

Abstract
In hippocampal neurons, a slowly inactivating aminopyridine-sensitive transient potassium current, D-current, influences the time course of action potential repolarization and therefore activity-dependent Ca2+ entry. We used high-speed optical recording techniques to study the effects of selectively inhibiting D-current with 4-AP (40 μm) on transmission at the Schaffer collateral (CA3)-CA1 synapse in rat hippocampal slices stained with the voltage-sensitive dye RH-155. We observed that addition of 4-AP to the bathing solution resulted in (1) augmentation of a fast component of the optical signal corresponding to the postsynaptic EPSP and action potential, and (2) the appearance of a delayed depolarization of CA1 neurons and other adjacent cells. 4-AP appeared to alter the presynaptic action potential and the dynamics of synaptic transmission to both reduce the sensitivity of the postsynaptic EPSP and action potential to ω-toxin calcium channel blockers (ω-conotoxin GVIA and ω-agatoxin IVA) and the Ca2+-dependent potassium channel blocker charybdotoxin, and to increase sensitivity to the dihydropyridine nifedipine, the NMDA receptor blocker aminophosphonopentanoic acid, and the intracellular Ca2+ release inhibitor thapsigargin. The delayed depolarization induced by 4-AP was inhibited in hyperosmotic extracellular solution, suggesting that enhanced transmitter release resulted in increased accumulation of K+ in the extracellular space. Because 4-AP is a convulsant at concentrations similar to those used here, we suggest that the 4-AP-targeted channel(s) carrying D-current may contribute to the hyperexcitability associated with epilepsy.
- hippocampus
- optical recording
- microelectrode recording
- voltage-gated potassium current
- A-current
- D-current
- 4-aminopyridine
- RH-155
- RH-482
- epilepsy
Potassium currents have long been targets of experimental interest because of their influence on neural excitability. In hippocampal pyramidal neurons, for which a rich variety of potassium currents have been described (for review, seeStorm, 1990), evidence suggests that action potential repolarization is driven by a combination of a Ca2+-dependent potassium current and a slowly inactivating transient potassium current termed D-current (or ID) (Storm, 1987, 1988). Although it is intuitively obvious and experimentally verified (Storm, 1987) that reducing repolarizing drive will increase the excitability of individual neurons, the precise links between alteration of action potential waveforms and the excitability of small ensembles of neurons have not been clear. The time course of hippocampal neuron repolarization is particularly important because action potential duration is an important determinant of activity-dependent Ca2+ entry and regulation of downstream Ca2+-dependent processes such as neurotransmitter release (see Discussion).
To selectively manipulate D-current, we used 4-aminopyridine (4-AP). In hippocampal neurons, millimolar concentrations of 4-AP block a rapidly inactivating transient potassium current (Gustafsson et al., 1982) similar to the A-current originally described in molluscan neurons (Hagiwara et al., 1961; Connor and Stevens, 1971; Neher, 1971); however, low concentrations of 4-AP (<200 μm) reduce the amplitude of a more slowly inactivating D-current while sparing A-current (Rudy, 1988; Storm, 1988; Ficker and Heinemann, 1992; Wu and Barish, 1992).
We examined the voltage responses of CA1 pyramidal cell neurons to stimulation of their Schaffer collateral input from area CA3 using recently developed optical recording techniques, and confirmed some of our results with intracellular microelectrode recordings from CA1 pyramidal neuron somata. We observed that addition of 4-AP (40 μm) to the bathing solution resulted in (1) augmentation of a fast component of the optical signal corresponding to the postsynaptic EPSP and action potential, and (2) the appearance of a delayed depolarization of CA1 neurons and other adjacent cells. Our data indicate that 4-AP changed the pharmacological sensitivities of transmission at this synapse in ways that could reflect alteration of the presynaptic action potential and prolongation of neurotransmitter release. Enhanced transmitter release, together with accumulation of K+ in the extracellular space, resulted in generation of the delayed depolarization of pre- and postsynaptic neural and glial membranes. The data thus indicate that the consequences of D-current regulation are manifest at multiple sites to affect the signaling characteristics of the CA3–CA1 synapse.
An intriguing property of 4-AP is its ability to increase action potential invasion of nerve terminal arbors (Obaid et al., 1987; Obaid and Salzberg, 1996), enhance synaptic transmission in a variety of preparations (Thesleff, 1980), and induce seizure in mammalian brainin vivo (Szente and Baranyi, 1987) and in experimental slice preparations (Rutecki et al., 1987; Perreault and Avoli, 1991, 1992). We therefore suggest that misregulation of the 4-AP-targeted channel(s) carrying D-current may be a source of the hyperexcitability associated with epilepsy.
We have published preliminary reports of some of these findings (Barish et al., 1994, 1995).
MATERIALS AND METHODS
The preparation. Hippocampal slices were prepared from 300–350 gm male rats decapitated under ether anesthesia. Brains were quickly cooled in iced artificial CSF (ACSF) continuously gassed with a mixture of 95% O2 and 5% CO2. After cooling for 5 min, hippocampi were dissected free, sliced at 400 or 500 μm thickness with a D.S.K. rotary tissue slicer, and transferred to a gassed ACSF bath where they were held at 30°C for 1–4 hr before use.
Artificial CSF consisted of (in mm): 124 NaCl, 5 KCl, 2 CaCl2, 1.25 MgSO4, 1.25 NaH2PO4, 22 NaHCO3, and 10 glucose, pH 7.4. A few experiments were performed using Gey’s Balanced Salt Solution (Gey and Gey, 1936), which consisted of (in mm): 120 NaCl, 5 KCl, 1.5 CaCl2, 1 MgCl2, 0.3 MgSO4, 0.85 Na2HPO4, 0.22 KH2PO4, 27 NaHCO3, and 5.6 glucose, pH 7.3–7.4. During recordings, ACSF was continuously bubbled with the O2/CO2 gas mixture and warmed to 30°C before entering the recording chamber. Drugs and other reagents were added to ACSF from stock solutions as indicated: 4-AP at 40 μm, bicuculline at 20 μm, CNQX at 10 μm, charybdotoxin (CbTX) at 100 nm, nifedipine at 50 μm (50 mm stock in ethanol), ω-conotoxin GVIA (ω-CgTX GVIA) at 100 nm to 1 μm, ω-agatoxin IVA (ω-AgaTX IVA) at 100 nm, thapsigargin (Tg) at 20 μm. Bovine serum albumin (1 mg/ml) was added to peptide-containing solutions to minimize nonspecific binding. Osmolarity of external solutions was measured using a Wescor 5500 vapor pressure osmometer and was adjusted for the experiment presented in Figure 15 by addition of sucrose. Nifedipine and Tg were obtained from Wako Pure Chemicals; channel blocking peptides were from Peptide Institute; all other reagents were from local suppliers.

