

Abstract
Throughout development of the vertebrate retina, progenitor cells are multipotential, producing a variety of distinctive cell types. Little is known of the molecular mechanisms directing the determination of cell fate. We have examined retinal progenitor cells for expression of receptor tyrosine kinases in an attempt to define receptors that could allow a progenitor to respond to its environment. We found that the receptor tyrosine kinase Flk-1, previously shown to be expressed in endothelial cells, is also expressed in neural progenitor cells of the mouse retina. Flk-1 RNA expression in the retinal progenitors commences with the onset of neuronal differentiation and persists throughout retinal neurogenesis. Flk-1 RNA and protein levels in the retina vary temporally during development, as shown by in situhybridization and Western blot analysis. Patterns of β-galactosidase expression in mice containing the lacZ gene in place of the Flk-1 gene are consistent with Flk-1 being expressed in retinal progenitors. In addition, we show that the ligand of Flk-1, vascular endothelial growth factor (VEGF), is expressed in the developing retina by differentiated cells and that a chimeric ligand of VEGF fused to alkaline phosphatase binds to proliferating retinal progenitors. Furthermore, the neural retina-derived Flk-1 protein kinase is activated by VEGF in vitro. Thus, the Flk-1 receptor protein kinase is expressed on the surface of neural progenitors in mouse retina and may play a critical role in neurogenesis as well as in vasculogenesis.
The vertebrate retina is an excellent model for studies of the development of the CNS. It is derived from the optic cup, a structure originating from an area of the neural tube near the junction of the diencephalon and telencephalon. As with other neural tube-derived structures, the cells abutting the luminal side of the optic cup form a ventricular zone (VZ) and are actively engaged in mitosis. As neurogenesis proceeds, progenitor cells remain associated with the ventricular surface while postmitotic progeny migrate to differentiate and form the laminar structure of the retina. The mature retina then receives light and processes visual signals to provide the brain with remarkably informative data concerning the visual scene.
Several types of investigations have provided some insight into the molecular and cellular mechanisms that govern neural progenitor behavior during development. Lineage analyses have demonstrated that individual progenitor cells from many areas of the CNS are capable of generating diverse progeny (Turner and Cepko, 1987; Bronner-Fraser and Fraser, 1988; Leber et al., 1990; Gray and Sanes, 1992; Walsh and Cepko, 1992; Golden and Cepko, 1996). The initiation of differentiation seems to involve cell–cell interactions mediated by the Notch family of receptors (Artavanis-Tsakonas et al., 1995; Austin et al., 1995;Chitnis et al., 1995; Dorsky et al., 1995), and the decision to enter a particular pathway seems to occur during or after the last mitotic cell cycle (Turner and Cepko, 1987; McConnell and Kaznowski, 1991). Extrinsic cues influencing progenitors presumably change both spatially and temporally, because each area of the CNS produces distinctive cell types in a particular order. In addition, the progenitor cells themselves may change in their intrinsic properties, as suggested by their ability to respond to these cues (Watanabe and Raff, 1990;Lillien and Cepko, 1992; Davis and Temple, 1994; Austin et al., 1995;Williams and Price, 1995; Cepko et al., 1996).
A large body of evidence indicates that cell-surface receptors with tyrosine kinase activity play critical roles in the development of both vertebrate and invertebrate organisms (Pawson and Bernstein, 1990;Schlessinger and Ullrich, 1992; Shilo, 1992; Fantl et al., 1993). These receptors and their cognate ligands participate in diverse biological processes, including formation of the fly body plan (Duffy and Perrimon, 1994; Perrimon, 1994), cell-fate decisions in the fly compound eye (Zipursky et al., 1992) and in the nematode vulva (Sternberg and Horvitz, 1991; Kenyon, 1995), vertebrate limb patterning (Laufer et al., 1994; Cohn et al., 1995), mesoderm induction (Amaya et al., 1991), germline formation, and hematopoiesis in mammals (Chabot et al., 1988; Geissier et al., 1988; Sherr, 1991). Numerous receptor tyrosine kinases seem to play a role in the development and maintenance of the vertebrate nervous system. The Trk family of receptor tyrosine kinases is activated by the neurotrophin family of ligands to affect survival and differentiation of a variety of neurons and glia (Glass and Yancopoulos, 1993; Snider, 1994; Temple and Qian, 1995). The Eph family of receptors (Tuzi and Gullick, 1994) includes members that may be involved in establishing segmental patterns in the hindbrain (Becker et al., 1994) and in establishing the topography of projections of retinal axons (Cheng and Flanagan, 1994; Cheng et al., 1995; Drescher et al., 1995; Tessier-Lavigne, 1995). The pleiotropic fibroblast growth factor (FGF) receptors and the endothelial growth factor (EGF) receptor seem to regulate proliferation of neural progenitors and bias cell-fate choices (Lillien and Cepko, 1992; Ghosh and Greenberg, 1995; Lillien, 1995; Vicario-Abejon et al., 1995).
The function and the subcellular location of receptor tyrosine kinases make them likely candidates to mediate extrinsic cues influencing the production of different types of progeny by neural progenitor cells. We have described an approach using RT-PCR to search for candidate tyrosine kinases expressed in the neural retina (Yang et al., 1993). Here we report that this approach allowed isolation of a previously identified protein kinase, Flk-1. Flk-1 initially was isolated from a population of primitive hematopoietic cells in a search for molecules involved in hematopoiesis (Matthews et al., 1991). In situhybridization experiments have revealed that Flk-1 is expressed by endothelial cells at all stages of development and established this kinase as the earliest known marker of the endothelial lineage (Millauer et al., 1993; Yamaguchi et al., 1993). Flk-1 receptor binds the endothelial cell-specific mitogen vascular endothelial growth factor (VEGF; Ferrara and Henzel, 1989; Gospodarowicz et al., 1989) with high affinity and is activated by VEGF in vitro(Millauer et al., 1993). Expression of a dominant negative Flk-1 gene inhibits angiogenesis in a glioblastoma in vivo (Millauer et al., 1994), and Flk-1-deficient mice die between embryonic day (E) 8.5 and 9.5 because of severe defects in the development of hematopoietic progenitors and endothelial cells (Shalaby et al., 1995). These data indicate that the Flk-1 receptor has a vital role in vasculogenesis and angiogenesis.
