

Abstract
Delivery of neurotrophic molecules to the CNS has gained considerable attention as a potential treatment strategy for neurological disorders. In the present study, a DHFR-based expression vector containing the human ciliary neurotrophic factor (hCNTF) was transfected into a baby hamster kidney fibroblast cell line (BHK). Using a polymeric device, encapsulated BHK-control cells and those secreting hCNTF (BHK-hCNTF) were transplanted unilaterally into the rat lateral ventricle. Twelve days later, the same animals received unilateral injections of quinolinic acid (QA; 225 nmol) into the ipsilateral striatum. After surgery, animals were behaviorally tested for apomorphine-induced rotation behavior and for skilled forelimb function using the staircase test. Rats receiving BHK-hCNTF cells rotated significantly less than animals receiving BHK-control cells. No behavioral effects of hCNTF were observed on the staircase test. Nissl-stained sections demonstrated that BHK-hCNTF cells significantly reduced the extent of striatal damage produced by QA. Quantitative analysis of striatal neurons further demonstrated that both choline acetyltransferase- and GAD-immunoreactive neurons were protected by BHK-hCNTF implants. In contrast, a similar loss of NADPH-diaphorase-positive cells was observed in the striatum of both implant groups. Analysis of retrieved capsules revealed numerous viable and mitotically active BHK cells that continued to secrete hCNTF. These results support the concepts that implants of polymer-encapsulated hCNTF-releasing cells can be used to protect striatal neurons from excitotoxic damage and that this strategy may ultimately prove relevant for the treatment of Huntington’s disease.
- polymer encapsulation
- Huntington’s disease
- CNTF
- neurotrophic factor
- transplantation
- quinolinic acid
- fibroblasts
- genetic modification
The delivery of neurotrophic molecules to the CNS has gained considerable attention as a potentially effective and rational strategy for the treatment of neurological disorders. Neurotrophic molecules have been shown to promote cell survivalin vitro (Dreyfus, 1989; Barnett et al., 1990; Lindholm et al., 1993; Lindsey et al., 1993) and to attenuate the behavioral and neurobiological consequences of CNS damage in animal models of Alzheimer’s, Parkinson’s, and Huntington’s diseases (HD) (Hefti et al., 1986; Fischer et al., 1987; Schumacher et al., 1991; Spina et al., 1992; Frim et al., 1993a,b; Emerich et al., 1994a,b; Hoffer et al., 1994; Kordower et al., 1994a,b; Levivier et al., 1995). Traditionally, neurotrophic molecules are thought to exert their effects on relatively specific populations of neurons. It has become increasing clear, however, that neurotrophic factors are capable of exerting effects on a much broader spectrum of neuronal populations than previously believed. One neurotrophic factor, ciliary neurotrophic factor (CNTF), is a member of the α-helical cytokine superfamily, which has well documented functions in the peripheral nervous system (Lin et al., 1989; Stockli et al., 1989; Arakawa et al., 1990; Sendtner et al., 1990; Oppenheim et al., 1991; Stockli et al., 1991; Sendtner et al., 1992; Apfel et al., 1993; Forger et al., 1993; Masu et al., 1993). Recently, it has become clear that CNTF also influences a wide range of CNS neurons. CNTF administration prevents the loss of cholinergic, dopaminergic, and GABAergic neurons in different CNS lesion paradigms (Clatterbuck et al., 1993; Hagg and Varon, 1993; Hagg et al., 1993), suggesting that delivery of CNTF to the damaged nervous system may be one means of treating the behavioral and neuroanatomical consequences of neurological disease.
One important aspect of neurotrophic factor therapy that has yet to be fully resolved is how to deliver safely a trophic factor such as CNTF to the brain for clinical applications. Recent efforts have concentrated on the transplantation of genetically modified cells releasing neurotrophic factors into normal or damaged brain regions (Gage et al., 1987; Rosenberg et al., 1988; Breakefield, 1989; Kawaja et al., 1991, 1992; Kordower et al., 1994a; Levivier et al., 1995). These cells can be delivered to the brain in a number of ways. Our group has focused on implanting trophic factor-secreting cells that have been encapsulated in a polymer membrane before transplantation. The pores of the membrane are sufficiently large to allow low-molecular-weight solutes such as trophic factors to cross the membrane and enter the host, but small enough to protect the encapsulated cells from host immune destruction (for review, seeEmerich et al., 1992). Using this approach, we have demonstrated that polymer-encapsulated fibroblasts genetically modified to secrete nerve growth factor (NGF) can survive for prolonged periods, prevent the loss of degenerating cholinergic basal forebrain neurons in rodents and nonhuman primates, and promote recovery of cognitive function in aged rodents (Hoffman et al., 1993; Emerich et al., 1994a; Kordower et al., 1994a; Winn et al., 1994; Lindner et al., 1996).
One neurodegenerative disease that might be amenable to encapsulated trophic factor therapy is HD. HD is an autosomal dominant neurodegenerative disease characterized by a relentlessly progressive movement disorder with devastating psychiatric and cognitive deterioration (for review, see Emerich and Sanberg, 1992). HD is associated with a consistent and severe atrophy of the neostriatum that is related to a marked loss of the GABAergic medium-sized spiny projection neurons, the major output neurons of the striatum (Reiner et al., 1988). Neurochemically, the loss of striatal neurons is associated with decreases in GABA, substance P, dynorphin, and enkephalin (Bird, 1980; Buck et al., 1981; Ferranti et al., 1987). On the other hand, local circuit aspiny neurons, reactive for NADPH-diaporase and somatostatin, are relatively spared (Beal et al., 1986, 1988, 1989). The large aspiny cholinergic interneurons, although spared in the early stages of the disorder, can exhibit degenerative changes as the disease progresses (Ferranti et al., 1987; Roberts and DiFiglia, 1989). The symptoms of HD result from this selective vulnerability of neostriatal neurons, possibly because of an endogenous disequilibrium of energy metabolism and excitotoxicity (Parker et al., 1990; Albin and Greenamyre, 1992; Beal, 1992; Wallace, 1992). Intrastriatal injections of excitotoxins such as quinolinic acid (QA) mimic the pattern of selective neuronal vulnerability seen in HD (Beal et al., 1986, 1988,1989). QA lesions result in motor and cognitive deficits that are among the major symptoms seen in HD (Sanberg et al., 1981, 1989; Emerich et al., 1991, 1992; Block et al., 1993). Thus, intrastriatal injections of QA have become a useful model of HD and can serve to evaluate novel therapeutic strategies aimed at preventing, attenuating, or reversing neuroanatomical and behavioral changes associated with HD. In this regard, infusions of trophic factors such as NGF or implants of cells genetically modified to secrete NGF have proven effective in preventing the neuropathological and behavioral sequelae resulting from intrastriatal injections of excitotoxins including QA (Schumacher et al., 1991; Frim et al., 1993a,b; Emerich et al., 1994b). Another trophic factor, CNTF, may be even more potent than NGF in protecting striatal neurons from excitotoxic degeneration, because it influences a wider variety of cell types (Clatterbuck et al., 1993; Hagg and Varon, 1993; Hagg et al., 1993). The present study evaluated whether implantation of polymer-encapsulated cells genetically modified to secrete human CNTF (hCNTF) could attenuate the behavioral and histological consequences of intra- striatal QA injections.