A, Increasing the osmolarity of the ACSF to 1.4 times normal by addition of sucrose reduced the EPSP/AP complex and completely eliminated the delayed depolarization. The effect was partially reversible. B, Time course of the effects of hyperosmotic ACSF showing that the reduction of the delayed depolarization occurred rapidly, whereas the EPSP/AP complex declined more slowly. The data presented are representative of two similar experiments and one additional experiment in the presence of CNQX and AP5.
Optical recordings. The MOS-based solid-state camera (128 × 128 elements) with frame memory and associated software has been described in detail in previous publications (Ichikawa et al., 1993; Iijima and Matsumoto, 1994). For the experiments presented here, pixels were binned in 2 × 2 blocks to enhance sensitivity, and images of 64 × 64 pixels were acquired at 0.6 msec/image in blocks of 256 images (252 data images, 1 reference image, and 3 additional frames for computational purposes); the total recording time was 151.2 msec/block. For each experiment, eight blocks of 256 frames were acquired in rapid succession, and the images were averaged and corrected for linear drift; each trace presented thus represents an averaged response to eight identical stimulations. The camera was mounted on a Zeiss Axioplan upright microscope, and the CA1 region was imaged using 10× Achroplan (water immersion, 0.3 numerical aperture; Zeiss) or Plan-neofluor (0.3 numerical aperture; Zeiss) objectives. The Achroplan water immersion objective permitted continuous perfusion with warmed and oxygenated ACSF; with the Plan-neofluor objective, perfusion was briefly halted during image acquisition to prevent movement artifacts. The imaged area was approximately 1 mm × 1 mm. Illumination was by a voltage-stabilized tungsten-halogen lamp filtered at 720 nm (±15 nm bandpass). The voltage-sensitive pyrazo-oxonol dye RH-155 increases absorption of 720 nm light as transmembrane voltages become more positive, and changes in membrane voltage recorded optically are presented here as the fractional change in light transmittance, f Δ T.
For all of the experiments presented in Results, slices were stained immediately before recording with RH-155 at 2.5 mg/ml (in ACSF) for 3–4 min, followed by >10 min of wash in ACSF. As with all similar dyes, a mixture of neural and non-neural membranes will be stained and therefore will contribute to the voltage signal (for review, seeGrinvald et al., 1988). Salzberg and coworkers (Konnerth et al., 1987) have suggested that different dyes may preferentially partition into neural and non-neural membranes, and therefore that it might be possible to separate signals originating in neurons and glia. To evaluate this possibility, we compared optical signals produced by RH-155 and the related RH-482 using two staining protocols, either 3 1/2 min at 2.5 mg/ml or 1 hr at 0.2 mg/ml. In the experiments reported by Konnerth et al. for skate cerebellum, the optical signals produced by RH-482 when applied according to the second protocol appeared to preferentially report axonal activity. However, for rat hippocampus, as shown in Figure 1, the optical signals obtained with both dyes and both loading protocols were very similar when compared for both control and 4-AP-containing solutions. We interpret these results to mean that voltage-dependent signals originated from similar populations of neural and glial membranes under all staining conditions, and thus that separation of signals based on cells of origin could not easily be achieved in these experiments.

Matrix of optical signals from area CA1 of rat hippocampal slice showing that similar differences in voltage-dependent signals obtained in control and 4-AP-containing solutions were observed using either of two related voltage-sensitive dyes, RH-155 or RH-482, and either of two different loading protocols, 0.2 mg/ml for 60 min or 2.5 mg/ml for 3 1/2 min. The variation between RH-155 and RH-482 evident in the ACSF relaxations, particularly at the lower staining concentration, was within the range observed in these experiments, and therefore separation of signals originating in neural or glial membranes may not be possible in this preparation (Konnerth et al., 1987). The data presented are representative of one experiment with RH-155 at 0.2 mg/ml, five experiments with RH-155 at 2.5 mg/ml, three experiments with RH-482 at 0.2 mg/ml, and two experiments with RH-482 at 2.5 mg/ml.
Orthodromic stimuli were delivered using electrically polished bipolar tungsten electrodes. Stimuli were 0.1–0.5 mA for 300 μsec; the amplitude was adjusted for each experiment to elicit a suprathreshold but not maximal response. Orthodromic stimuli were delivered to the Schaffer collateral pathway in stratum (st.) radiatum.
Microelectrode recordings. Intracellular recordings were made from somata of CA1 pyramidal neurons using sharp electrodes and standard techniques. Electrodes were filled with 4 mK-acetate plus 20 mm KCl (to provide a nonpolarizing junction with the Ag-AgCl wire) and had resistances of 120–140 MΩ in ACSF. Voltage was measured and recorded using an Axoclamp 2A amplifier and pClamp software (both from Axon Instruments). The intracellular recordings were made in the same ACSF solutions, on the same microscope with the same optics and stimulating electrodes, and in some experiments at the same time as the optical recordings.
Data analysis and presentation. The data presented here are drawn from a pool of 92 successful experiments. The experiments presented here are representative of the manipulations performed (numbers of experiments are indicated in the figure legends), because although qualitatively consistent data could be obtained, statistical analysis was complicated by variation in absolute values off Δ T because of differences in slice thickness, dye staining, camera sensitivity (which is temperature-dependent), and other not-well-understood factors. A further complication was the irreversible nature of the manipulations performed. The effects of 4-AP were only partially reversed after >40 min of continuous wash, and many of the peptides used bind irreversibly to their targets.
RESULTS
4-Aminopyridine altered signaling at the CA3–CA1 synapse
The optical signals recorded from the CA1 region originated in all the membranes stained by RH-155 (Grinvald et al., 1988). Thus, the changes in voltage recorded will reflect the combination of the Schaffer collateral action potential, the EPSP and subsequent postsynaptic action potential(s) in pyramidal neurons, and additional voltage signals derived from interneurons as well as non-neuronal membranes.
Figure 2A shows the propagation of a region of membrane depolarization through the CA1 region of a rat hippocampal slice stained with RH-155 as described in Materials and Methods. The orientation of the slice is such that the CA3 region is below the portion imaged, and the Schaffer collateral input to CA1 was stimulated with a brief (300 μsec) pulse delivered just outside the field imaged and near the location marked with the arrowhead. The area of the image is approximately 1 × 1 mm, and each pixel thus represents a square approximately 16 × 16 μm in the plane of the slice. The stimulus was delivered between the first and second frames, and resulting depolarization (indicated inpseudocolor) can be seen entering the slice from below. The maximum depolarization (in optical terms) was seen ∼8 msec after stimulation (frame 12), and the entire depolarization returned to baseline after ∼30 msec.

