolated hippocampal slices respond to neuronally released neurotransmitter(s) or simply to the increase in [K+]oassociated with neuronal activity. Our results indicate that hippocampal astrocytes in situ respond to neuronally released glutamate with increases in [Ca2+]i.
MATERIALS AND METHODS
Dye loading of acutely isolated hippocampal slices.Transverse hippocampal slices (300 μm thick) were prepared from rats (postnatal days 10–13) with a Vibroslice (Campden Instruments, Sileby, UK). To load the astrocytes with the calcium-sensitive fluorescent dye, the slices were incubated in aerated (95% O2/5% CO2) artificial CSF (aCSF) containing 0.06% pluronic acid and 10 μm Calcium Green-1 AM for 1.5–3 hr at 34°C. Experiments were done at room temperature. As reported previously (Porter and McCarthy, 1995a,b), the astrocytes were preferentially loaded with the dyes. There was little if any loading of the neurons. Details of the loading procedure have been reported previously (Porter and McCarthy, 1995a).
Image acquisition. The dye-loaded slices were secured in a perfusion chamber (Warner Instruments, Hamden, CT) on the stage of an inverted confocal microscope (Olympus, Lake Success, NY) and continuously perfused with aCSF. All experiments were done within 9 hr after removing the slices from the animals.
4-Aminopyridine (4-AP) and tetrodotoxin treatments were carried out by dissolving the drugs in aCSF and placing them in reservoirs suspended above the microscope stage. Treatments involving ω-conotoxin-MVIIC (MVIIC), α-methyl-4-carboxyphenylglycine (MCPG), and kynurenic acid (KY) were carried out by dissolving the compound in 2 ml of aerated aCSF and manually exchanging the bath once every minute for 20–30 min. When the bath was manually exchanged with aCSF alone in control slices, 88% (141/160 astrocytes/7 slices) of the astrocytes that responded to neuronal stimulation before the treatment responded after the treatment control. Also, this treatment did not block the field potentials. These results indicate that the method of drug treatment could not account for the changes in astrocytic and neuronal responses seen with the various drug treatments.
4-AP, TTX, and KY were purchased from Sigma (St. Louis, MO). MCPG and MVIIC were purchased from Research Biochemicals (Natick, MA), and Calcium Green-1 AM was obtained from Molecular Probes (Eugene, OR).
A krypton–argon laser (Omnichrome, Chino, CA) excited the Calcium Green-1 at 488 nm, and emission of >515 nm was collected for analysis. During the treatments, the fluorescent intensities of the loaded astrocytes were sampled every 1–2 sec. After each treatment, there was a 30 min washout period. The relative changes in fluorescence were then graphed to illustrate the relative changes in [Ca2+]i in response to the various treatments. The actual [Ca2+]i was not calculated because the complexity of the slice preparation makes even the calculation of approximate [Ca2+]i questionable. Measurement of changes in relative [Ca2+]i was sufficient to address the aims of this study. Details of image acquisition have been reported previously (Porter and McCarthy, 1995a).
Electrophysiology. Figure 1 illustrates the approximate electrode arrangement and the location of the examined astrocytes. Bipolar concentric electrodes connected to a constant-current isolation source (A360 Acupulser; WPI, Sarasota, FL) were placed in the stratum radiatum of the CA1 region of the hippocampal slice to stimulate the Schaffer collateral axons. Trains of orthodromic stimuli (200–400 μA pulses, 200 μsec) were given (A310 Acupulser; WPI), and the relative changes in [Ca2+]i were monitored in the astrocytes of the stratum radiatum 200–600 μm from the stimulating electrode.

This diagram illustrates the relative positions of the stimulating (Stim) and recording (Record) electrodes and the studied astrocytes within the hippocampal slice. The Schaffer collaterals (SC) were electrically stimulated, and the changes in [Ca2+]iwere monitored in the astrocytes in the stratum radiatum of CA1 (white area) with a confocal microscope. The recording electrode was used to record the field potentials in response to single test pulses.
Throughout the experiment, test pulses of equal amplitude were given and the evoked field potentials were measured with glass microelectrodes. The field potentials were used as a measure of the relative neuronal activity during the various treatments. Field potentials were amplified, filtered (2 kHz; Warner Instruments, Hamden, CT), digitized with an analog-to-digital converter, and stored using commercially available software (Atari, Instrutech, Greatneck, NY).