MATERIALS AND METHODS
Animal subjects
Adult male Sprague–Dawley rats (Taconic Breeders, Germantown, NY), ∼3 months old and weighing 300–350 gm, were used in these studies. The animals were housed in groups of 3–4 in a temperature- and humidity-controlled colony room that was maintained on a 12 hr light/dark cycle with lights on at 7:00 A.M. Food and water were available ad libitum throughout the experiment. All experimentation was conducted in accord with National Institutes of Health guidelines.
pNUT-hCNTF-TK plasmid construction
The CNTF expression vector pNUT-hCNTF-TK was constructed as follows. A linker generating an SmaI site was introduced at +7 of the mouse metallothionein-I promoter (MT-1) (−650 to +7; Hoyle et al., 1993). This SmaI site was fused to a Klenow-filledXbaI site at the 5′ end of a 150 bp murine Ig region containing parts of exons 1 and 2 encoding the signal peptide and the small intervening intron A sequences. The second exon had anEcoRI site at amino acid 18 of the signal peptide. The human CNTF gene was obtained by PCR amplification of human genomic DNA with primers #405 (5′-CGG AAT TCG GCT TTC ACA GAG CAT TCA CCG-′3) and #406 (5′-GAA GAT CTT AAC TGC TAC ATT TTC TTG TTG TTA G-3′), whereas the former included an EcoRI site at the position of the natural hCNTF initiation codon, and the latter a BglII site 7 bp 3′ to its termination codon. Dideoxynucleotide sequencing of the cloned PCR above confirmed the nucleotide sequence of the exons to be identical to the published sequence of the human CNTF cDNA (Genbank accession number X60542). This PCR fragment was fused to the Ig sequence at the EcoRI site such that the sequence starting with amino acid 18 was Asn-Ser- Ala-Phe-Thr-Glu-His-Ser, where the underlined amino acids represent the N-terminal sequence of hCNTF after the initiating methionine is removed. The signal peptidase likely cleaves between Ser and Ala, generating a secreted protein identical to normal hCNTF. A 325 bp AvaI fragment containing the human growth hormone polyadenylation sequence (hGH pA) (Hoyle et al., 1993) was cloned into the SpeI site of the Bluescript cloning vector (Stratagene, La Jolla, CA) such that it was flanked withBamHI at the 5′ end and the NotI site at the 3′ end. This fragment was then fused to the BglII site engineered into the 3′ end of the hCNTF-coding region via aBglII/BamHI fusion. The 3050 bpKpnI–NotI fragment containing MT-1/Ig/hCNTF/hGH pA was then inserted into pNUT (Baetge et al., 1986) at theKpnI and NotI sites, where the NotI site was generated by inserting a linker into a Klenow-filledEcoRI site. The resulting plasmid was named pNUT-hCNTF. A 2 kb PvuII fragment containing the herpes simplex virus-thymidine kinase (HSV-TK) gene was cloned into theEcoRI site of Bluescript, and the XhoI site was converted to NotI such that a NotI fragment containing the HSV-TK gene could be isolated and then inserted into the NotI site of pNUT-hCNTF to generate pNUT-hCNTF-TK.
BHK-hCNTF cell line production
The pNUT-hCNTF-TK construct was introduced into BHK cells using a standard calcium phosphate-mediated transfection method (Baetge et al., 1986). BHK cells were grown in DMEM containing 10% fetal bovine serum and 2 mml-glutamine (Life Technologies, Grand Island, NY) in 5% CO2at 37°C. Transfected BHK cells were selected in medium containing 200 μm methotrexate (Sigma, St. Louis, MO) for 3–4 weeks, and resistant cells were maintained as a polyclonal population either with or without 200 μm methotrexate. Mock-transfected cells served as controls in these experiments.
Encapsulation procedure
Asymmetric hollow fibers of poly [acrylonitrile-co-vinyl chloride (PAN-PVC)] copolymer were fabricated by a dry/wet (jet) spinning technique according to Cabasso (1980). Encapsulation devices were manufactured by mounting a section of dry hollow fiber onto hubs with a septal fixture at the proximal end providing loading access for the BHK cells. BHK cells ± hCNTF were removed from culture flasks using Ca2+- and Mg2+-free HBSS and trypsin/EDTA and prepared as a single-cell suspension. The BHK cell suspensions, at a density of 2 × 107cells/ml, were mixed with collagen and loaded into the encapsulation devices as described previously (Emerich et al., 1994a; Winn et al., 1994). After infusion of the cellular suspensions, the final assembled device had an outer diameter of ∼750 μm with a length of 7 mm. All cell-loaded devices were maintained in serum-free medium 2–3 d before hCNTF analysis by ELISA. For the ELISA, the cell-loaded capsules were rinsed in HBSS and placed in 1 ml of fresh PC-1 medium, and the conditioned medium (CM) was collected and assayed after a 20 hr assay time.
hCNTF ELISA
Quantification of hCNTF released from encapsulated BHK-hCNTF cells was performed using a commercially available (Quantikine, R&D Systems, Minneapolis, MN) quantitative sandwich enzyme immunoassay kit. Analysis of hCNTF release was performed before implantation and after implant retrieval. Briefly, a monoclonal antibody specific for CNTF was coated onto a microtiter plate provided by the manufacturer. CNTF standards and samples were pipetted into the wells, and any CNTF present was bound by the immobilized antibody. After washing away any unbound proteins, a horseradish peroxidase-linked polyclonal antibody specific for CNTF was added to the wells to sandwich the CNTF immobilized during the first incubation. After a wash to remove any unbound antibody/enzyme reagent, a substrate solution of tetramethylbenzidine and hydrogen peroxide was added to the wells. The resulting color development occurred in proportion to the amount of CNTF bound in the initial step. The color development was then stopped with sulfuric acid, and the intensity of the color was measured on a microplate reader at 450 nm.
Surgery
Immediately before surgery, rats were anesthetized with sodium pentobarbital (45 mg/kg, i.p.) and positioned in a Kopf stereotaxic instrument. A midline incision was made in the scalp, and a hole was drilled for the placement of a cell-loaded capsule into the lateral ventricle. Rats were unilaterally implanted into the lateral ventricle with a single polymer capsule containing BHK cells that were (BHK-hCNTF; n = 8) or were not (BHK-control;n = 8) transfected with the human CNTF construct. The capsule was placed within an 18 gauge Teflon catheter mounted to the stereotaxic frame as described previously (Emerich et al., 1993). The stereotaxic coordinates for implantation into the lateral ventricle were: 0.5 mm anterior to Bregma, 1.5 mm lateral to the sagittal suture, and 7.5 mm below the cortical surface (Paxinos and Watson, 1980). After implantation, the skin was sutured closed.