Propagation of depolarization through area CA1 of rat hippocampal slice before (A) and during (B) application of 4-AP (40 μm), illustrating potentiation of the initial voltage transient (EPSP/AP complex) and induction of a delayed depolarization by 4-AP (see Fig.4). The slice was stained with RH-155 and imaged at 0.6 msec/frame as described in Materials and Methods. The major layers of the slice are labeled in the first panel. The depolarization was measured as the fractional change in transmittance (f Δ T) in each pixel; this value is encoded inpseudocolor as indicated in the scale and is superimposed on a transmitted light image of the slice. The Schaffer collateral input from CA3 to CA1 was stimulated using bipolar tungsten electrodes positioned just below the lower edge of the image (black arrowhead), and in each sequence the stimulus was delivered between the first and second frames. The two finger-like projections in the top right portion of each image are from an electrode used for antidromic stimulation. The images inB were acquired after the slice had been exposed to 4-AP for 20 min; in general, we allowed 15–30 min for the effects of 4-AP to reach a steady state.
The effects of application of 40 μm 4-AP are apparent in Figure 2B (see also Fig. 4). At this concentration, 4-AP will inhibit D-current while sparing A-current in hippocampal neurons (Storm, 1988; Ficker and Heinemann, 1992; Wu and Barish, 1992). Two effects of 4-AP on the optical signals were noted. First, 4-AP enhanced the initial rapid depolarization that reached a maximum 6–8 msec after stimulation. Second, after a plateau 9–12 msec after stimulation, a delayed depolarization was seen, the amplitude of which (in optical terms) was greater than the initial sequence recorded immediately after Schaffer collateral stimulation. This delayed depolarization was largest in the mid-dendritic region (st. radiatum), but was seen throughout the CA1 region (from the hippocampal fissure to the alveus; see Fig. 3). The duration of this delayed long-lasting depolarization was typically 70–110 msec.

Deconstruction of the optical signal into its constituent components. An initial depolarization was attributable to impulse activity in the Schaffer collateral pathway because it was preserved when synaptic transmission was blocked in zero Ca2+ ACSF (illustrated) and in the presence of CNQX (10 μm) and AP5 (50 μm) (data not shown). The EPSP/AP complex was evident as a large peak seen when transmission was preserved. In the presence of 4-AP, this EPSP/AP complex was potentiated, and a secondary delayed depolarization was observed. Theinset shows the initial phase of the depolarization on an expanded time scale. Note that the slope of the small initial rise was increased in the two traces taken in the presence of 4-AP; this could reflect a change in the Schaffer collateral action potential (Kuhnt and Grinvald, 1982; Kocsis et al., 1983).

Time course of voltage signals in ACSF and in the presence of 4-AP. Voltage changes in the one-dimensional stripe indicated in the top panel were extracted from each frame and arranged in a time series in the bottom panels. Note the prominence of the 4-AP-induced delayed depolarization in the st. radiatum and its bidirectional lateral expansion.
Another view of the image data from Figure 2, A andB, emphasizing the changes in the spatial distribution of depolarization induced by 4-AP is shown in Figure 3. The bottom panels show f Δ T within the band indicated above, arrayed in a time series and superimposed on the slice image from the same region. A small secondary depolarization is evident under control conditions. In the presence of 4-AP, as described above, the 4-AP-induced delayed depolarization was most evident in the st. radiatum, the cell body layer (st. pyramidale) was relatively spared, and the st. oriens showed a smaller depolarization. Notable here is the slow rise of depolarization in the st. radiatum (compared with the st. pyramidale), followed by the bidirectional lateral spread of depolarization from the central area of the st. radiatum toward both the st. pyramidale and the st. lacunosum-moleculare (Grinvald et al., 1982; Albowitz and Kuhnt, 1991).
The depolarization signal recorded during this period can be decomposed into the following distinct elements (Grinvald et al., 1982;Albowitz and Kuhnt, 1991; Iijima and Matsumoto, 1994) (Fig.4): (1) an initial brief deflection originating in the Schaffer collateral pathway since it is preserved after block of the CA3 to CA1 synapse; (2) an immediately following rapidly rising phase composed of the EPSP and the action potential originating in the dendrites of CA1 pyramidal neurons (EPSP/AP complex); and (3) after a delay, the depolarization seen only in the presence of 4-AP. In this figure and the other figures showing plots of f Δ T versus time (Figs. 1, 5, 6, and 8, 9, 10, 11, 12, 13, 14, 15), the measurements shown are taken from a single image element (a 2 × 2 pixel average) in the mid-dendritic region of the st. radiatum, except as indicated. These figures thus illustrate maximal changes in light transmittance, and thus maximal total depolarization of the population of stained neural and non-neural membranes.