Here we report that the Flk-1 receptor is also expressed in the mouse CNS. Progenitor cells in the retina expressed Flk-1 mRNA, and levels of Flk-1 protein varied with time of development. β-galactosidase was expressed in the retina of mice in which one Flk-1 allele was replaced with the lacZ gene. In addition, proliferating retinal cells were able to bind to a chimeric ligand of VEGF fused to alkaline phosphatase. Furthermore, Flk-1 receptors on retinal progenitors were activated by VEGF in vitro. These results indicate that Flk-1 receptor is expressed by retinal progenitor cells and may play an important role in retinal neurogenesis.
MATERIALS AND METHODS
Unless otherwise noted, all standard cloning techniques were performed according to Ausubel et al. (1989), and all enzymes were obtained from Boehringer Mannheim Biochemicals (Indianapolis, IN).
RT-PCRs, cDNA isolation, and DNA sequence analysis. RT-PCRs were performed as described (Yang et al., 1993), using oligonucleotides with the following sequences: XY81, 5′ CGC, GGA, TCC, A(C/T)(C/A), GNG, A(C/T)(C/T), TNG, CNG, CN(C/A), G and XY82, 5′ CCG, GAA, TTC, A(T/C)N, CC(G/A), (A/T)AN, (G/C)(A/T)C, CAN, AC(G/A), TC. All PCR products were subcloned into pcDNAII (Invitrogen, San Diego, CA). DNA sequences were determined using Sequenase v2.0 (US Biochemical, Cleveland, OH). A 210 bp PCR clone N27, which later was determined to share homology with murine Flk-1, was used to screen a mouse postnatal (P) P0–3 eye cDNA library, as previously described (Yang et al., 1993). Four individual cDNA clones were isolated, the longest of which (3.7 kb), N27-22, was sequenced completely.
Ribonuclease protection assays. Total cellular RNAs were purified from BOSC cells (Pear et al., 1993) and dissected P0 mouse retinae by the single-step RNA extraction method (Ausubel et al., 1989). An α-32P-labeled riboprobe 280 nucleotides long, encoding from +2417 to +2625 of the Flk-1 sequence, was synthesized with the MAXIscript kit (Ambion, Austin, TX). Ribonuclease protection assays were performed using the RPAII kit (Ambion) and ∼5 × 104 cpm of gel-purified probe per reaction. RNAs were separated by denaturing PAGE and visualized by autoradiography.
Whole-mount and section in situ hybridization.Digoxigenin-labeled riboprobes were synthesized by using linearized DNA templates in pcDNA I or pcDNA II vectors (Invitrogen). Transcription reactions were performed according to the manufacturer’s instructions, using either T7 or SP6 RNA polymerase in the presence of 11-digoxigenin UTP. The Flk-1 probes used for in situhybridization are shown in Figure 1A. The VEGF probe was derived from the entire murine VEGF1 cDNA coding sequence (Breier et al., 1992).

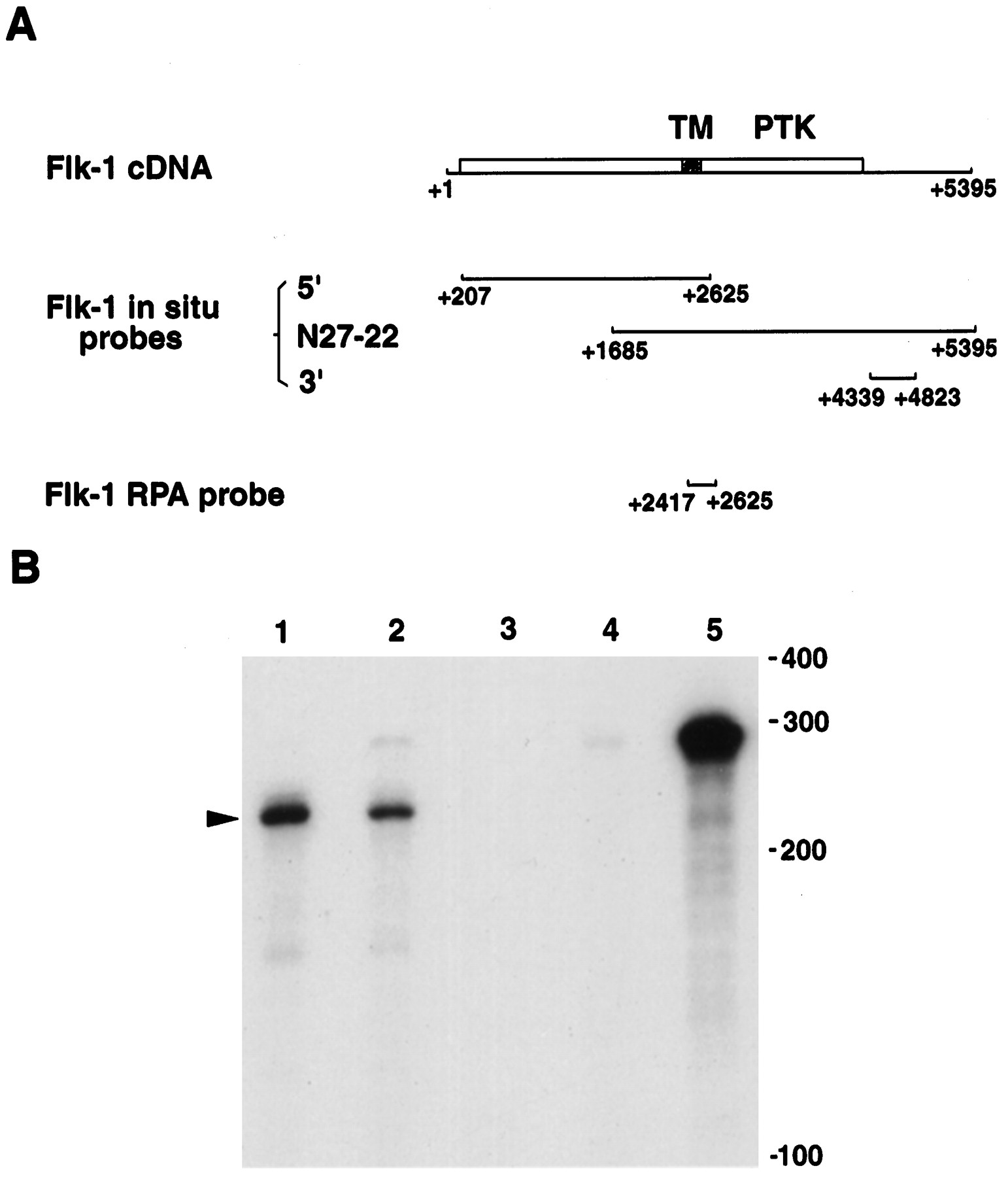
A, Schematic maps of murine Flk-1 cDNA and Flk-1 probes used for in situ hybridization and ribonuclease protection assays (RPA). The full-length Flk-1 cDNA, including the 5′ and 3′ untranslated regions, is shown. The open reading frame is indicated by a box, which begins at +208 and ends at +4310. The location of probes used for in situ hybridization and RPA are indicated. TM, Transmembrane domain; PTK, protein tyrosine kinase catalytic domain. B, Ribonuclease protection assays examining Flk-1 expression. Total