Twelve days after implantation of cell-loaded capsules, all animals were anesthetized, placed in the stereotaxic instrument, and injected with 225 nmol of QA (Sigma) into the striatum at the following coordinates: 1.2 mm anterior to Bregma, 2.6 mm lateral to the sagittal suture, and 5.5 mm ventral to the surface of the brain (Paxinos and Watson, 1980). QA was infused into the striatum using a 28 gauge Hamilton syringe. Injections were made in a 1 μl volume over 5 min. The injection cannula was left in place for an additional 2 min to allow the QA to diffuse from the needle tip. The skin was then resutured.
Immediately after the lesion, animals were injected intraperitoneally with 10 ml of a lactated Ringer’s solution. Animals were housed postoperatively with food mash and water available ad libitum. At the conclusion of behavioral testing 70 d after implantation, animals were anesthetized and placed into the stereotaxic instrument. A craniotomy was performed over the implantation site, and the dural scar surrounding the implant site was excised. The cortical surface was cut to expose the underlying capsule, which was retrieved with a pair of Dumont (#5) forceps.
Histology
Immediately after capsule retrieval and while still under deep anesthesia, rats were transcardially perfused using a peristaltic pump with 20 ml of saline followed by 500 ml of 4% paraformaldehyde. All solutions were ice-cold (4°C) and prepared in 50 mm PBS (pH 7.4). Brains were removed after fixation, placed in 25% buffered sucrose (pH 7.4), and refrigerated for ∼48 hr. Sections throughout the entire striatum were cut at 40 μm intervals on a cryostat and stored in a cryoprotectant solution. Adjacent sections through the striatum were processed for the immunocytochemical localization of choline acetyltransferase (ChAT; 1:1000, Chemicon, Temecula, CA) and glial fibrillary acidic protein (GFAP; 1:5000, Dakopatts, Copenhagen, Denmark) as described previously (Emerich et al., 1994a; Kordower et al., 1996) using the labeled antibody procedure (Hsu et al., 1981). One series of sections was stained for glutamic acid decarboxylase (GAD; 1:7500, Oncogene) with nickel intensification using a modification of the above procedure (for details, see Kordower et al., 1996). All immunohistochemical reactions were terminated by three 1 min rinses in PBS. Sections were mounted, dehydrated, and coverslipped. Control sections were processed in an identical manner except that the primary antibody solvent or an irrelevant IgG was substituted for the primary antibody. It is important to note that even though staining was eliminated in sections in which the primary antibody was deleted or an irrelevant IgG substituted, the potential for antiserum to react with structurally related proteins cannot be excluded. Thus, a degree of caution that is inherent to immunohistochemical procedures is warranted. In this regard, the terms ChAT-, GAD-, or GFAP immunoreactivity in this study refer to -“like” immunoreactivity (ir). A separate series of sections through the striatum were stained for NADPH-diaphorase (NADPH-d; Sigma) as described previously (Beal et al., 1989). Adjacent sections were stained for Nissl to aid in cytoarchitechtonic delineation.
After the ELISA analysis for CNTF secretion, the retrieved capsules were fixed in a 4% paraformaldehyde and 0.5% glutaraldehyde solution, rinsed in PBS, and dehydrated up with to 95% ethanol. A 1:1 solution of glycol methacrylate (Historesin, Reichert-Jung, Cambridge Instruments) was then added to the capsules for 1 hr. Pure infiltration solution replaced the 1:1 mixture and remained for a minimum of 2 hr. The capsules were then rinsed with the embedding solution, transferred to flat molds, and embedded in glycol methacrylate. Sections (5-μm-thick, Reichert-Jung, Supercut microtome 2065) were mounted on glass slides and stained for hemotoxylin and eosin (H+E).
Quantitative morphometric analysis
The extent of the lesion produced by QA was determined in all animals using a semiautomated image analysis system (NIH-Image). For determination of lesion size, five equidistant (200 μm) sections were evaluated. Two sections were anterior to the lesion, one section was at the level of the QA injection, and two sections were posterior to the lesion. The levels of analysis were ∼0.1, 0.3, 0.5, 0.7, and 0.9 mm anterior to Bregma. The border of the lesion was traced on Nissl-stained sections throughout the five sections, and the average size of the lesioned area in those five sections was determined and then expressed in mm2. Counts of ChAT-, GAD-, and NADPH-d-stained neurons were performed within the striatum using the sections adjacent to those analyzed for lesion size. For each stain, five equidistantly spaced sections (200 μm) anterior to the lesion, at the level of the lesion, and posterior to the lesion were used. Every ChAT-, GAD-, or NADPH-d-positive neuron in each of those sections was counted bilaterally by an individual blind to the animal’s experimental condition.
Behavioral testing
Rotometry. Apomorphine-induced rotation provides a sensitive and rapid behavioral correlate of striatal damage and has been used to evaluate the potential efficacy of tissue/cell grafts into the excitotoxic-lesioned striatum (Norman et al., 1988; Bjorklund et al., 1994). Beginning 4 weeks after QA injections, animals were tested for apomorphine-induced rotation (1.0 mg/kg in normal saline containing 0.2% ascorbate) in one of eight rotation devices (Rotoscan, Omnitech Instruments). These rotometry devices were connected to an IBM computer for automated data collection. Animals were injected with apomorphine and placed into the test chamber for a 5 min habituation period before a 30 min test session. Sensitization of apomorphine-induced rotation behavior occurs after excitotoxin lesions of the striatum (Norman et al., 1988). Therefore, animals were tested four times, with each session separated by a 3–4 d interval. Rotations were defined as complete 360° ipsilateral turns and were reported as the net difference between the two directions.
Staircase testing. After rotation testing, animals were examined for their ability to retrieve food pellets using a staircase apparatus as described by Montoya et al. (1991). The staircase test is highly sensitive to excitotoxic lesions of the striatum and has also been used in studies examining the effects of transplants into the lesioned striatum (Bjorklund et al., 1994). The deficits observed in this task are severe, stable for long periods of time, and can be used in unilaterally lesioned animals with the intact limb providing an internal control. Animals were food-deprived to 85% of their ad libitum body weight and were maintained at this weight until the conclusion of testing. In this test, rats were placed on a narrow central platform. To retrieve food pellets, they were allowed to reach into a trough on the left side of the central platform with only their left forepaw and on the right side with only their right forepaw (45 mg, Formula A/I, PJ Noyes Co., Lancaster, NH). There were six wells on each side, and each well was located 13 mm deeper and 14 mm further caudal than the previous well, in staircase fashion. The top stair was at snout level and did not require forepaw use. The apparatus was constructed so that pellets had to be grasped, held, and lifted in the forepaw. Four pellets were placed into each of the six wells of the double staircase. During the test session, rats were placed in the test boxes for 10 min. At the conclusion of each session, the staircases were removed and the number of pellets remaining on each side of the apparatus was recorded (maximum 24 pellets). Animals received a total of 10 trials over 11 d.