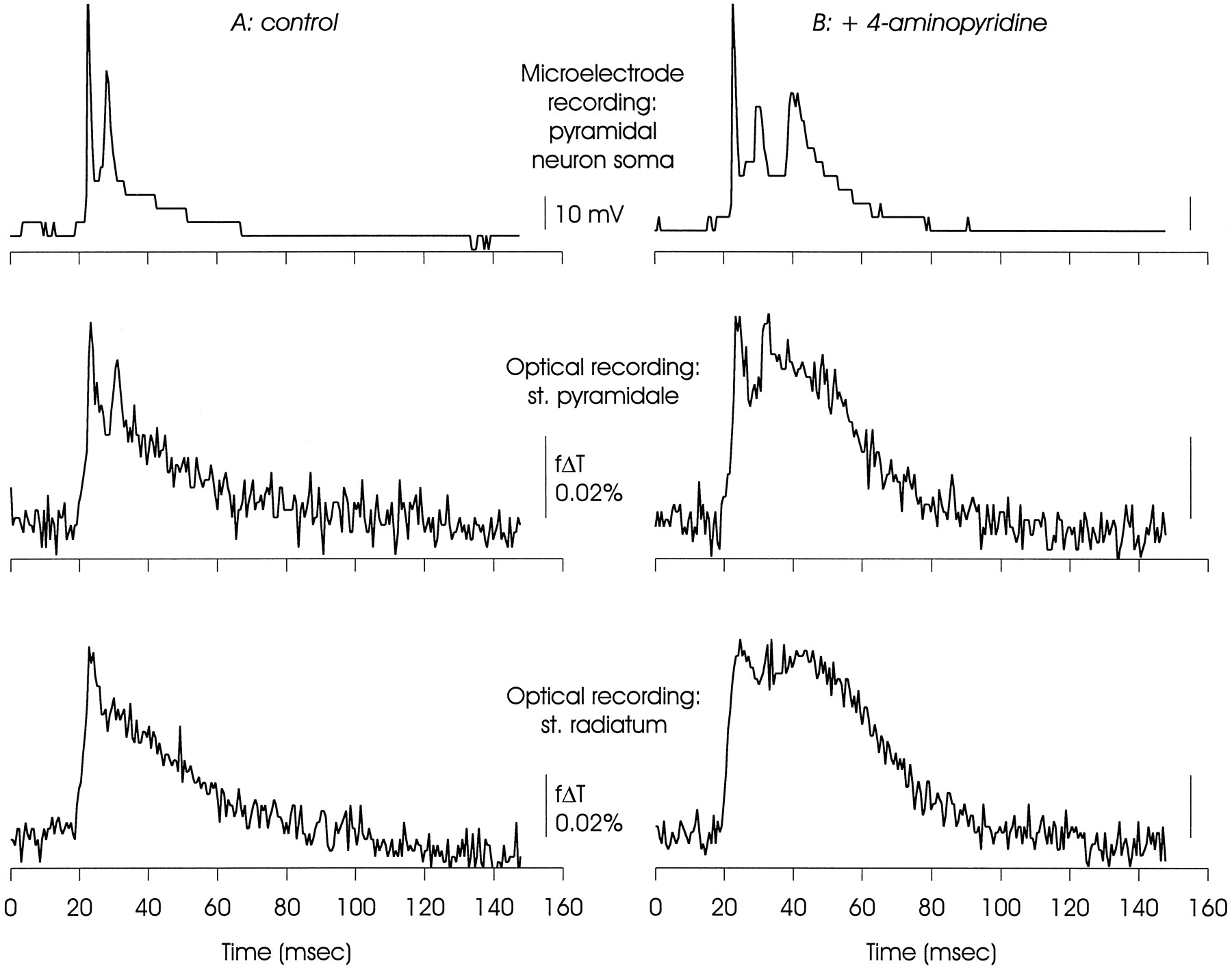
Comparison of electrical and optical measurements of membrane voltage made simultaneously in CA1, illustrating that the delayed depolarization was a property of neural as well as other membranes. The microelectrode recording was made from a pyramidal neuron soma, and one of the optical signals was taken from the same region of st. pyramidale. Note that although the optical signals were clearly related to the microelectrode signal, the action potential peaks were less prominent relative to the slower depolarizations. This may reflect temporal dispersion of the action potentials as well as contributions of optical signals originating in glial membranes that will depolarize but not support action potentials. The delayed depolarization was most evident in optical recordings from st. radiatum (see also Figs. 2, 3), and discrete secondary action potentials were not always recorded from this region. The large steps in the microelectrode recording are the result of encoding this signal in an image pixel without conditioning amplification; this was done to ensure temporal registration.

Block of GABAA receptors with bicuculline (20 μm) did not occlude generation of the optically recorded delayed depolarization by 4-AP (A), or the reverse (B). Rather, each appeared to potentiate the actions of the other. The data presented are representative of 15 experiments with bicuculline used at 10–20 μm.