RNA from P0 mouse retina (lane 1) and from BOSC cells transiently expressing Flk-1 (lane 2) protected 210 nucleotides of probe, as indicated by the arrowhead. Lane 1, 10 μg of P0 retina total RNA; lane 2, 2 μg of total RNA from BOSC cells transfected with Flk-1 expression vector; lane 3, 2 μg of total RNA from nontransfected BOSC cells;lane 4, 10 μg of total yeast RNA; lane 5, the 280 nucleotides long probe without RNase treatment. Positions of RNA size markers are shown on the right. The autoradiograph shown was exposed for 4 hr.
Whole-mount in situ hybridization was performed as described by Riddle et al. (1993), except that mouse embryo powders were used to preabsorb the anti-digoxigenin antibody. For in situhybridization using tissue sections, tissues were fixed with 4% paraformaldehyde/PBS, cryoprotected with 30% sucrose/PBS, and embedded in OCT (Miles, Elkhart, IN). Cryosections (10–20 μm thick) were collected on Super Frost Plus slides (Fisher Scientific, Pittsburgh, PA), dried, and kept at −80°C until used. The in situhybridization protocol used for tissue sections was similar to procedures described for whole-mount embryos except that the proteinase K treatment was reduced to 1 μg/ml at room temperature for 15 min.
For the digoxigenin in situ hybridization and anti-BrdU antibody double-detection experiments, retinae were dissected free of surrounding tissues and cultured for 1 hr in DMEM/10% fetal calf serum containing 30 μm BrdU. Then retinae were dissociated with trypsin into single-cell suspensions and allowed to attach to poly-d-lysine-coated glass slides for 2 hr at 37°C (Altshuler and Lillien, 1992). Slides were fixed with 4% paraformaldehyde and stored in 100% methanol at −20°C. The in situ hybridization protocol described for tissue sections was used with these dissociated cells, except that glutaraldehyde fixation was omitted. Alkaline phosphatase detection using X-phos as substrate was performed first, followed by a 1 hr incubation with the anti-BrdU antibody (Amersham, Arlington Heights, IL), and then a 1 hr incubation with an anti-mouse antibody conjugated with Texas Red (Jackson ImmunoResearch, West Grove, PA).
Histochemical staining of β-galactosidase of Flk-1 +/− mice.Males of Flk-1-deficient mice were crossed with CD1 females. Eyes from F1 embryos and pups of timed pregnant females were harvested and fixed on ice in 0.5% glutaraldehyde/PBS for 10–20 min. The retinae were dissected from surrounding tissues in PBS and stained as whole mounts at 37°C overnight, as described (Turner and Cepko, 1987). After staining, retinae were processed for cryosectioning at 25 μm.
Transient expression by transfection. The full-length murine Flk-1 cDNA (Matthews et al., 1991) was a generous gift from Dr. Lemischka (Princeton University, Princeton, NJ) and was subcloned into a modified pBABE vector (Morgenstern and Land, 1990) under control of the Moloney virus LTR promoter. BOSC cells (Pear et al., 1993) were cultured in DMEM/10% fetal calf serum and transfected with the pBABE vector expressing Flk-1 using Lipofectamine (Life Technologies, Gaithersburg, MD) as per the manufacturer’s instructions. Routinely, 50% transfection efficiency was achieved. Cell extracts were harvested at 48 or 72 hr after transfection.