Data analyses
Data were analyzed with an SAS-PC statistics program. ANOVAs were conducted using the procedures for general linear models with options for repeated measures where appropriate (SAS Institute, Inc., 1989). ω2 was computed as a measure of effect size (Dodd and Schultz, 1973; Kirk, 1982). Data are presented in the text and in all figures as mean ± SEM.
RESULTS
General
No overt signs of behavioral or neurological toxicity were observed in any animals after implantation of either BHK-hCNTF or BHK-control capsules. During the postoperative recovery period after QA injections, the BHK-control group exhibited whole-body barrel rotations that persisted for 2–4 hr. These same animals had a transient period of weight loss, piloerection, and diarrhea that subsided within 3–4 d after QA. Animals that received QA together with BHK-hCNTF capsules did not show whole-body rotations but did exhibit a slight motor asymmetry after QA. This asymmetry was transient, and recovery was seen within several hours. No additional signs of systemic toxicity, including changes in body weight, were noted.
Behavioral testing
Rotometry
The number of rotations per session was analyzed using a two-way ANOVA with Groups (BHK-control and BHK-hCNTF) and Trials (4) included as factors in the analysis. After QA injections, animals displayed apomorphine-induced rotations ipsilateral to the lesion with the extent of rotation behavior increasing with repeated tests. As shown in Figure1, the number of rotations increased across trials in both groups, but this increase was significantly attenuated in BHK-hCNTF-treated rats. The two-way ANOVA revealed a significant main effect of Group (ω2 = 0.28, F(1,14) = 35.67, p = 0.0001), a significant main effect of Trials (ω2 = 0.43, F(3,42) = 66.64, p = 0.0001), and a significant Group × Trials interaction (ω2 = 0.16, F(3,42) = 25.45, p = 0.0001).

Implants of encapsulated BHK-hCNTF cells reduce apomorphine-induced (1.0 mg/kg) rotation in rats after unilateral intrastriatal injections of QA. This figure presents the mean ± SEM number of rotations in a 30 min test period.
Behavioral testing
Staircase test
The number of food pellets eaten in the staircase test was analyzed in a three-way ANOVA using Groups (BHK-control and BHK-hCNTF), Side (Left vs Right forelimb), and Trials (10) as factors in the analysis. Rats acquired the task rapidly, and the number of food pellets eaten increased dramatically over the course of the 10 trials; the main effect for Trials was statistically significant (ω2 = 0.68, F(9,126) = 93.75, p = 0.0001; Fig. 2). As the rats acquired the task, an asymmetry emerged between the nonaffected limb ipsilateral to the QA lesion and the affected limb contralateral to the QA lesion. In this regard, the Side × Trials interaction was statistically significant (ω2 = 0.04, F(9,126) = 16.88, p = 0.0001). However, the Group × Side × Trials interaction was not statistically significant (ω2 = 0.00, F(9,126) = 0.26, p = 0.98), indicating that performance on the staircase task using either the affected or the nonaffected limb was not significantly different in BHK-hCNTF- and BHK-control-implanted animals.

Implants of encapsulated BHK-hCNTF cells do not influence QA lesion-induced deficits in the retrieval of food pellets on the staircase test. This figure presents the mean ± SEM number of pellets eaten for both the left and right sides in a 10 min test period.
hCNTF ELISA
Before implantation and after retrieval (immediately before perfusion), the encapsulated BHK cells were incubated in CM, which was then assayed for hCNTF by ELISA. Before implantation, the encapsulated BHK-hCNTF cells released 17.26 ± 1.0 ng hCNTF/capsule/24 hr. BHK-control cells showed hCNTF levels no different than measured in control medium. Postexplant values of hCNTF from capsules averaged 11.15 ± 2.8 ng hCNTF/capsule/24 hr. hCNTF was undetectable in CM incubated with explanted BHK-control capsules.
Histology
The cell-loaded devices were easily retrieved with little or no host tissue adhering to the capsule wall. As seen consistently in previous studies (Emerich et al., 1993, 1994a,b), Nissl staining and GFAP immunohistochemistry revealed that the intraventricular implants induced minimal damage to the host brain (data not shown). All implants were located within the lateral ventricle. Dorsally, the devices extended through the corpus callosum and overlying cortex. The capsules typically expanded the size of the lateral ventricle but did not significantly impact on the septum medially or the striatum laterally. All capsules were removed intact, and there was no evidence that any capsule broke either in situ or during the retrieval procedure.
The morphology of BHK-hCNTF cells within a retrieved capsule is shown in Figure 3. Seventy days after transplantation, all capsules contained a large number of viable BHK cells. BHK cells were evenly distributed at high density within the polymeric device. Numerous mitotic figures were observed throughout all of the cell-loaded capsules (Fig. 3C). Areas of focal cell debris were occasionally observed within the core of large viable cell aggregates. BHK cell viability within the capsule was similar in BHK-control- and BHK-hCNTF-implanted animals.

Low (A)-, medium (B)-, and high (C)-power photomicrographs of hemotoxylin- and eosin-stained sections through a capsule containing BHK-hCNTF cells, which was retrieved from a rat 70 d after transplantation. Note the large number of BHK cells that filled the capsule. C, Some cells within the capsule are in active stages of mitosis (arrowhead). Scale bar (shown in C):A, 1000 μm; B, 250 μm; C, 50 μm.
Within the host striatum, QA induced a characteristic lesion of intrinsic neurons while sparing glia and fibers of passage. In agreement with previous studies (Emerich et al., 1994b), QA administration in animals receiving BHK-control-loaded devices produced a substantial atrophy of the striatum, resulting in a marked ventricular dilation (Figs. 4, 5B). In some cases, moderate cell loss was observed in the nucleus accumbens and cortical regions adjacent to the injection site. In BHK-control-implanted rats, the QA-induced lesion was elliptical in shape and encompassed much of the striatum at the level of the injection (Fig. 5B). The core of the lesion was filled with glial cells, and few Nissl-stained neurons were observed (Fig. 5D). Those neurons that could be identified were shrunken and displayed a dystrophic morphology. In contrast, the size of the lesion was significantly reduced in BHK-hCNTF-implanted rats (Figs. 4, 5A). Often, the core of the lesion was limited to a small area adjacent to the needle tract (Figs. 4,5A). Numerous healthy appearing Nissl-stained neurons were observed within the striatum of BHK-hCNTF-implanted rats after the QA lesion, even in regions proximal to the needle tract (Fig.5C). Quantitative analysis confirmed that the size of the lesion was significantly reduced in those animals receiving BHK-hCNTF implants (1.44 ± 0.34 mm2) compared with those animals receiving BHK-control implants (2.81 ± 0.25 mm2; F(1,13) = 9.96, p = 0.008).