ω-CgTX GVIA (100 nm) reduced transmission at the Schaffer collateral–CA1 synapse under control conditions (evident as reduction in the EPSP/AP complex). In the presence of 4-AP, it had minimal effect on the EPSP/AP complex and slightly reduced the delayed depolarization. In this and the following figures, the initial depolarizations are shown on an expanded time scale in the insets. The data presented are representative of nine experiments with ω-CgTX GVIA used at 100 nm to 1 μm.
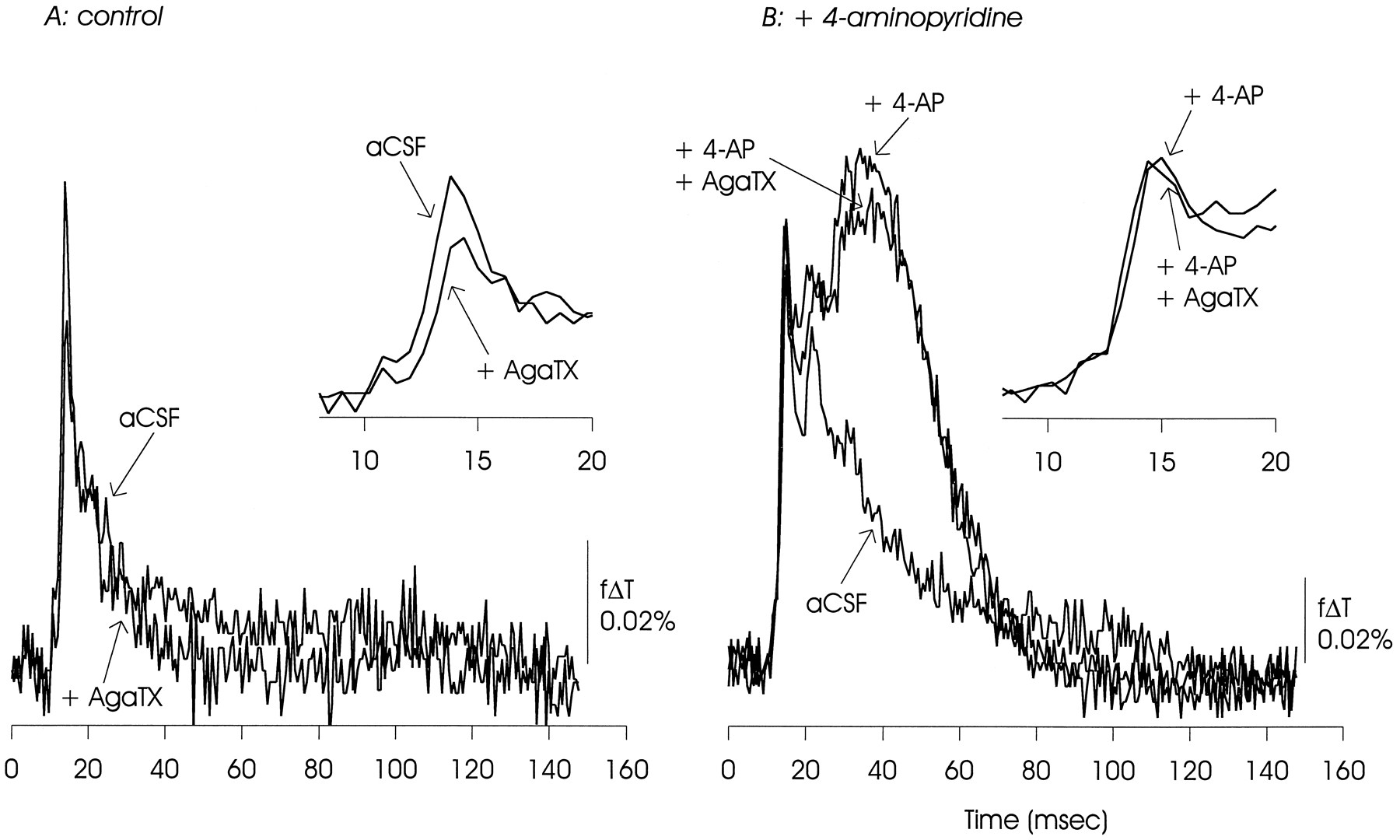
ω-AgaTX IVA (100 nm) also reduced the initial EPSP/AP complex under control conditions, but had minimal effect in the presence of 4-AP. There was a small reduction of the delayed depolarization. The data presented are representative of two experiments.

Nickel (50 μm) had no apparent effects on the EPSP/AP complex under control conditions or in the presence of 4-AP. The delayed depolarization was also unaffected. The data presented are representative of three similar experiments.

Nifedipine (50 μm) had no apparent effect on the EPSP/AP complex under control conditions. In the presence of 4-AP, nifedipine caused a small reduction in the EPSP/AP complex, and a large reduction in the delayed depolarization. The data presented are representative of 12 experiments with nifedipine used at 50–200 μm.

CbTX (10 μm) augmented the initial EPSP/AP complex under control conditions and appeared to hasten the subsequent return of voltage to baseline. In the presence of 4-AP, CbTX had only small effects on either the EPSP/AP complex or the delayed depolarization. The data presented are representative of three similar experiments.

Tg (20 μm) had minimal effects on the EPSP/AP complex under control conditions. In the presence of 4-AP, Tg appeared to slightly reduce the EPSP/AP complex and cause a significant reduction in the delayed depolarization. The data presented are representative of six experiments with Tg used at 10–20 μm.

AP5 reduced the EPSP/AP complex under control conditions, but the majority of this signal was sensitive to CNQX (not shown). In the presence of 4-AP, AP5 had a similar effect on the EPSP/AP complex, and sharply reduced the delayed depolarization. The data presented are representative of six similar experiments.
Optical signals reflected electrical activity as measured in simultaneous microelectrode recordings
Figure 5 shows records obtained during a microelectrode recording made from a CA1 pyramidal neuron soma, and optical recordings of voltage from two regions of the same hippocampal slice. Overall, waveforms of electrical and optical signals originating in the cell body layer were similar. Two action potentials were detected by the microelectrode in the soma under control conditions, and two deflections were seen in the optical record taken from a four-pixel cluster positioned over the st. pyramidale. In the presence of 4-AP, an additional action potential was generated in the soma, and the optical recording from st. pyramidale showed a third late peak. However, in the optical recordings the relative amplitudes of the fast action potentials were reduced in relation to the slower events, as can be seen by comparing the microelectrode soma recordings with the optical records from the st. pyramidale. This could be a consequence of asynchronous generation of somatic action potentials (which would result in a smaller peak amplitude in optical recordings, attributable to averaging across many somata) as well as a mixing of optical signals originating in neurons and non-neuronal cells because only the neurons will generate action potentials (Sontheimer and Waxman, 1993). Note in these records the lack of discrete action potentials in the optical signals originating from the st. radiatum.
Generation of the delayed depolarization required glutamatergic synaptic transmission
The synapse of the Schaffer collaterals originating in CA3 onto dendrites of CA1 pyramidal neurons is glutamatergic (Davies and Collingridge, 1989; Lambert et al., 1989), with the fastest components of the EPSC sensitive to CNQX (Honoré et al., 1988) and other blockers of non-NMDA-type glutamate receptors (Hestrin et al., 1990). Generation of the EPSP/AP complex and the delayed depolarization in the presence of 4-AP required functional synaptic transmission, because they both were eliminated by CNQX (10 μm; data not shown) as well as by removal of external Ca2+ (Fig. 4). Also, the delayed depolarization, when recorded in the presence of 4-AP, was differentially sensitive to blockade of NMDA receptors (see below).
Generation of the delayed depolarization did not require input from area CA3 itself, only activation of the Schaffer collateral input to CA1, as the delayed depolarization was observed when a cut was made severing CA3 from CA1 and stimulation was made distal to the cut (data not shown). This suggests that the delayed depolarization was not a result of repetitive activity in CA3 pyramidal neuron somata (see Discussion).
Inhibition of GABAergic synaptic transmission enhanced the EPSP/AP complex and the delayed depolarization
Stimulation of the Schaffer collateral input to CA1 also results in activation of GABA-releasing interneurons and generation of IPSPs in CA1 pyramidal neurons. Relief of inhibition by the GABAAreceptor blocker bicuculline will thus enhance CA1 pyramidal neuron excitability (Andersen, 1990).
In optical recordings, 4-AP and bicuculline (20 μm) displayed mutual and nonoccluding enhancement of the delayed depolarization (Fig. 6). This pattern was seen if bicuculline preceded (A) or followed (B) exposure to 4-AP. Notable was enhancement of the EPSP/AP complex when bicuculline preceded 4-AP (A).
In microelectrode recordings, bicuculline also enhanced the delayed depolarization seen in the presence of 4-AP (Fig. 7). Note the apparent increase in amplitude of the initial action potential in the averaged traces in the presence of 4-AP, which could be a reflection of increased synchronicity.