VEGF–alkaline phosphatase fusion ligand binding. The VEGF2–alkaline phosphatase chimeric ligand was constructed by amplifying the portion of VEGF1 cDNA that contains the VEGF2 coding sequence (Breier et al., 1992) using PCR with the following primers: XY280 5′ GCG, AAG, CTT, ATG, AAC, TTT, CTG, CTG and XY284 5′ CCT, AGA, TCT, CCT, CGG, CTT, GTC, ACA, TTT, TTC, TGG, CTT, TGT. The 480 bp amplified fragment was then cloned into the HindIII andBglII restriction sites of APtag-2 (Cheng et al., 1995). The coding region of VEGF2 and the fusion site were verified by DNA sequencing. The fusion resulted in an insertion of three amino acid residues, Ser-Ser-Gly, at the junction. The VEGF2–alkaline phosphatase fusion protein and the control alkaline phosphatase protein (from APtag-4) were produced by transient transfection into BOSC cells as described (see above). Supernatants of transfected BOSC cells were collected, concentrated, and stored as described (Cheng et al., 1995).
P0 mouse retinal cells were dissociated, plated on Lab-Tech slides as described above, and incubated overnight in 50% F12/5% fetal calf serum/DMEM containing 20 μm BrdU. The VEGF2–alkaline phosphatase fusion protein or alkaline phosphatase protein was applied as described (Cheng et al., 1995). The cells were washed six times at room temperature with 10% fetal calf serum/20 mm HEPES, pH 7.0/0.1% sodium azide/DMEM, fixed with 50% acetone/2% paraformaldehyde for 2.5 min, and followed by three washes with 150 mm NaCl/20 mm HEPES, pH 7.0. Then cells were heated at 65°C for 15 min, rinsed once with alkaline phosphatase detection buffer, and reacted with X-phos substrates in the dark at room temperature. At the end of the alkaline phosphatase reaction, the anti-BrdU antibody was applied, followed by staining with DAPI and a secondary antibody conjugated to Texas Red.
Immunocytochemistry. Cryosections 10–20 μm thick were collected on gelatin-coated slides and dried. Sections were post-fixed with 4% paraformaldehyde/PBS for 15 min at room temperature, washed with PBS, and dehydrated in 50, 75, and 100% methanol. Then sections were incubated in 5% H2O2 in methanol for 15 min to eliminate endogenous HRP activity and washed in PBS. Blocking solution (10% fetal calf serum/0.1% Triton X-100/DMEM), supplemented with 2% normal serum of the species from which the secondary antibody is derived, was applied for 1 hr, followed by a 2 hr incubation with primary antibodies diluted in blocking solution. Biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA) and HRP detection (ABC Elite, Vector Laboratories) were used to visualize signals. Control experiments omitting primary and/or secondary antibodies were also performed.
The anti-mouse CD31 antibody MEC 13.3 was purchased from PharMingen (San Diego, CA). The anti-VEGF antibody, the VEGF A20 antigen peptide, and the JAK2 C20 peptide were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). For competition experiments, VEGF A20 and JAK2 C20 peptides were incubated at 20 mg/ml with the anti-VEGF antibody (at 0.2 mg/ml) before tissue application. Preincubation with the JAK2 C20 peptide did not affect the staining of the anti-VEGF antibody (data not shown).
Immunoprecipitation and Western blot analysis. Different stage chick and mouse retinae were dissected in cold PBS. Retinal cell and BOSC cell extracts were prepared by lysing cells with lysis buffer (50 mm Tris-Cl, pH 8.0/250 mm NaCl/0.5% NP40/2 mg per ml aprotinin/2 mg per ml leupeptin/ 0.1 mg per ml PMSF) on ice for 30 min. Cell lysates were spun for 4 min at 4°C in a microfuge, and supernatants were used for immunoprecipitation or Western blot assays. Protein concentrations were determined by the Bradford method, using Bio-Rad (Richmond, CA) reagents.
To assay phosphorylation, dissociated mouse P0 retinal cells were plated as a subconfluent monolayer in 60 mm tissue culture dishes in serum-free medium (Altshuler and Lillien, 1992) without insulin for 4 hr at 37°C. Recombinant human VEGF1 or EGF (R&D System) was then added to 100 ng/ml for 10 min. Cells were washed twice with cold PBS and lysed in lysis buffer containing 1 mm sodium vanadate. An anti-Flk-1 CT128 serum (kindly provided by Dr. Cord Brakebusch, Max Planck Institute, Germany; Millauer et al., 1993) was incubated with the lysates for 1 hr at 4°C, followed by incubation with Protein A Sepharose beads (Pharmacia, Piscataway, NJ) for an additional hour. The beads were then pelleted, washed three times with lysis buffer, and resuspended in SDS gel-loading buffer. Proteins were electrophoresed on 7% SDS polyacrylamide gels and blotted to nitrocellulose membrane by the semi-dry transfer method. Blots were probed with anti-phosphotyrosine antibody 4G10 (Upstate Biotechnology, Lake Placid, NY) and visualized using an HRP-conjugated anti-mouse antibody (Amersham) and ECL (Amersham) detection. The same filter was stripped by incubating with 100 mm glycine pH 2.5, washed in PBS, and reprobed with the CT128 antibody.
RESULTS
Flk-1 RNA is expressed in the neural retina as well as in endothelial cells
To identify receptor tyrosine kinases expressed during mouse retinal neurogenesis, we used RT-PCR with degenerate oligonucleotides to amplify tyrosine kinases from mouse postnatal day 0 (P0) eye cDNAs. PCR fragments 210 bp in length were then subcloned, sequenced, and used to screen a P0–3 mouse eye cDNA library. Several cDNA clones sharing identical sequences were obtained by using PCR clone N27 as probe. The longest clone (clone N27-22) was 3.7 kb in length and completely co-linear with the previously characterized Flk-1 kinase cDNA (Fig.1A; Matthews et al., 1991).