A, Schematic representation of the extent of the QA lesion at the level of the injection site in individual animals in both control and CNTF-treated groups.B, Schematic reconstruction of the extent of QA-induced striatal damage from one animal in each experimental group (CONTROL, CNTF, denoted by asterisksin A). Sections in A are from 0.48 mm anterior to Bregma, whereas sections in B are +1.0, +0.7, +0.48, +0.2, and −0.2 mm from Bregma. All sections are from Paxinos and Watson (1986). The extent of ventricular dilation is illustrated in each animal by black coloring, whereas the extent of striatal damage is illustrated by black lines.

Low (A, B)- and high (C, D)-power photomicrographs of Nissl-stained sections through the striatum of rats receiving implants of encapsulated BHK-hCNTF (A, C) or BHK-control (B, D) cells. The arrowheads inA and B illustrate the size of the lesion. Note the dramatic reduction in lesion size in BHK-hCNTF-implanted animals even in sections with the needle tract (open arrowheads). The large lesion in BHK-control-implanted rats resulted in striatal atrophy and ventricular dilation. C, In BHK-hCNTF-implanted animals, numerous healthy appearing Nissl-stained neurons were evident adjacent to the needle tract (arrowheads). D, In contrast, only glial cells were observed adjacent to the needle tract (arrowheads) in BHK-control-implanted animals. Scale bar (shown in A): A, B, 1000 μm;C, D, 50 μm.
Cell counts were performed to quantitate the extent of cell loss produced by QA and the subsequent protection mediated by hCNTF delivery. It is important to note that a lack of immunolabeling does not necessarily equal a loss of neurons. In this regard, the loss of ChAT-ir, GAD-ir, and NADPH-d-positive neurons refers to a loss of immunolabeling and may not reflect a true loss of neurons. ChAT-, GAD-, and NADPH-d-stained sections within the intact contralateral striatum revealed a general pattern of labeled perikarya, consistent with previous observations (Beal et al., 1986, 1989; Emerich et al., 1994;Kordower et al., 1996). The number of ChAT-ir, GAD-ir, and NADPH-positive striatal neurons was analyzed with a 2 × 2 ANOVA including Groups (BHK-control vs BHK-hCNTF) and Side (Lesioned vs Nonlesioned) as factors in the analyses. Cell counts for each type of stain (ChAT, GAD, or NADPH-d) were analyzed separately.
Qualitatively, the QA lesion resulted in a dramatic loss of ChAT-ir neurons within the striatum (Fig.6A,B), similar to that reported previously (Schumacher et al., 1991; Emerich et al., 1994b;Kordower et al., 1994b). In sections proximal to the needle tract, there was almost a complete loss of ChAT-ir cells. Those few neurons that did remain after the lesion appeared atrophic with a stunted dendritic morphology (Fig. 6C). In contrast, there were numerous ChAT-ir neurons within the striatum of QA-injected rats that received intraventricular implants of hCNTF-secreting cells. Even in sections that contained the needle tract, many ChAT-ir neurons were seen (Figs. 6D,E). These neurons were large in size (25–35 μm in diameter) with long neuritic processes (Fig.6F). They displayed the typical morphological profile of healthy cholinergic striatal interneurons. Quantitatively, the number of ChAT-positive cells was reduced on the lesioned side relative to the nonlesioned side; the main effect for Side was statistically significant (ω2 = 0.43, F(1,13) = 38.56, p = 0.0001; Fig. 7). This loss of ChAT-ir neurons was significantly attenuated in rats implanted with BHK-hCNTF cells (12%) relative to rats implanted with BHK-control cells (81%). The Side × Group interaction was statistically significant (ω2 = 0.21, F(1,13) = 19.16, p = 0.0007). Interestingly, when the number of ChAT-positive neurons only on the contralateral sides was compared, there was a significant difference; the main effect for Group was statistically significant (F(1,13) = 9.83, p = 0.008). Whether this difference is an anomaly or an actual consequence of CNTF treatment is unclear, but future studies should evaluate these possibilities.

Low-, medium-, and high-power photomicrographs of ChAT-ir sections from BHK-control (A–C)- and BHK-hCNTF (D–F)-implanted rats. In BHK-control-implanted animals, there was a significant loss of ChAT-ir neurons. The remaining dark images on the photomicrographs are principally cellular debris. C, Of the few ChAT-ir neurons that survived the lesion, most appeared atrophic. D,E, In contrast, numerous ChAT-ir neurons were observed within the striatum of BHK-hCNTF-implanted animals. F, ChAT-ir neurons were large with healthy appearing morphological profiles. Arrowheads in A, B,D, and E depict the needle tract. Scale bar (shown in B): A, B, D,E, 500 μm; C, F, 50 μm.

Quantification of ChAT-ir neurons within the striatum of QA-lesioned rats.
BHK-hCNTF implants also prevented the degeneration of GAD-ir striatal neurons. In BHK-control-implanted rats, there was a dramatic loss of GAD-ir striatal neurons. Indeed, in sections through the lesion site, GAD-ir neurons were rarely observed (Fig.8A,B). Albeit relatively few, more GAD-ir neurons were observed in sections 400 μm away from the injection. In contrast, GAD-ir neurons were spared in BHK-hCNTF-implanted animals. The QA lesion resulted in a small spherical loss of GAD-ir neurons and neuropil that appeared restricted to a striatal region at the tip of the injection site (Fig.8C). Outside this immediate center core, numerous GAD-ir neurons were observed within the striatum of BHK-hCNTF-implanted rats. Quantitative analysis confirmed this qualitative assessment. A two-way ANOVA revealed that the number of GAD-ir cells was significantly reduced in all animals on the lesioned side relative to the nonlesioned side; the main effect for Side was statistically significant (ω2 = 0.50, F(1,13) = 78.98, p = 0.0001). However, rats implanted with BHK-hCNTF cells displayed an attenuated loss of GAD-ir cells (20%) relative to rats implanted with control BHK cells (72%). The Side × Group interaction was statistically significant (ω2 = 0.18, F(1,13) = 29.70, p = 0.0001; Fig. 9).