Astrocyte identification. After the experiment, the astrocytes were identified by fixing the hippocampal slices, immunostaining them for the presence of glial fibrillary acidic protein (GFAP) immunoreactivity, and then imaging the slices with a confocal microscope as described previously (Porter and McCarthy, 1995a). In the absence of the primary antibody or when the primary antibody was replaced with normal rat serum, there was essentially no staining of the astrocytes. Because our examination of hippocampal slices immunostained for GFAP indicates that essentially all of the cell bodies loaded with Calcium Green-1 are GFAP+astrocytes, not every slice was subsequently immunostained for GFAP. In Results, the number of responsive astrocytes includes astrocytes both from slices that were immunostained for GFAP and from slices that were not immunostained. For each treatment paradigm, at least one slice was immunostained for GFAP to confirm the identity of the astrocytes. All of the data presented in the graphs are from astrocytes that were identified by immunocytochemistry for GFAP.
RESULTS
Astrocytes in situ respond to stimulation of the Schaffer collaterals
In acutely isolated hippocampal slices, electrical stimulation of the Schaffer collaterals produced increases in [Ca2+]i in 62% (375/601 astrocytes, 20 slices) of the astrocytes examined in the CA1 stratum radiatum (Figs. 2, 3). The increases in [Ca2+]i occurred in both the cell bodies and in the processes. Astrocytic responses were not induced by single stimuli, whereas trains of stimuli (50 Hz, 2 sec) were very effective in eliciting responses in the astrocytes. These same stimulus parameters have been used to induce increases in [Ca2+]i in hippocampal pyramidal cells in situ (Regehr et al., 1989; Regehr and Tank, 1990). The responses generally began at the beginning of the stimulus train, reached a maximum at the end of the stimulation, and then slowly decayed toward baseline over an average of 14 ± 7 sec (n = 71). Subsequent trains of stimuli produced astrocytic responses of similar shape, amplitude, and duration (data not shown).

Electrical stimulation of Schaffer collaterals induces increases in [Ca2+]i in the hippocampal astrocytes in situ. The pseudocolorized confocal images of hippocampal astrocytes in situ loaded with Calcium Green-1 illustrate the relative fluorescence of the astrocytes before stimulation (a), at the end of the stimulation (200 μA, 50 Hz, 2 sec) (b, c), and 28 sec after the stimulation (d). The color bar on the right indicates that the relative [Ca2+]iincreases as the colors change from violet to red. Scale bar, 20 μm. The cells studied in a–d were identified as astrocytes by subsequent immunostaining for the astrocytic marker GFAP (e). The astrocytic nuclei were stained with propidium iodide (f).

Graphical representation of the experiment shown in Figure 2. A, This image is the black and white equivalent of image b in Figure 2. The relative fluorescence of the indicated regions (1–5) of the astrocytes was monitored. B, This graph illustrates the relative changes in calcium for the regions labeled in A. The lettersa–d correspond to imagesa–d of Figure 2. The position of the letters indicates where in the time sequence the image was taken. Theopen bar at the bottom indicates when the stimulation (200 μA, 50 Hz, 2 sec) was applied to the Schaffer collaterals. The traces in this graph and all subsequent graphs were arbitrarily shifted along the ordinate axis for clarity. C, This field potential was induced by a single test pulse of 200 μA just before the experiment. The presynaptic and postsynaptic portions of the field potential are indicated by the open arrow and the filled arrow, respectively.