In situ hybridization using the N27-22 clone as probe on whole-mount embryos and tissue sections detected Flk-1 transcripts in the developing vasculature of the embryo, including the perineural vasculature and in vessels invading the developing spinal cord as well as in vessels derived from the meninges of cortex and cerebellum (data not shown). These results are consistent with the previously reported Flk-1 expression in endothelial cells (Millauer et al., 1993; Yamaguchi et al., 1993).
In addition to endothelial cell expression, Flk-1 transcripts were detected in whole-mount retinae from different stages of development (data not shown). To confirm that this signal in the neural retina was attributable to expression of Flk-1 and not a related gene, ribonuclease protection assays were performed. Total RNA from both mouse P0 retina and transfected BOSC cells expressing the full-length Flk-1 cDNA (Matthews et al., 1991) protected a 210-nucleotide-long fragment (Fig. 1B, lanes 1 and 2). Because at P0 the neural retina does not contain blood vessels (see below), this result confirmed that Flk-1 RNA was expressed in neural cells.
To determine which retinal cell populations expressed Flk-1, in situ hybridizations were performed using tissue sections from different stages of mouse development. The earliest stage with detectable Flk-1 transcripts in the retina was E11.5. At this stage, the majority of Flk-1 expression was associated with the developing vasculature of the eye, i.e., the blood vessel network surrounding the retinal pigmented epithelium (the choroid) and the blood vessels entering the vitreal space through the optic stalk (the hyaloid; Fig.2A). However, Flk-1 hybridization signal was also detected in a group of cells located in the center of the neural retina (Fig. 2A). This early Flk-1 expression at E11.5 coincides with the onset of ganglion cell differentiation in the retinal neuroepithelium (Young, 1985). By E15, Flk-1 expression had expanded to the periphery of the retina (Fig.2B), reflecting the center-to-periphery gradient of retinal development (Young, 1985; LaVail et al., 1991). During the perinatal periods of development when retinal neurogenesis yields a large number of postmitotic neurons, Flk-1 transcripts were detected almost exclusively in the VZ where progenitor cells reside (Fig.2C, E17; 2D, P0). By P7, when neurogenesis in the retina is nearly complete, Flk-1 transcripts were present in the future inner nuclear layer, where residual progenitors may persist (Fig. 2E). In the adult retina, Flk-1 transcripts were expressed in a row of cells within the inner nuclear layer (Fig.2F), most likely Muller glial cells (also, see below).

Expression of Flk-1 and CD31 in the developing eye. A–F, Tissue sections processed for in situ hybridizations using probes of Flk-1. G–I, Immunohistochemical staining of the endothelial cell-surface antigen CD31. A cross section of an E11.5 eye (A) displays the Flk-1 signal in the center of the neural retina, as well as in blood vessel networks of the eye (choroid and hyaloid). A cross section of an E15 eye (B) shows the spread of Flk-1 expression into the periphery of the retina. Cross sections of E17 eye (C) and P0 retina (D) display Flk-1 signal in the VZ of the retina. A cross section of a P7 retina (E) reveals more restricted expression of Flk-1 in the future inner nuclear layer. A cross section of an adult retina (F) shows limited Flk-1 expression in cells of the inner nuclear layer. Cross sections of E18 (G), P2 (H), and adult eye (I) stained with an anti-CD31 antibody reveal blood vessel formation in the mouse eye. Note that, at E18 and P2, blood vessels were seen in the choroid and hyaloid networks, but no endothelial cells were detected in the neural retina proper. Arrowheads indicate in situ hybridization signals in endothelial cells.le, Lens; vs, ventricular surface;a, differentiating amacrine cell layer;g, retinal ganglion cell layer; onl, outer nuclear layer; pe, pigmented epithelium.
Two additional probes directed against the 5′ and 3′ regions of Flk-1 RNA (5′ and 3′ probes, Fig. 1A) were used to verify that the hybridization signals in retina depicted authentic Flk-1 expression. Identical expression patterns for both endothelial and neural cell signals were obtained with all three probes (data not shown). Because the 3′ probe encodes sequences in an untranslated region of the Flk-1 cDNA that is not likely to be highly conserved among different genes and because in situ hybridizations were performed under high stringency conditions, it is unlikely that the patterns of expression were attributable to cross-hybridization with a homologous kinase.
Blood vessel patterns at different stages of eye development were characterized by using the endothelial cell surface marker CD31 (Baldwin et al., 1994). At E18 and P2, anti-CD31 antibody detected the choroid and hyaloid vessel networks in the eye (Fig.2G,H). However, no endothelial cells were detected in the neural retina (Fig. 2G,H). In the mature eye, capillaries were seen within the retina in all layers except the photoreceptor cell layer (Fig. 2I). Thus, as expected, Flk-1 expression overlapped with the endothelial-specific expression of CD31. However, Flk-1 clearly showed additional patterns of expression in the embryonic and perinatal neural retina, where no endothelial cells were detected, confirming that Flk-1 is expressed in neural tissues.