Low (A, C)-, medium (B, D)-, and high (E)-power photomicrographs of GAD-ir sections. A, In BHK-control-implanted rats, the lesion area was extensive. In the section illustrated, arrowheads demarcate the borders of the lesion dorsally. This lesion extended to the nucleus accumbens ventrally. B, Within the lesion area, virtually no GAD-ir neurons were observed. C, In contrast, only a small spherical lesion was created by the QA in BHK-hCNTF-implanted rats (arrowheads) even in sections directly through the needle tract (open arrow). D, Outside this lesion core were numerous GAD-ir neurons (E) that displayed healthy morphological features. Scale bar (shown in E):A, C, 500 μm; B, D, 100 μm; E, 17 μm.

Quantification of GAD-ir neurons within the striatum of QA-lesioned rats.
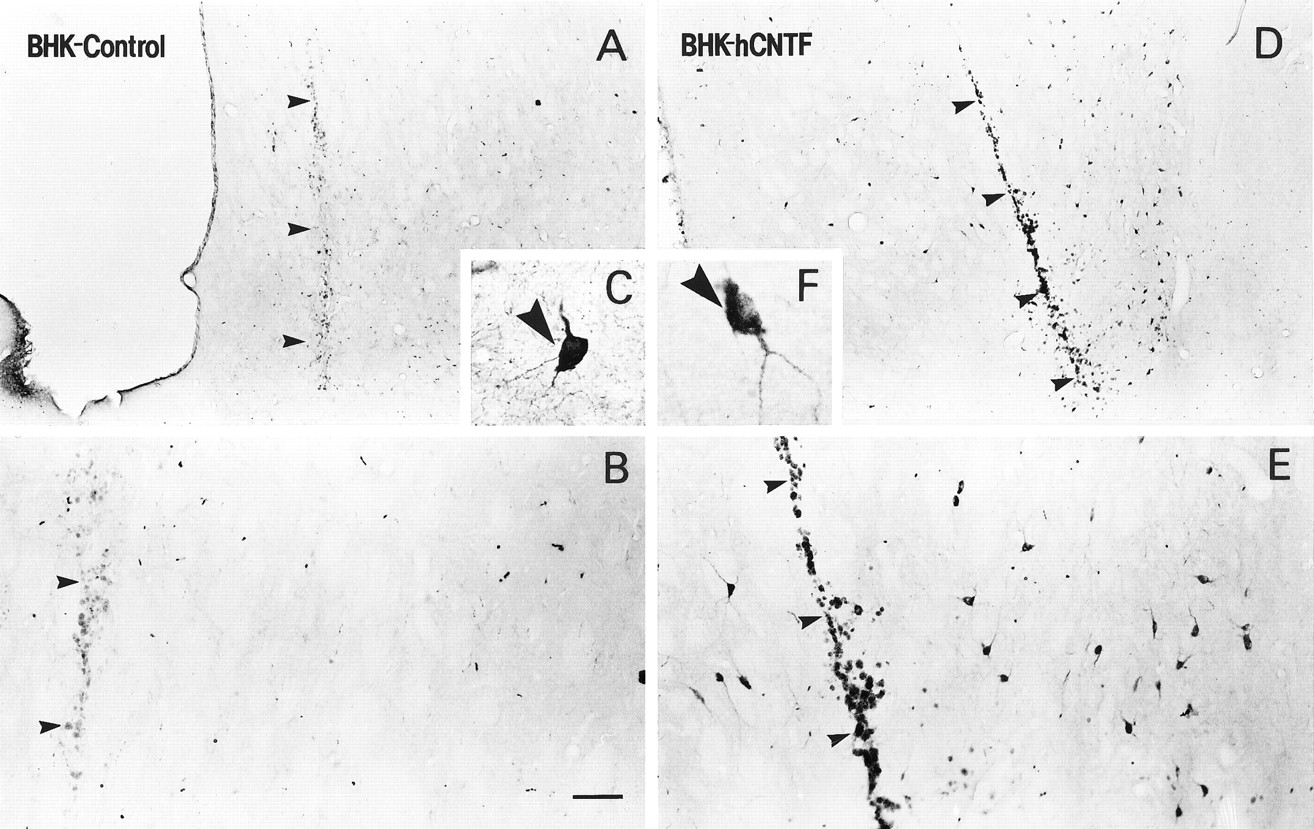
In contrast to ChAT-ir and GAD-ir neurons, implants of BHK-hCNTF-secreting cells failed to protect NADPH-d-positive striatal neurons. Within the intact striatum, numerous small NADPH-d-positive cells with long varicose processes were observed scattered throughout the striatum (Fig. 10A,B). In QA-lesioned rats, the number of these cells was dramatically reduced on the lesioned side relative to the intact side. The main effect for Side was statistically significant (ω2 = 0.87, F(1,13) = 170.00, p = 0.0001; Fig. 11). The magnitude of this effect was similar in BHK-control (78% cell loss; Fig. 9C)- and BHK-hCNTF (65% cell loss; Fig. 10D)-implanted animals. The Side × Group interaction was not statistically significant (ω2 = 0.01, F(1,13) = 2.67, p = 0.13; Fig. 11).

Low (A, C, D)- and high (B, E)-power photomicrographs of NADPH-d-stained sections. A, B, On the intact side, numerous NADPH-d-stained neurons were observed scattered throughout the striatum. A similar extensive loss of NADPH-d-positive neurons was observed in (C) BHK-control- and (D) BHK-hCNTF-implanted rats. E, In both groups, many remaining NADPH-d-stained neurons were atrophic in appearance.