Bicuculline (20 μm) also potentiated the 4-AP-induced delayed depolarization in a microelectrode recording from a CA1 pyramidal neuron soma. Sixteen consecutive individual traces are shown in the top panels. They are averaged in thebottom panels to illustrate how temporal dispersion of the action potentials riding on the delayed depolarization can reduce their apparent amplitude. Such averaging may occur in the optical signals, which originate in many neurons simultaneously (see Fig. 5). The data presented are representative of three similar experiments.
Potential for pre- and postsynaptic actions of 4-AP
In principle, the actions of 4-AP could be on invasion of the Schaffer collateral action potentials into terminal arbors, action potentials in presynaptic terminals, excitability of postsynaptic membranes, or all three. Consistent with the possibility of three or more sites of action, effects of 4-AP on conduction in the Schaffer collateral pathway have been reported (Kuhnt and Grinvald, 1982), 4-AP-sensitive potassium channels have been found in other CNS presynaptic terminals (Forsythe, 1994), and D-currents have been recorded from CA1 neurons in situ (Storm, 1987, 1988) and after acute dissociation (R.-L. Wu and M. E. Barish, unpublished observations). Data presented below indicate that 4-AP in fact has consequences for both pre- and postsynaptic membranes.
4-Aminopyridine altered the sensitivity of the EPSP/AP complex and the delayed depolarization to manipulations that affect neurotransmitter release
The identity of the calcium channels mediating neurotransmitter release at the Schaffer collateral–CA1 pyramidal cell synapse have been extensively investigated. From a potential pool of T-, N-, P-, Q-, R-, and L-type channels that have been defined kinetically and pharmacologically (Fox et al., 1987; Llinás et al., 1989; Randall and Tsien, 1995), presently available evidence suggests that fast transmitter release at this and other central synapses is mediated by multiple classes of channels (N-type, Q-type, and other types), some of which are selectively sensitive to ω-CgTX GVIA and ω-AgaTX IVA (Obaid et al., 1989; Luebke et al., 1993; Takahashi and Momiyama, 1993;Wheeler et al., 1994; Wu and Saggau, 1994). Normal transmission at this synapse has also been observed to be insensitive to inhibition of T-type channels with Ni2+ or to manipulation of L-type channels with dihydropyridines.
Optical measurements of the EPSP/AP complex made under control conditions showed the sensitivity to inhibition of calcium channels expected from these previous electrophysiological measurements. As illustrated in Figures8A, 9, 10, 11A, postsynaptic responses to Schaffer collateral stimulation were substantially inhibited by ω-CgTX GVIA (100 nm to 1 μm), were reduced (although to a lesser extent) by ω-AgaTX IVA (100 nm), and were insensitive to Ni2+ (100 μm) and the dihydropyridine nifedipine (50 μm).
In the presence of 4-AP, this pattern of pharmacological sensitivities was altered, as shown in Figures 8B, 9, 10, 11B. In contrast to observations made under control conditions, the EPSP/AP complex was insensitive to ω-CgTX GVIA and ω-AgaTX IVA, and slightly reduced by nifedipine. The EPSP/AP complex remained insensitive to Ni2+. The 4-AP-induced delayed depolarization showed minimal sensitivity to ω-CgTX GVIA and ω-AgaTX IVA, and was substantially reduced, but not eliminated, by nifedipine. It also was not affected by Ni2+.
Repolarization of hippocampal neuron action potentials is driven in part by activation of Ca2+-dependent potassium channels blocked by CbTX (Storm, 1987; Goh et al., 1992), and CbTX (and other blockers of Ca2+-dependent potassium channels) increases EPSP amplitudes at other synapses (Augustine et al., 1988; Robitaille and Charlton, 1992). In optical measurements made under control conditions, CbTX (100 nm), as expected, enhanced the EPSP/AP complex and accelerated a slow component of repolarization (Fig. 12A). However, in the presence of 4-AP, the EPSP/AP complex and the delayed depolarization were minimally affected by CbTX (Fig. 12B).
Sensitivity to inhibition of intracellular Ca2+ release
The endoplasmic reticulum of hippocampal and other neurons sequesters and releases Ca2+. Both ryanodine- and InsP3-sensitive Ca2+ release channels are found in hippocampal neurons (Sharp et al., 1993; Seymour-Laurent and Barish, 1995), and intracellular Ca2+ responses to activation of the CA3–CA1 synapse are reduced by inhibition of Ca2+-induced Ca2+ release (Alford et al., 1993). The SERCA family of Ca2+ sequestering ATPases is inhibited by Tg (Thastrup et al., 1990), and Tg thus can reduce or eliminate intracellular Ca2+ release. We observed that under control conditions Tg (20 μm) had little effect on the EPSP/AP complex (Fig. 13A). In the presence of 4-AP, Tg reduced the amplitude of the delayed depolarization (Fig. 13B).
Generation of the delayed depolarization was sensitive to inhibition of NMDA receptors
Aminopyridines can enhance and prolong postsynaptic responses at hippocampal and other synapses (Thesleff, 1980;Buckle and Haas, 1982; Rutecki et al., 1987; Wheeler et al., 1996). At the Schaffer collateral–CA1 synapse, the NMDA component of the postsynaptic response activates and decays more slowly than the non-NMDA component (Collingridge et al., 1988; Forsythe and Westbrook, 1988; Hestrin et al., 1990), and therefore might be expected to be involved in generating a delayed depolarization induced by prolonged transmitter release.
We observed that the NMDA receptor blocker aminophosphonopentanoic acid (AP5; 50 μm) reduced the amplitude of the fast EPSP/AP complex under control conditions and in the presence of 4-AP (Fig.14). AP5 also substantially inhibited generation of the delayed depolarization.
Accumulation of extracellular K+ and generation of the delayed depolarization
An increase in K+ concentration in the space separating neurons and glia will depolarize neuronal and other adjacent membranes. K+ accumulation can be reduced by expanding extracellular space with hypertonic medium, and, in optical recordings of membrane voltage in skate cerebellum, a delayed depolarization similar to that seen here was reduced by raising the external osmotic strength to 1.4 times normal (Konnerth et al., 1987).