The lacZ gene inserted into the Flk-1 gene is also expressed in neural retina
To verify further the presence of Flk-1 receptor in the retina, β-galactosidase expression was analyzed in mice carrying the lacZ gene, which replaced the translated portion of the first coding exon and part of the adjacent intron of the Flk-1 gene (Shalaby et al., 1995). Because homozygous Flk-1-deficient mice die early during embryogenesis, cross sections of retinae from different stages of heterozygous mice were stained histochemically. β-galactosidase staining was first detected in E17 mice in the center portion of the retina and in the hyaloid vessels of the eye (Fig.3A). This expression pattern was spatially similar, but temporally delayed, relative to the expression of Flk-1 RNA detected by in situ hybridization (Fig. 2). At early postnatal stages when retinal progenitors remain mitotically active, β-galactosidase expression was observed in both central and peripheral retina (Fig. 3B, P0; 3C, P3) and in vessels attached to the ganglion cell fiber layer. This pattern of expression overlapped with the expression of Flk-1 RNA in retinal progenitors at these ages. At P7, when retinal proliferation has declined and the last cell types of the retina, the bipolar interneurons and Muller glia, are generated, the strongest expression of β-galactosidase was found in the middle and outer portions of the inner nuclear layer (Fig. 3D). Similar patterns were observed in P14 retina (Fig. 3E). In the adult retina, β-galactosidase staining was detected in the inner nuclear layer, the ganglion cell layer, and in the outer nuclear layer in which the cell bodies of the photoreceptors reside (Fig.3F). No β-galactosidase staining was found in the inner and outer segment layers of photoreceptors. This staining pattern is characteristic of Muller glia, the cell bodies of which occupy the center portion of the inner nuclear layer but the processes of which extend through a large portion of the neural retina. The outer processes terminate at the outer limiting membrane, located between the outer nuclear layer and the layer of inner segments, and the inner processes define the inner limiting membrane just inside the fiber layer. Flk-1 RNA expression during gliogenesis (Fig.2E) and in the adult retina (Fig.2F) is consistent with Muller cell expression, because RNA is localized predominantly within cell bodies.

Expression of β-galactosidase in retinae of heterozygous Flk-1-deficient mice. Cross sections of retinae were processed for histochemical staining of β-galactosidase using X-gal. A cross section of an E17 retina (A) through the optic nerve shows the onset of β-galactosidase expression in the VZ of the central neural retina and in the hyaloid vessels. Partial views of P0 (B) and P3 (C) retinal sections indicate the spread of β-galactosidase activity toward the periphery and in vessels attached to the ganglion cell layer. Cross sections of P7 (D) and P14 (E) retinae demonstrate intense β-galactosidase staining in some cells of the inner nuclear layer and weaker staining in the outer nuclear layer and the ganglion cell layer. Staining in the adult retina (F) shows a defined boundary between the photoreceptor cell bodies and their inner segments and reaches the ganglion cell fiber layer.Arrowheads indicate blood vessels. pe, Pigmented epithelium; vs, ventricular surface;g, retinal ganglion cell layer; onl, outer nuclear layer; inl, inner nuclear layer;is, inner segments; os, outer segments;op, optic nerve head.
Thus, patterns of β-galactosidase expression in the retina of heterozygous Flk-1-deficient mice primarily coincided with the patterns of Flk-1 RNA expression as detected by in situ hybridization and strongly suggest that Flk-1 receptor is in both neural and endothelial tissues during development.
Flk-1 RNA is expressed in retinal progenitor cells during neurogenesis
Cells expressing Flk-1 were located almost exclusively in the VZ of the retina from the onset of expression at E11.5 through the active period of retinal neurogenesis (Fig. 2A–D). BrdU labeling of a P0 retina revealed staining patterns that were very similar to Flk-1 expression patterns at this stage (Fig.4A,B), suggesting that Flk-1 is expressed in the progenitor cells residing in the VZ. A few cells in the differentiating areas of the retina were also Flk-1 positive at P0 (Figs. 3D, 4B). These were likely to be newly differentiated amacrine cells that had just left the proliferative zone. Flk-1 transcripts were not evenly distributed in the VZ of the retina throughout neurogenesis. Cells located at the ventricular surface consistently showed lower levels of signal (Fig.2C,D). This heterogeneity may reflect regulation of Flk-1 mRNA during the cell cycle, because progenitor cells entering metaphase move toward the ventricular surface to divide, or it may be attributable to some postmitotic, presumptive cones occupying this position before their differentiation (Bruhn and Cepko, 1996).

Expression of Flk-1 RNA in retinal progenitor cells. A, Anti-BrdU immunocytochemical staining of a mouse P0 retina-labeled for 6 hr defines the proliferative zone.B, Flk-1 in situ hybridization of a P0 retina section reveals signals within the progenitor cell population and in newly differentiated amacrine cells. Black arrowheads in A and B indicate the ventricular surface of the retina. C,D, The same field of dissociated E15 retinal cells double-labeled for BrdU (C) and Flk-1 transcripts (D). Blue arrowheads indicate examples of costaining cells.
To demonstrate unequivocally that Flk-1 transcripts were expressed in proliferating cells, E15.5 retinal cells were labeled with BrdU for 1 hr and then assayed for BrdU incorporation and Flk-1 transcript expression on dissociated cells. Of the E15.5 cells, ∼50% were Flk-1 positive by in situ hybridization (Fig.4D), and ∼25% of the total population were labeled by the anti-BrdU antibody (Fig. 4C). Although the intense alkaline phosphatase signal from in situ hybridization seemed to quench the BrdU fluorescent signal in some cells, cells that were positively stained by both assays could be detected and were numerous. Similar results were obtained for P0 retinal cells (data not shown). Thus, Flk-1 transcripts are expressed during S phase in cycling retinal progenitors.