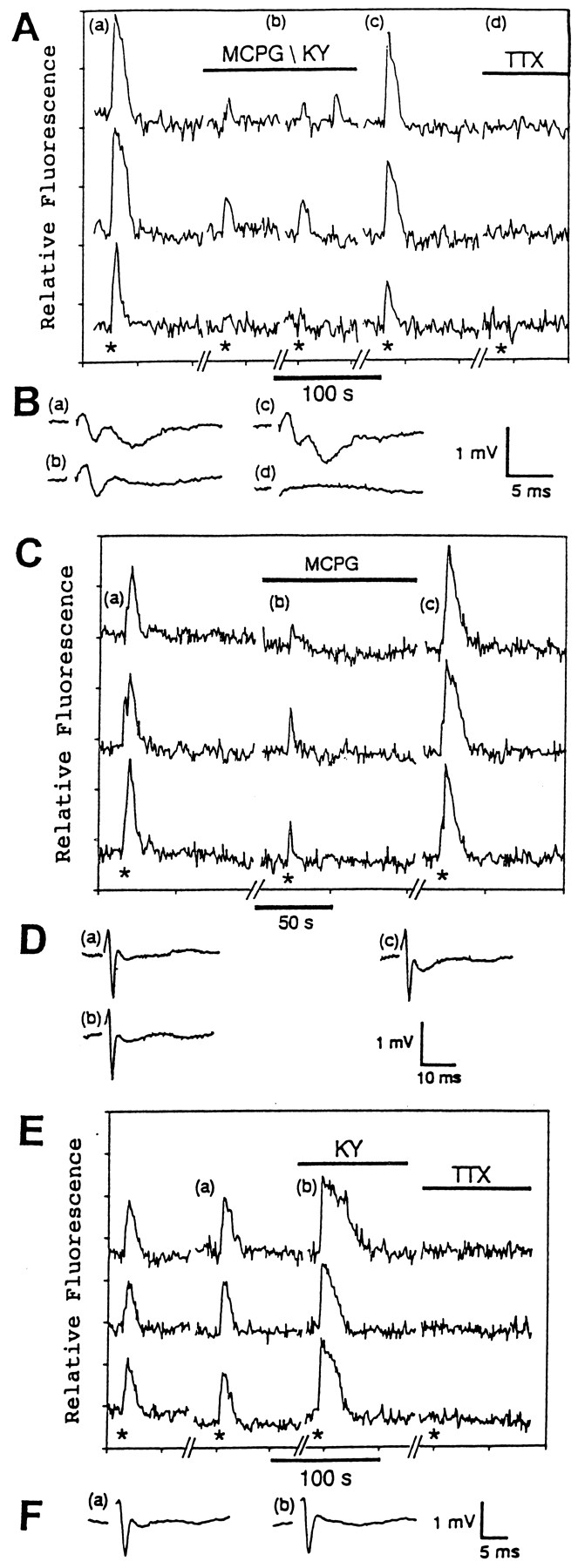
Because most of the astrocytic responses occurred within 200–500 μm of the stimulating electrode, several controls were done to control for direct stimulation of the astrocytes by the electrical field. First, 100% of the astrocytic responses (419/419 astrocytes, 20 slices) were blocked by the sodium channel antagonist TTX (Figs.4, 5, 6), which prevents axonal action potential propagation (Dingledine, 1983). The field potential was also eliminated by the TTX (Figs. 4B, 5B). Because a majority of the GFAP+ astrocytes in the CA1 stratum radiatum do not appear to contain TTX-sensitive sodium channels in situ (Sontheimer and Waxman, 1993; Kressin et al., 1995), it is unlikely that TTX is interacting with astrocytic channels to block the astrocytic responses. These results indicate that neuronal activity is necessary for the induction of the astrocytic responses.

The astrocytic responses to stimulation of the Schaffer collaterals are enhanced by the K+channel antagonist 4-AP and are inhibited by the Na+ channel antagonist TTX and the voltage-dependent Ca2+ channel antagonist MVIIC.A, This graph illustrates the responses of three different astrocytes in situ to electrical stimulation of the Schaffer collaterals (200 μA, 50 Hz, 2 sec) in the presence or absence of 100 μm 4-AP and 1 μm TTX. The asterisks indicate when the electrical stimulations were given. The letters a–c indicate the approximate times when the field potentials shown in B were recorded.B, The field potentials (a–c) indicate the relative level of neuronal activity in the absence of 4-AP and TTX (a), in the presence of 4-AP (b), and in the presence of 4-AP and TTX (c). C, This graph demonstrates that the responses of three different hippocampal astrocytes in situ to Schaffer collateral stimulation (400 μA, 50 Hz, 2 sec; asterisks) in the presence of 100 μm 4-AP are blocked by 5 μm MVIIC. The letters a andb indicate when the field potentials shown in Dwere recorded. D, The field potentials indicate the relative level of neuronal activity in the presence of 4-AP before (a) and after (b) MVIIC treatment.