The records in Figure 15A illustrate that increasing the ionic strength of the external solution to 1.4 times normal reduced the 4-AP-induced delayed depolarization to control (pre-4-AP) levels and the fast EPSP/AP component to below control levels. The delayed depolarization appeared differentially sensitive to an increase in external ionic strength in that, as can be seen in Figure 15B, the delayed depolarization declined rapidly after the bath was exchanged for the high ionic strength solution, whereas the reduction in the EPSP/AP component was more gradual and incomplete.
Loss of the delayed depolarization was not simply a consequence of reducing the fast EPSP/AP complex because this component was only partially reduced by the increase in osmolarity, and in the presence of 4-AP, we observed the delayed depolarization at all subthreshold stimulus intensities (data not shown).
DISCUSSION
Comparison with previous optical and microelectrode recordings from hippocampal slice
In early studies concerned with synaptic signaling in hippocampal slice, Grinvald et al. (1982) and Albowitz and Kuhnt (1991) used optical recording techniques to record, as in the present study (see also Ichikawa et al., 1993), (1) a small fast signal in the st. radiatum, interpreted to represent activity in the Schaffer collateral axons; (2) a larger depolarization with longer latency taken to represent the EPSP and action potential in apical dendrites; and (3) a rapidly rising signal in the st. pyramidale interpreted to be action potentials in the somata of pyramidal cells. Albowitz and Kuhnt (1991)also induced epileptiform activity using 4-AP and reported repetitive firing in st. pyramidale of CA1 and the appearance of a delayed depolarization most prominent in st. radiatum after stimulation of the Schaffer collateral pathway. Aminopyridine-induced changes in synaptic efficacy were also accompanied by enhanced impulse propagation in the Schaffer collateral pathway (Kuhnt and Grinvald, 1982).
In a microelectrode recording study, Perreault and Avoli (1989)observed that 4-AP (5–100 μm) induced two late potentials at the CA3–CA1 synapse: a delayed EPSP with latency of 25–120 msec and a long-lasting depolarization with latency of 100–600 msec. This delayed EPSP was most evident in the presence of bicuculline and most likely corresponds to the delayed depolarization described here. However, because their delayed EPSP was insensitive to blockade of NMDA receptors but was sensitive to TTX applied locally to the CA2–CA3 region, Perreault and Avoli (1989) concluded that it was driven by repetitive firing of the CA3 input into CA1. The long-lasting depolarization of Perreault and Avoli (1989) is less likely to correspond to the delayed depolarization reported here because the long-lasting depolarization was affected by manipulations of GABAA receptor function and was not sensitive to glutamate receptor antagonists (see also Perreault and Avoli, 1991, 1992).
In another study, Rutecki et al. (1987) examined the mossy fiber input to CA3 pyramidal neurons and observed that 4-AP (5–10 μm) enhanced and prolonged excitatory synaptic currents independently of any effect on GABAA-mediated synaptic conductances. Their observations are thus broadly consistent with those reported here, particularly regarding enhancement and prolongation of transmitter release from presynaptic terminals.
Most recently, Wheeler et al. (1986) reported a 4-AP-induced increase in EPSP slope in field recordings from hippocampal slices; these results are discussed in more detail below.
Mechanisms by which 4-AP may alter transmission at the CA3–CA1 synapse
4-Aminopyridine was applied at concentrations suggested by several studies to selectively block D-current in hippocampal neurons (Storm, 1988; Ficker and Heinemann, 1992; Wu and Barish, 1992). How might D-current inhibition enhance synaptic efficacy?
Enhancement of transmitter release from presynaptic terminals
Aminopyridine and toxin sensitivities of membrane currents in mouse motor neuron terminals (Brigant and Mallart, 1982; Dreyer and Penner, 1987), identification of D-like potassium currents in CNS presynaptic terminals (Forsythe, 1994), prolongation of the presynaptic fiber volley in hippocampal slice by micromolar 4-AP (Kuhnt and Grinvald, 1982), and the ability of 4-AP to enhance and prolong postsynaptic responses at hippocampal and other synapses (Llinás et al., 1976; Thesleff, 1980; Buckle and Haas, 1982; Galvan et al., 1982; Rutecki et al., 1987; Perreault and Avoli, 1989; Wheeler et al., 1996) all suggest involvement of D-current in regulation of transmitter release. Therefore, changes in the terminal action potential waveform and consequent increases in neurotransmitter release induced by 4-AP may underlie enhancement of the EPSP/AP component of the optical signal. A lengthening of the period of neurotransmitter release as a result of a prolonged terminal action potential is consistent with the sensitivity of the delayed depolarization to blockade of NMDA receptors. However, several investigations have suggested that 4-AP may also induce repetitive firing of presynaptic terminals (Heuser et al., 1979; Perreault and Avoli, 1989).
Increased invasion of presynaptic terminal arbors
The EPSP/AP complex also may be enhanced by more complete invasion of terminal arbors (Wall, 1995; Obaid and Salzberg, 1996), as first suggested by Obaid et al. (1987) for vertebrate neurohypophysis, in which 4-AP can increase the amplitude and horizontal extent of depolarization in axonal arborizations after stimulation of the infundibular stalk. In hippocampus, potassium channel inhibition could result in enhanced invasion of CA3 axon branches (Tamamaki and Nojyo, 1991; Li et al., 1994) and/or dihydropyridine-sensitive Ca2+ entry in axonal regions close to, but separate from, vesicle release sites closely apposed to postsynaptic dendrites (Sorra and Harris, 1993).