A chimeric ligand of VEGF fused to alkaline phosphatase binds to retinal progenitor cells
Flk-1 receptor has been reported to bind VEGF with high affinity. To investigate the distribution of VEGF binding activity in the retina, a modified ligand containing VEGF fused to alkaline phosphatase was constructed and used in binding assays. Dissociated P0 retinal cells grown as a monolayer were labeled with BrdU, exposed to VEGF–alkaline phosphatase fusion protein, and then assayed for BrdU incorporation and alkaline phosphatase activity. Of the P0 cells, ∼50% were alkaline phosphatase positive (Fig. 5E), and ∼20–25% of the cells were labeled by BrdU (Fig.5F). Although intense alkaline phosphatase staining quenched BrdU-dependent fluorescent signal, the majority of cells that had detectable BrdU labeling was also alkaline phosphatase positive. In contrast, alkaline phosphatase alone did not bind to P0 retinal cells (Fig. 5B). Therefore, in addition to expressing Flk-1 RNA, proliferating retinal progenitor cells possess the ability to bind VEGF.

Binding of the VEGF–alkaline phosphatase fusion protein to neural retinal cells. Monolayers of P0 retinal cells were labeled with BrdU, bound to the VEGF–alkaline phosphatase fusion protein or alkaline phosphatase alone, and then processed to detect cell nuclei (A, D), alkaline phosphatase activity (B, E), and BrdU incorporation (C, F). A–C, The same field of cells exposed to the alkaline phosphatase protein alone. D–F, The same field of cells exposed to the VEGF–alkaline phosphatase fusion protein. Arrowheads indicate cells staining for both alkaline phosphatase and BrdU.
VEGF expression in the developing retina is complementary to Flk-1 expression
VEGF can promote proliferation and differentiation of endothelial cells. To determine whether VEGF colocalizes with progenitors in the retina, we performed immunohistochemical staining with an anti-VEGF antibody. At P0, when Flk-1 kinase was abundant in the retina, VEGF was localized mostly in the differentiated ganglion and amacrine cells and in the developing inner plexiform layer (Fig.6A). Preincubation with the peptide antigen abolished this staining (Fig. 6B), indicating that the antibody specifically recognized VEGF or a protein sharing similar sequence motifs. Consistent with the distribution of VEGF protein at this stage, VEGF RNA was detected in the differentiated areas of the retina by in situ hybridization (Fig.6C; Stone et al., 1995). As retinal development progressed, more VEGF RNA was detected in the inner nuclear layer (Fig.6D,E). At P12, a row of cells within the inner nuclear layer, most likely Muller glia, expressed the highest amount of VEGF RNA.

Expression of VEGF protein and RNA in the retina.A, A cross section of a P0 retina stained with an anti-VEGF (A20) antibody shows VEGF in the differentiated ganglion cell layer and perhaps in differentiating amacrine cells. B, A competition experiment in which a cross section of a P0 retina was incubated with the anti-VEGF antibody in the presence of the peptide used as antigen. In situ hybridizations displaying VEGF RNA are shown for P0 (C), P3 (D), P6 (E), and P12 (F) retinae.vs, Ventricular surface; g, ganglion cell layer; inl, inner nuclear layer; onl, outer nuclear layer.
Thus, in P0 retina VEGF ligand(s) was concentrated in a region directly opposing the proliferative zone of the retina. As neurogenesis proceeded, cells expressing VEGF RNA gradually colocalized with cells expressing Flk-1 receptor. In the adult retina, the population of cells producing VEGF may be the Muller glia.
Flk-1 protein level changes during retinal neurogenesis
Flk-1 protein levels at different stages of development were examined on Western blots. A doublet of protein bands with molecular weights expected for the Flk-1 protein (∼180 kDa) was detected in mouse retinal extracts throughout development (Fig.7A). These bands displayed mobilities similar to the murine Flk-1 protein derived from extracts of BOSC cells transfected with the full-length Flk-1 cDNA (Matthews et al., 1991;Millauer et al., 1993). It is likely that the doublet represents different post-translationally modified forms of Flk-1 protein. Levels of Flk-1 protein increased between embryonic stages and early postnatal stages and then decreased as retinal neurogenesis declined (Fig.7A). In adult retina, a residual level of Flk-1 protein was present, which is consistent with in situ hybridization results showing persistent RNA expression in a subset of retinal cells (Fig. 3F). Changes in Flk-1 protein levels were not likely attributable to contaminating endothelial components of the eye, because the retinal-to-endothelial cell ratio was high (estimated to be at least 100:1; compare Fig. 2G–I with Fig.2A–F), and dissections were carefully monitored to minimize contamination from the vascular network lining the inner retinal surface. More Flk-1 protein (∼5- to 10-fold) was detected in early postnatal retina than in the E15 retina. This increase during development may be attributable to increased amounts of Flk-1 protein per cell and/or an increased percentage of retinal cells expressing Flk-1.

A, Levels of Flk-1 protein change during mouse and chick retinal development. Equal amounts (50 ng) of total cell extracts from different stages of chick and mouse retina and from BOSC cells untransfected (−) and transfected (+) with a Flk-1-expressing vector were assayed by Western blot analysis using the anti-Flk-1 antibody CT128. The arrowhead on theleft indicates the position of Flk-1 protein detected in chick retinal extracts (∼210 kDa). Arrowheads on theright indicate positions of Flk-1 protein doublets present in mouse retinal and BOSC cell extracts (∼180 kDa).B, VEGF activates the Flk-1 protein tyrosine kinasein vitro. The top panel shows a Western blot analysis of P0 mouse retinal cell extracts. Monolayer cultures were treated with vehicle only (−), with VEGF (V), or with EGF (E) and immunoprecipitated with the anti-Flk-1 antibody (CT128). Western blots then were probed with an anti-phosphotyrosine antibody. Only the VEGF treatment resulted in phosphorylation of the Flk-1 kinase (arrowhead). The bottom panel shows the same filter stripped and reprobed with the anti-Flk-1 antibody, confirming that approximately equal amounts of Flk-1 protein had been immunoprecipitated from the three extracts (arrowhead).