In the absence of 4-AP, the astrocytic responses are attributable to the stimulation of metabotropic glutamate receptors. A, This graph demonstrates the effect of the selective metabotropic glutamate receptor antagonist MCPG (1 mm) and the ionotropic glutamate receptor antagonists KY (3 mm) and TTX (1 μm) on the responses of three different hippocampal astrocytes in situ to Schaffer collateral stimulation (200 μA, 50 Hz, 2 sec; asterisks). The lettersa–d indicate when the field potentials inB were measured. B, The field potentials indicate the relative levels of neuronal activity before (a), during (b), and after (c) treatment with MCPG and KY. TTX treatment blocked the field potential (d). C, This graph illustrates the effect of MCPG (1 mm) on the responses of three different astrocytes in situ to Schaffer collateral stimulation (200 μA, 50 Hz, 2 sec;asterisks). The letters a–c indicate when the field potentials in D were recorded. D, These field potentials were recorded before (a), during (b), and after (c) treatment with MCPG.E, This graph illustrates the effect of KY (3 mm) and TTX (1 μm) on the responses of three different hippocampal astrocytes in situto stimulation of the Schaffer collaterals (300 μA, 50 Hz, 2 sec;asterisks). The letters a and bindicate when the field potentials shown in F were measured.F, These field potentials were recorded before (a) and during (b) the KY treatment.

In the presence of 4-AP, the astrocytic responses are attributable to stimulation of both metabotropic and ionotropic glutamate receptors. A, This graph illustrates the effect of MCPG (1 mm) and KY (3 mm) on the responses of three different hippocampal astrocytes in situ to Schaffer collateral stimulation (400 μA, 50 Hz, 2 sec; asterisks) in the presence of 4-AP (100 μm). The lettersa–c indicate when the field potentials inB were measured. B, The field potentials were measured before (a), during (b), and after (c) treatment with MCPG and KY. C, This graph demonstrates the effect of MCPG (1 mm) and TTX (1 μm) on the responses of three different hippocampal astrocytes in situ to Schaffer collateral stimulation (200 μA, 50 Hz, 2 sec; asterisks) in the presence of 4-AP (100 μm). The lettersa–c indicate when the field potentials inD were measured. D, The field potentials were recorded in the absence of 4-AP (a), in the presence of 4-AP (b), and in the presence of 4-AP and MCPG (c).E, This graph illustrates the effect of KY (3 mm) on the responses of three different hippocampal astrocytes in situ to Schaffer collateral stimulation (200 μA, 50 Hz, 2 sec; asterisks) in the presence of 4-AP (100 μm). The lettersa–d indicate when the field potentials inF were recorded. F, The field potentials were recorded in the absence of 4-AP (a), in the presence of 4-AP (b), in the presence of 4-AP and KY (c), and after washout of KY (d).
Second, astrocytic responses could be blocked by making a lesion perpendicular to the pyramidal cell layer between the stimulating electrode and the astrocytes being examined (data not shown). In these lesioned slices, 75% (45/60 astrocytes, 10 slices) of the astrocytes on the proximal side of the lesion responded to electrical stimulation of the Schaffer collaterals, whereas <1% (1/111 astrocytes, 11 slices) of the astrocytes on the distal side of the lesion responded to the same stimulation; 86% (18/21 astrocytes, 2 slices) of the astrocytes on the distal side of the lesion that did not respond to stimulation of the Schaffer collaterals did respond to depolarization with 55 mm K+ with a rise in [Ca2+]i. This observation indicates that the astrocytes were not simply damaged by the lesion. These results indicate that a physical connection was necessary between the stimulating electrode and the examined astrocytes, further indicating that the astrocytic responses required propagation of neuronal action potentials along the Schaffer collateral axons. However, such astrocytic responses could be caused by either an increase in extracellular K+ or a release of a neurotransmitter(s).
4-AP potentiates the astrocytic responses
In the presence of the potassium channel antagonist 4-AP, the astrocytic responses to stimulation of the Schaffer collaterals were potentiated (Figs. 4A, 6C,E); 81% (505/626 astrocytes, 25 slices) of the astrocytes examined responded to Schaffer collateral stimulation in the presence of 4-AP. The average duration of the astrocytic responses was 51 ± 26 sec (n = 71). In hippocampal slices, 4-AP causes a large enhancement of Ca2+ influx into the presynaptic terminals with only a slight increase in Ca2+influx into the postsynaptic sites (Jones and Heinemann, 1987). In cerebral cortical synaptosomes, 4-AP induces a calcium-dependent release of glutamate that can be blocked by TTX (Tibbs et al., 1989). The increase in glutamate release enhances field potentials in hippocampal slices (Buckle and Haas, 1982) and, as indicated in Figures4B and 6, D and F, the field potentials were potentiated in the presence of 4-AP. These results indicate that the magnitudes of the astrocytic responses are correlated with the level of synaptic activity.