Enhanced dendritic excitability
It is not clear whether 4-AP at the concentration used (40 μm) could increase the intrinsic excitability of postsynaptic dendrites. Concentrations ≥100 μm were required to affect CA1 dendritic action potentials in one recent study (Andreasen and Lambert, 1995), and this possibility should be investigated in more detail.
Enhanced accumulation of external K+
The increased accumulation of K+ in the extracellular space that accompanies application of 4-AP (Galvan et al., 1982) could affect presynaptic function as well as postsynaptic neural and glial membranes. The CA3–CA1 synapse EPSP is enhanced by both elevated extracellular K+ (Hablitz and Lundervold, 1981) and activity-dependent K+ accumulation (Poolos et al., 1987), and action potential-driven K+ accumulation can generate a delayed depolarization in axons and periaxonal glia of skate cerebellum (Konnerth et al., 1987).
Reduction of the delayed depolarization by Tg could derive from sensitivity of K+ accumulation to activation of Ca2+-dependent potassium channels, which in turn could be reduced by inhibition of intracellular Ca2+ release. K+ accumulation in this model would reflect balances among depolarizing calcium currents, intracellular Ca2+sequestration and release, and the Ca2+-sensitive and -insensitive repolarizing potassium currents contributing K+ to the extracellular space. Consistent with this model,Konnerth et al. (1987) observed that calcium channel blockers reduced the delayed depolarization in skate cerebellum dependent on K+ accumulation.
Changes in the pharmacological sensitivities of transmission as reflected in the EPSP/AP complex
4-Aminopyridine reduced the sensitivity of the EPSP/AP complex to ω-toxins (ω-CgTX GVIA and ω-AgaTX IVA) and increased its sensitivity to a dihydropyridine (nifedipine). Observations reminiscent of these have been reported from other studies. For example, Yawo and Chuhma (1994) reported for chick ciliary ganglion synapse that 4-AP (400 μm) can expose a small component of ω-CgTX GVIA-insensitive release, and several other groups have noted that sustained stimuli such as elevated external K+ can elicit dihydropyridine-sensitive transmitter release in preparations in which nerve-driven release is dihydropyridine-insensitive (Perney et al., 1986; Rane et al., 1987; Holz et al., 1988; Feuerstein et al., 1991;Momiyama and Takahashi, 1994). Most recently, Wheeler et al. (1996)reported a 4-AP-induced loss of sensitivity to ω-CgTX GVIA and ω-AgaTX IVA for the CA3–CA1 EPSP as recorded in field potential recordings, but they did not observe an effect of a dihydropyridine on the field EPSP in the presence of 4-AP.
Wheeler et al. (1996) interpreted the 4-AP-induced loss of ω-toxin sensitivity in the context of a “spare channel” model of secretion (Dunlap et al., 1995) in which action potential prolongation results in Ca2+ entry through either ω-CgTX GVIA- or ω-AgaTX IVA-sensitive calcium channels that is sufficient to saturate vesicle release mechanisms. In the Wheeler et al. experiment, only the combined application of ω-CgTX GVIA and ω-AgaTX IVA was able to substantially block the field EPSP in the presence of 4-AP. Such saturation of the release mechanism may have been operating in our experiment; we did not test this combination of toxins on the EPSP/AP complex.
Properties of neurotransmitter release also may relate to the spatial distribution of Ca2+ in the presynaptic terminal and the arrangement of calcium channels in the presynaptic membrane (Miller, 1987). In this model, which is not exclusive of the spare channel model described above, dihydropyridine-sensitive channels could be localized to portions of presynaptic terminals remote from the N-, Q-, and other-type channels localized to release sites, and they could be recruited by longer-lasting terminal action potentials (Wheeler et al., 1996) and/or more efficient invasion of terminals (Obaid and Salzberg, 1996). In other systems, different patterns of depolarization recruit different classes of calcium channels (references above) and/or release particular populations of transmitter-filled vesicles (Verhage et al., 1991; Pocock et al., 1995). Sensitivity to dihydropyridines could also be enhanced by depolarization of calcium channels (Rane et al., 1987) in presynaptic axons as a result of activity-dependent K+accumulation (Poolos et al., 1987).
The additional possibility that 4-AP-enhanced EPSPs may elicit dendritic depolarizations and/or action potentials (Wong et al., 1979) sensitive to dihydropyridines such as nifedipine (Regehr and Tank, 1992; Andreasen and Lambert, 1995; Christie et al., 1995) seems less likely. Ca2+ influx in both proximal and distal dendrites is reported to be reduced by Ni2+ (with distal dendrites more sensitive) and is reported to be relatively insensitive to dihydropyridines (Christie et al., 1995; Johnston et al., 1996).
Relevance to mechanisms underlying epileptogenesis and seizure
Taken together, these data suggest that the 4-AP-sensitive channel or channels that carry D-current may control hippocampal excitability by effects on the presynaptic action potential and K+accumulation in extracellular space. Therefore, these potassium channels could have a direct role in epileptogenesis that is independent of any simultaneously occurring processes of disinhibition (Rutecki et al., 1985; Stasheff et al., 1993).
Footnotes
This work was supported by Agency of Industrial Science and Technology, Ministry of International Trade and Industry, and Ministry of Education, Science, and Culture of Japan (T.I.), and the National Institute of Neurological Diseases and Stroke of National Institutes of Health (NS23857 and NS34581 to M.E.B.). We thank Dr. B. M. Salzberg for his discussions of optical recording and comments on this manuscript.
Correspondence should be addressed to Michael E. Barish at his permanent address: Division of Neurosciences, Beckman Research Institute of the City of Hope, Duarte, CA 91010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}