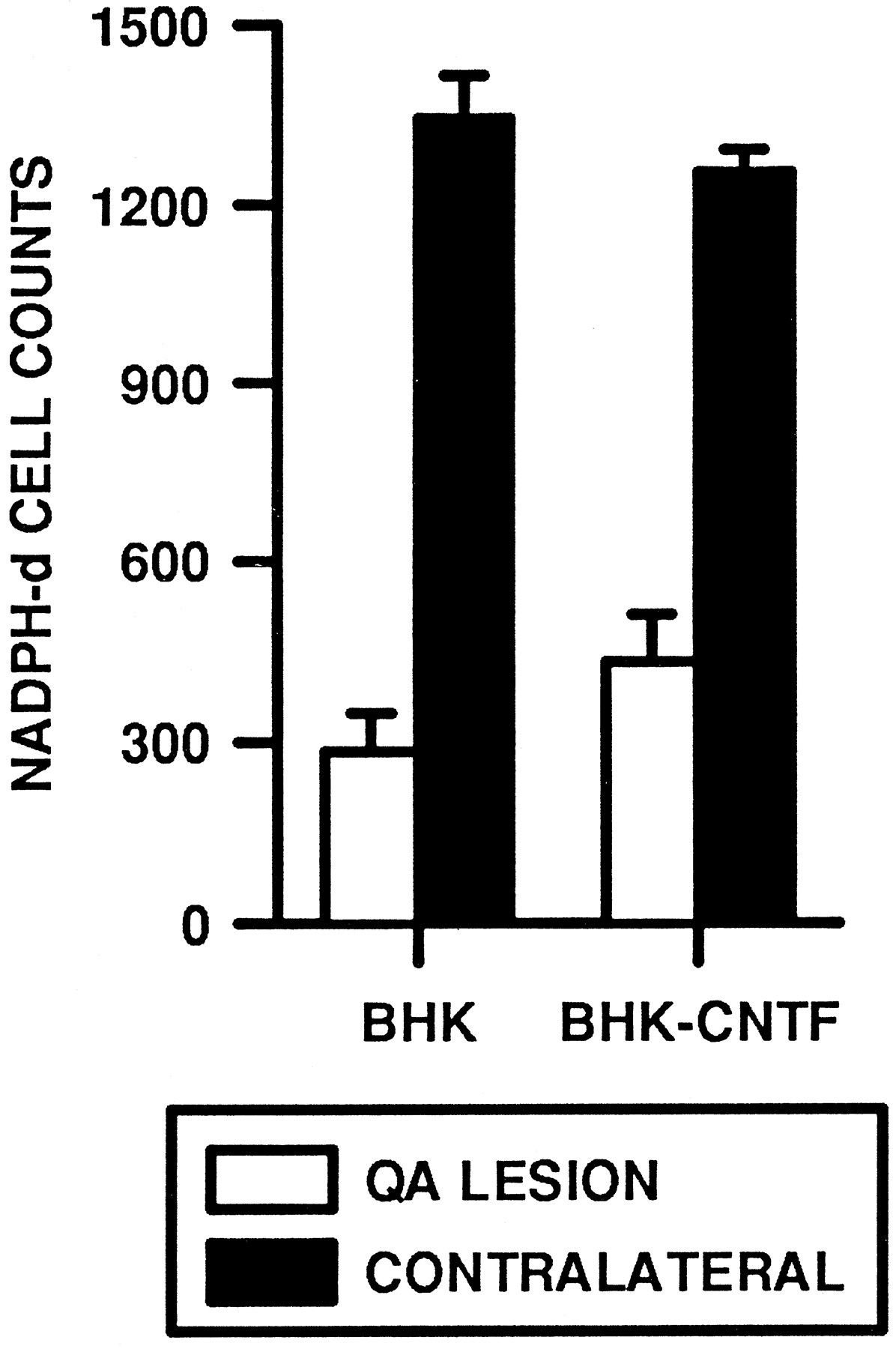
Quantification of NADPH-d-positive neurons within the striatum of QA-lesioned rats.
DISCUSSION
The present series of experiments demonstrates that polymer-encapsulated cells genetically modified to secrete hCNTF can prevent, in part, the neuroanatomical and behavioral sequelae seen in an animal model of HD. In rats implanted with BHK-control cells, QA injections produced a marked striatal atrophy together with a loss of ChAT-, GAD-, and NADPH-d-positive neurons. In contrast, implantation of BHK-hCNTF cells decreased the overall extent of the lesion produced by QA and preserved ChAT-ir and GAD-ir striatal neurons destined to die after the lesion. This effect appears to be selective for specific populations of vulnerable neurons because the BHK-hCNTF implants did not prevent the degeneration of NADPH-d-positive striatal neurons. The rescue of specific striatal cell populations was associated with the attenuation of lesion-induced behaviors. These data are similar to findings obtained by implanting fibroblasts genetically modified to secrete hNGF (Emerich et al., 1993; Frim et al., 1993a,b). NGF can prevent drug-induced rotation behavior and reduce the loss of ChAT- and NADPH-d-positive neurons after QA. Although the protection produced by both NGF and CNTF appears to be similar in magnitude, the specific cell types affected differ. NGF protected ChAT-ir and NADPH-d-positive (GABAergic cells were not evaluated) neurons, whereas CNTF protected ChAT-ir and GAD-ir but not diaphorase-positive neurons after QA. These different profiles of protection may be related to distinct underlying mechanisms. Importantly, although multiple trophic factors may be potent in this model system, the effects of both hNGF and hCNTF were specific, because neither mock-transfected (this study) nor BDNF-transfected fibroblasts (Schumacher et al., 1991) prevent the striatal degeneration after intrastriatal QA. Future studies are clearly needed to determine the exact mechanisms by which different trophic factors are exerting these protective effects.
The implantation of hCNTF-producing fibroblasts produced a significant behavioral protection when the animals were tested for apomorphine-induced rotation behavior. However, this behavioral protection was not observed when the animals were tested on a staircase test that required the skilled use of the normal and impaired forelimbs. Therefore, the behavioral protection resulting from hCNTF was task-specific; these animals recovered on the drug-induced task assessing whole-body motor asymmetries but not on the more complex task requiring fine motor control. The most straightforward explanation for the limited behavioral protection observed in the present study relates to the pattern and extent of neuroanatomical protection provided by the hCNTF implants. Although few studies have been conducted to examine the relationship between the degree of neuronal damage and behavioral deficits produced by QA, Sanberg et al. (1989) did report that hyperactivity in rats was related to the dose of QA injected and the resulting striatal damage. In these studies, intrastriatal injections of 150–225 nmol, but not 75 nmol, of QA produced significant effects on activity levels and feeding behavior. Associated with the behavioral changes in these animals was an increase in lesion size, suggesting a direct relationship between neuronal loss and behavioral disruption. In accordance with these studies, extensive damage of the striatum was observed in animals receiving BHK-control implants. The concomitant loss of striatal neurons was significantly attenuated by hCNTF treatment. It seems reasonable, therefore, that the diminished response to apomorphine is a reflection of this neuronal protection. However, the threshold for behavioral changes may be quite different depending on the type of behavior in question. The amount of neuronal protection that maintains a simple drug-induced behavior such as apomorphine-induced rotations may be insufficient to alter a more complex task such as the staircase test, which requires the skilled use of the animals forelimbs. Additionally, the presence or absence of neurons as determined by cell counts may be too simplistic a measure for correlative studies using a complex task such as the staircase test. Preservation of neurons by a trophic factor does not ensure that normal striatal circuitry has been preserved. Indeed, although the loss of GAD-ir neurons was dramatically attenuated by BHK-hCNTF implants, some animals still displayed a significant loss of GAD-ir neuropil staining, suggesting that although GAD-ir neurons remained viable after the transplant and lesion, they still may be functioning in an abnormal manner. Support for this concept comes from experiments in which fetal striatal tissue grafted into the lesioned striatum produces recovery of skilled limb use that is associated with the extent of reconstruction of the striatal circuitry (Bjorklund et al., 1994). The level of normal striatal circuitry maintained in the present model system may not be sufficient to maintain all forms of behavior.