MVIIC blocks the astrocytic responses
Although the above results indicate that the hippocampal astrocytes in situ are responsive to neuronal activity, they do not indicate whether the release of neurotransmitter(s) is required. Experiments indicate that the release of neurotransmitter in the hippocampus involves the activation of several voltage-dependent calcium channels including N-, P-, and Q-type channels (Luebke et al., 1993; Wheeler et al., 1994). The nonselective calcium channel antagonist MVIIC (Hillyard et al., 1992) blocks the postsynaptic portions of field potentials elicited in CA1 of the hippocampal slice by stimulation of the Schaffer collaterals, indicating that neurotransmitter release is blocked (Wheeler et al., 1994). Even in the presence of 4-AP, the addition of 5 μm MVIIC inhibited the astrocytic responses to Schaffer collateral stimulation in 92% (78/85 astrocytes, 4 slices) of the astrocytes examined (Fig.4C). MVIIC also blocked the postsynaptic portion of the field potential but not the presynaptic portion (Fig. 4D). Therefore, action potentials and depolarization of the presynaptic neuronal elements are insufficient to cause the astrocytic responses. These results indicate that Ca2+ influx into the presynaptic terminals is necessary for the astrocytic responses and suggest that neurotransmitter release is involved in the responses.
The astrocytic responses are attributable to neuronal glutamate release
Hippocampal astrocytes in situ have been shown to contain ionotropic and metabotropic glutamate receptors (Jabs et al., 1994; Porter and McCarthy, 1995a), adenosine receptors (Porter and McCarthy, 1995b), and α1-adrenergic receptors (Duffy and MacVicar, 1995) coupled to increases in [Ca2+]i. Because glutamate is the major neurotransmitter released at Schaffer collateral synapses (Collingridge et al., 1983), we first examined whether glutamatergic antagonists were able to block the astrocytic responses to Schaffer collateral stimulation. In the presence of both 3 mm kynurenic acid (KY), which blocks ionotropic glutamate receptors, and 1 mm MCPG, which selectively blocks metabotropic glutamate receptors (Eaton et al., 1993), the astrocytic responses were reversibly inhibited in 67% (51/76 astrocytes, 2 slices; Fig. 5A). The blockade of glutamate receptors also blocked the postsynaptic portions of the field potentials (Fig. 5B). These results indicate that stimulation of glutamate receptors is required for the astrocytic responses to Schaffer collateral stimulation. To determine whether the astrocytic responses required stimulation of both ionotropic and metabotropic glutamate receptors, KY and MCPG were tested individually. A concentration of 1 mm MCPG alone reversibly inhibited the astrocytic responses in 72% (39/54 astrocytes, 3 slices) of the astrocytes (Fig. 5). As reported by others (Bashir et al., 1993), MCPG alone had little if any effect on the field potentials (Figs. 5D, 6D). This indicates that the astrocytic responses can be prevented without blocking synaptic transmission. These results suggest that the astrocytic responses are attributable to the stimulation of metabotropic receptors on the astrocytes. This conclusion is supported further by the finding that 3 mm KY alone did not inhibit the astrocytic responses in 98% (44/45 astrocytes, 2 slices) of the astrocytes (Fig.5E). KY, which is known to block synaptic transmission in the hippocampus (Ganong et al., 1983; Robinson et al., 1984), blocked the postsynaptic portion of the field potentials while leaving the presynaptic portion intact (Figs. 5F, 6F). This indicates that the astrocytes can respond even if the firing of the postsynaptic CA1 pyramidal neurons is prevented. This further indicates that the astrocytic responses are independent of fast synaptic responses in the CA1 pyramidal neurons. These results argue against the possibility that the astrocytes are responding to the depolarization-induced release of something from the CA1 pyramidal neuron dendrites. However, an effect on the astrocytes from a slow response in the CA1 neurons caused by stimulation of metabotropic glutamate receptors cannot be excluded. In contrast, the astrocytic responses in the presence of 4-AP were unaffected or only slightly inhibited by either 1 mm MCPG or 3 mm KY alone in 92% (45/49 astrocytes, 2 slices; Fig. 6C) and 85% (38/45 astrocytes, 2 slices; Fig. 6E) of the astrocytes, respectively. In slices treated with 4-AP, the combination of MCPG and KY blocked the astrocytic responses in 91% (61/67 astrocytes, 3 slices) of the astrocytes examined and also blocked synaptic transmission (Fig.6A,B).
DISCUSSION
Hippocampal astrocytes in situ respond to neuronal release of glutamate
Our results indicate that hippocampal astrocytes,in situ, associated with the synaptic terminals of the Schaffer collateral afferents in CA1 can be activated by the release of the neurotransmitter glutamate. Although it has been hypothesized for a long time that astrocytes are responsive to released neurotransmitters, this is the first actual demonstration that protoplasmic astrocytes of the gray matter can respond to neuronal neurotransmitter release. Fundamentally, these findings have very important implications. They indicate that not only do astrocytes in situ have neurotransmitter receptors, but that these receptors are functional and can be activated by neuronal activity. The results also suggest that during neuronal activity sufficient quantities of glutamate can escape the synaptic cleft and activate glutamate receptors outside of the synapse. Such extrasynaptic communication may be a very common and important form of communication in the CNS.
Electrical stimulation of the hippocampus induces the release of adenosine (Mitchell et al., 1993), norepinephrine (Jonzon and Fredholm, 1985), and ATP (Wieraszko et al., 1989) in addition to glutamate. Because the hippocampal astrocytes in situ also appear to express adenosine (Porter and McCarthy, 1995b) and α-adrenergic receptors (Duffy and MacVicar, 1995) coupled to increases in [Ca2+]i, it is also likely that the astrocytes could also respond to neuronal release of adenosine, ATP, or norepinephrine. Under our stimulation conditions, the astrocytic responses appeared to be mainly attributable to the neuronal release of glutamate. However, 29% (29/99 astrocytes, 3 slices) of the astrocytes tested responded in the presence of both MCPG and KY, and many of the astrocytes that were affected by the combination of MCPG and KY were not completely blocked. These findings may suggest that agents other than glutamate such as adenosine or norepinephrine contributed to the responses. However, such differences may also be attributable to insufficient antagonist concentrations to compete with the released glutamate at the astrocytic receptors.
The results of Dani et al. (1992) demonstrated that astroglia in hippocampal slice cultures respond to electrical stimulation of the mossy fibers with increases in [Ca2+]i. Their results indicate that astroglia in hippocampal slice cultures are responsive to neuronal activity, but do not distinguish between astroglial responses to a neuronally released neurotransmitter versus an increase in [K+]o. Furthermore, it is uncertain what effect culture conditions have on astrocyte receptor expression and neuronal–astrocytic interactions.
The responsiveness of astrocytes to released neurotransmitters is not restricted to hippocampal astrocytes. The specialized astrocyte-like cells found in the pituitary also respond to neuronally released neurotransmitters (Mudrick-Donnon et al., 1993). Stimulation of the pituitary stalk resulted in depolarization of the stellate (astrocyte-like) cells in situ, which was antagonized by GABA and dopamine receptor antagonists. These experiments provide evidence that specialized glial cells can respond to neuronally released GABA and dopamine. The pituitary is a very specialized structure with axonal projections making synapse-like contacts on the stellate glial cells (van Leeuwen et al., 1983; Buijs et al., 1987). Our results indicate that astrocytes within the general confines of the gray matter without such specialized neuronal contacts also respond to neuronally released neurotransmitters.
There is also evidence that glial cells in the peripheral nervous system (Jahromi et al., 1992; Reist and Smith, 1992) and in white matter (Kriegler and Chiu, 1993) can respond to neuronal activity with increases in [Ca2+]i. Although the exact cause of the glial responses was not determined, these results demonstrate that amphibian Schwann cells and white matter glia are also responsive to neuronal activity.
Sensitivity of astrocytes to neuronal activity
In our experiments, we were unable to monitor changes simultaneously in neuronal and astrocytic [Ca2+]i because the neurons did not load with the calcium-sensitive dye. However, because we used stimulation parameters (200–400 μA, 50 Hz, 2 sec) similar to those used to induce increases in [Ca2+]i in CA1 pyramidal neurons (400–1000 μA, 50 Hz, 2 sec; Regehr et al., 1989; Regehr and Tank, 1990), our results suggest that the astrocytes are able to respond to stimulation in a range similar to the neurons. Although we did not systematically test the astrocytic responses to increasing levels of synaptic activity, the magnitudes of the astrocytic responses were correlated with the amount of neuronal activity. When the neuronal activity was blocked with TTX, the astrocytes did not respond. In the absence of any treatment, the neuronal responses were moderate, as were the astrocytic responses. When the neuronal activity was enhanced with 4-AP, the astrocytic responses were also larger. This suggests that different levels of synaptic activity are able to induce varying levels of astrocytic responses. The astrocytic responses to neuronal activity appear to be graded rather than simply all-or-none. Such graded responses are likely to be critical for modulation of astrocytic functions.