The mechanism by which hCNTF exerts its protective effect after excitotoxicity is unclear, but several possibilities exist. One possibility is that the mode of action of hCNTF is related to neurotrophic effects via CNTF-α receptors. However, a recent detailed study by MacLennan et al. (1996) failed to detect CNTF receptor immunoreactivity within the adult striatum of rodents. It seems unlikely, therefore, that the neuroprotective effects observed in the present study are related to direct actions at the level of the CNTF receptor. Grafts of NGF-producing fibroblasts similarly influence striatal neurons that do not express the appropriate Trk receptor (Frim et al., 1992; Emerich et al., 1994b; Kordower et al., 1996). A second possibility is that hCNTF is altering the toxic effects of QA by interacting with the NMDA receptor through which QA binds and produces its toxic effects. In this regard, hCNTF delivered into the striatum may exert a direct effect on glutamate receptor synthesis, number, or binding kinetics. A third possibility is that hCNTF protects neurons from oxidative damage or changes in ionic balance including calcium stores, NA+/K+ ATPase, or chloride fluxes (Rothman et al., 1985; Choi, 1988; Sendtner et al., 1988; Mattson et al., 1989, 1993). Fibroblasts producing various trophic factors including NGF prevent striatal excitotoxicity and induce the expression of free radical-detoxifying enzymes such as catalase (Jackson et al., 1990; Frim et al., 1992). Thus, trophic factors may be acting to increase endogenous levels of antioxidant enzymes or perhaps decrease the production of oxidative agents after excitotoxicity in addition to providing classical trophic effects.
HD proceeds over a protracted course, resulting in mental deterioration and motor abnormalities, ultimately resulting in death, which usually occurs within 17 years from the onset of symptoms. There are currently no effective treatments, although several experimental approaches are being considered for evaluation in HD. The rationale underlying many of these forms of treatment is to decrease or prevent the neuronal loss and behavioral effects in HD. Strategies directed toward preventing the degeneration of host systems have a unique appeal for HD because all patients who ultimately will get this disease can be positively identified before the onset of symptoms and neural degeneration. This suggests that a successful approach can have a dramatic impact on the course of the disease. The symptoms of HD result from the degeneration of selectively vulnerable neurons within the striatum. In particular, the GABAergic medium spiny neurons, which are the cells of origin for the direct and indirect basal ganglia pathway, are devastated (Reiner et al., 1988). A number of different strategies have demonstrated protection of striatal neurons from excitotoxicity and mitochondrial dysfunction, including grafts of adrenal chromaffin cells (Sanberg et al., 1991), fetal striatum (Tulipan et al., 1988), and NGF-producing fibroblasts (Frim et al., 1993; Emerich et al., 1994b). However, none of these strategies has demonstrated protection of the GABAergic neurons, the cell type most vulnerable in HD and the cell type most central in basal ganglia circuitry. The present study demonstrates that implants of hCNTF-producing cells can prevent the degeneration of GABAergic striatal neurons. In this regard, QA-lesioned rats receiving BHK-control implants displayed a 72% reduction in GAD-ir neurons. In contrast, rats treated in an identical manner except for the single addition of the hCNTF construct displayed only a 20% loss of GAD-ir neurons. These data indicate that hCNTF can protect this essential cell population. The protection of GABAergic neurons, along with protection of less vulnerable cell populations, suggests that implants of hCNTF-producing cells may be a viable treatment strategy for patients with HD.
If trophic factors prove to be a worthwhile therapeutic strategy, the method of delivery may be critical. To date, long-term administration of growth factors has been limited to intraventricular infusions using cannulae or pumps. These routes of administration require repeated injections or refilling of pump reservoirs to maintain specific drug levels and avoid the degradation of the therapeutic agent in solution. Additionally, chronic low-dose infusion of compounds is difficult to sustain using current pump technology. Current pump technology is also suitable for ventricular but not parenchymal delivery. An alternative method is the implantation of cells that have been genetically modified to produce a therapeutic molecule (Gage et al., 1987; Rosenberg et al., 1988; Breakefield, 1989; Kawaja et al., 1991, 1992; Levivier et al., 1995). This avoids the problem of degradation and repeated refilling while allowing a localized distribution within the CSF or parenchyma. The use of immortalized cell lines for delivery of trophic molecules avoids many of these concerns by providing a continuous de novo cellular source of the desired molecule, the dose of which theoretically can be adjusted with specific promoters. Despite its potential, several obstacles remain before clinical evaluation of hCNTF or any growth factor in HD. The location of trophic factor delivery is critical, especially because systemic administration may produce serious adverse side effects. In the present study, hCNTF was delivered to the striatum from the lateral ventricle. Because the human striatum is massive relative to the rodent, the extent of hCNTF diffusion from either ventricular or parenchymal implant sites still remains to be determined. Once obtained, this information will likely dictate the optimal implant site, the number of implants required, and the amount of CNTF that needs to be delivered. A detailed behavioral analysis including motor and cognitive function should be conducted both in rodents and in nonhuman primates to understand the extent and persistence of behavioral that might be expected. Studies in nonhuman primates are particularly important given the similar organization of the human and nonhuman primate basal ganglia. Behavioral deficits in lesioned primates may also be more analogous to HD (Isacson et al., 1989, 1990; Hantraye et al., 1990). The timing of treatment also deserves considerable attention. It appears that trophic factor therapy would be most effective early in the course of HD, thus requiring long-term administration. Long-term secretion of trophic factors from transfected cells has been achieved both in vitro andin vivo. Primary cells genetically modified using retroviral vectors can survive for up to 6 months in rat striatum (Kawaja et al., 1991, 1992). Encapsulated BHK cells continue to survive and to secrete hNGF for at least 13.5 months in rodent lateral ventricles (Winn et al., 1995). Although these data are encouraging, the long-term expression of hCNTF secreting cells still needs to be determined empirically. Finally, clinical trials cannot proceed without careful attention to the possible toxicological profile of hCNTF or any trophic factor.
In summary, the present results indicate that delivery of hCNTF from polymer-encapsulated BHK cells dramatically attenuates both the pathological and the behavioral consequences of QA administration. In particular, hCNTF-secreting implants prevent the degeneration of GABAergic striatal neurons. Excitotoxicity has been implicated in a variety of pathological conditions including ischemia and neurodegenerative diseases such as Huntington’s, Parkinson’s, and Alzheimer’s (Choi, 1988; Olney, 1989). Accordingly, biologically delivered hCNTF may provide one means of preventing the cell loss and associated behavioral abnormalities of these and possibly other human disorders. The implantation of encapsulated engineered cells may provide a safe and practical therapeutic approach for the chronic delivery of factors for a number of human neurodegenerative disorders.
Footnotes
Correspondence should be addressed to Dr. Dwaine F. Emerich, CytoTherapeutics, Inc., 2 Richmond Square, Providence, RI 02906.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}