Our results also indicate that the subtypes of glutamate receptors activated by the release of glutamate vary with the amount of neuronal stimulation. At lower levels of stimulation (absence of 4-AP), mainly metabotropic glutamate receptors are activated on the astrocytes, whereas high levels of stimulation (presence of 4-AP) result in the activation of both metabotropic and ionotropic glutamate receptors on the astrocytes. These results indicate that the metabotropic glutamate receptors can be activated independent of the ionotropic glutamate receptors. Such selective receptor activation may be important for selective modulation of different astrocytic functions. For example, the smaller calcium responses generated by only metabotropic receptor activation may be important for short-term changes such as modulation of calcium-sensitive K+ channels, whereas the larger calcium responses generated by activation of both metabotropic and ionotropic glutamate receptors may be necessary for long-term changes such as changes in gene transcription.
Although 4-AP can cause spreading depression in hippocampal slices (Psarropoulou and Avoli, 1993), it is unlikely that the astrocytic responses were caused by spreading depression. Spreading depression is reported to be blocked by NMDA antagonists (Psarropoulou and Avoli, 1993) and unaffected by TTX (Sugaya et al., 1975; Tobiasz and Nicholson, 1982), whereas the astrocytic responses reported here were not blocked by KY and were blocked by TTX.
Possible significance of astrocytic responses to released glutamate
Although the functional end points of stimulation of glial glutamate receptors in situ are unknown, several possible outcomes can be inferred from previous experiments. From in vitro experiments, it is known that glutamate stimulation of astroglial cultures results in the release of arachidonic acid (Stella et al., 1994), extension of astroglial filopodia (Cornell-Bell et al., 1990), and astrocytic swelling (Hansson, 1994). Because arachidonic acid has been shown to cause a prolonged inhibition of glutamate uptake (Yu et al., 1986), stimulation of astrocytic glutamate receptorsin situ could modulate neuronal excitation by regulating the duration of glutamate in the synaptic cleft. Both the extension of astrocytic filopodia and the astrocytic swelling could also affect neuronal excitability by changing the volume of the extracellular space (Traynelis and Dingledine, 1989; McBain et al., 1990). Because these experiments were done in vitro, it remains to be determined whether such events occur in vivo. Experiments on immature hippocampal astrocytes in situ suggest that stimulation of ionotropic glutamate receptors on astrocytes may regulate astrocytic clearance of extracellular K+ by modulating calcium-sensitive K+ channels (Jabs et al., 1994). Clearly, further work is needed to determine the functional consequences of stimulation of astrocytic glutamate receptors in situ and in vivo.
Are astrocytes involved in long-term potentiation?
Interestingly, the astrocytes responded to stimulation levels that have been used to induce long-term potentiation (LTP) of the synaptic activity in the hippocampal slice (Regehr et al., 1989). Furthermore, stimulation of metabotropic glutamate receptors appears to be involved in the production of LTP of the Schaffer collateral synapses in CA1 (Bashir et al., 1993). This raises the question of whether stimulation of astrocytic metabotropic glutamate receptors is involved in LTP. Although astrocytic involvement in hippocampal LTP has been reported previously (Sastry et al., 1988, 1990) and inhibition of astrocytic metabolism disrupts hippocampal synaptic transmission (Keyser and Pellmar, 1994), more experiments are needed to determine what role if any astrocytes play in LTP.
In summary, our results indicate that hippocampal astrocytes in situ respond to the neuronal release of glutamate with increases in [Ca2+]i. Therefore, astrocytes in situ have functional ionotropic and metabotropic glutamate receptors that can be activated by neuronal activity. Because the astrocytic responses were observed within hours of removal from the animal, it is likely that hippocampal astrocytesin vivo also respond to the neuronal release of glutamate.
Footnotes
This work was supported by National Institutes of Health Grant NS 20212.
Correspondence should be addressed to Ken D. McCarthy, Department of Pharmacology, CB #7365, FLOB Building, University of North Carolina-Charlotte School of Medicine, Chapel Hill, NC 27599-7365.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}