Chick retinal cell extracts were also examined for the expression of Flk-1 to ascertain whether a role for Flk-1 in neurogenesis was conserved among vertebrate species. Western blots revealed a single protein with a slightly higher molecular weight (∼200 kDa) than murine Flk-1 protein. The level of this protein had already reached a plateau by E4, the stage at which the predominant retinal neurons being generated are ganglion cells (Prada et al., 1991). The decline of Flk-1 protein in the chick retina, however, primarily resembled its decline in the mouse, given that chick retinal neurogenesis terminates at ∼E10 (Dutting et al., 1983; Prada et al., 1991). The cellular location of Flk-1 in the chick retina is presently unknown, but because the chick retina is avascular (Coulombre and Coulombre, 1965), the presence of Flk-1 in this tissue further supports a role for this kinase in neurogenesis.
Flk-1 kinase in the retina is activated by VEGFin vitro
We used a phosphorylation assay to determine whether Flk-1 kinase in the retina could be activated by VEGF. P0 mouse retinal cells were treated with VEGF, cell lysates were immunoprecipitated by an anti-Flk-1 antibody, and a Western blot was used to detect increased phosphorylation of Flk-1. Flk-1 protein was phosphorylated in the presence of exogenous VEGF, but not EGF (Fig. 7B). Identical results were obtained by immunoprecipitating with an anti-phosphotyrosine antibody and then probing the blot with an anti-Flk-1 antibody (data not shown). These results demonstrate that the receptor tyrosine kinase Flk-1 in neural retina can be activated by the ligand VEGF.
DISCUSSION
Multiple lines of evidence indicate that the Flk-1 receptor tyrosine kinase is expressed in the neural retina during mouse embryogenesis. First, three probes were used for in situhybridizations, including one from the 3′ untranslated region, and each produced identical patterns of expression in endothelial cells and neural retina. Second, ribonuclease protection assays indicated that the Flk-1 gene, as opposed to a close relative, is transcribed in retinal cells. Third, the binding of an alkaline phosphatase–VEGF fusion protein to approximately one-half of the dissociated cells from P0 retina demonstrated the presence of Flk-1 receptor on the surface of these cells. Fourth, the anti-Flk-1 antibody recognized a protein in retinal cell extracts with a molecular weight equal to the predicted molecular weight of Flk-1, and this protein was phosphorylated in vitro in response to treatment with VEGF. Finally, Flk-1 expression patterns overlapped with, but were distinct from, patterns of blood vessels in the eye identified with an independent endothelial cell marker, CD31. Because the ratio of retinal to endothelial cells in the developing eye is very high, the abundant signals detected in ribonuclease protection assays, Western blot analyses, and phosphorylation assays were not likely to be endothelial cell-derived.
Additional evidence for the presence of Flk-1 receptor in the retina was provided by an analysis of β-galactosidase expression in Flk-1-deficient mice, in which part of the Flk-1 gene was replaced by a promoterless lacZ gene (Shalaby et al., 1995). During embryogenesis, the lacZ gene faithfully recapitulated the expression of Flk-1 in endothelial cells in the vasculature of the embryo (Shalaby et al., 1995), including blood vessels of the neonatal brain (data not shown). Therefore, the genetic manipulation that resulted in a null mutation of Flk-1 did not seem to interfere with regulatory elements controlling its expression in the endothelial lineage. In the neural retina, β-galactosidase expression was primarily consistent with the in situ hybridization pattern of Flk-1 RNA, with some minor discrepancies. First, although spatial patterns of β-galactosidase expression mimicked patterns of Flk-1 RNA expression in embryonic stages, i.e., the expression followed a center-to-peripheral gradient and was mostly in VZ cells, the earliest detection of β-galactosidase by X-gal staining was delayed by ∼6 d relative to detection byin situ hybridization. This is likely attributable to disruption of some control elements specific for neural tissues in the Flk-1-deficient mice. In situ hybridization using retinal sections, dissociated retinal cells, and Western blot analysis all support an earlier onset of Flk-1 expression in the neural retina. Second, postnatal β-galactosidase expression in the retina was broader than Flk-1 in situ hybridization patterns. This simply may be attributable to differences in the subcellular locations and/or half-lives of Flk-1 RNA and β-galactosidase. Despite these differences, results obtained from characterizing Flk-1-deficient mice strongly support the expression of Flk-1 receptor in the neural retina as well as in endothelial cells.
Expression of the Flk-1 receptor in retina suggests that it plays a role in neurogenesis in addition to its role(s) in hematopoiesis, vasculogenesis, and angiogenesis. Further, its expression in mature Muller glial cells might indicate a role in maintenance of glial cell function and/or in allowing glial cells to respond to some changes in the environment. In both vertebrate and invertebrate systems, there are precedents in which a receptor and its ligand participate in multiple and distinct biological processes during development. For example, the kit receptor and its ligand are involved in neural crest development, germline differentiation, and hematopoiesis in mammals (Chabot et al., 1988; Geissier et al., 1988). In Drosophila, the DER receptor participates in the establishment of the body plan and the development of the eye disk (Shilo, 1992). It is intriguing that the Flk-1 receptor is expressed in two progenitor cell pools during mouse development, initially in the common progenitors of the hematopoietic and endothelial lineage and subsequently in a neural progenitor pool that gives rise to neurons and glia. It is conceivable that Flk-1 plays a similar role in the two progenitor pools. Alternatively, activation of the Flk-1 receptor in these two progenitor pools could result in different cellular responses because of previously existing differences between these two lineages.
Although VEGF is expressed in neural retina and can activate retina-derived Flk-1 kinase in vitro, it remains to be determined whether VEGF is the endogenous ligand for Flk-1 in the CNS. In the developing brain, VEGF transcripts are found in the VZ (Breier et al., 1992). It has been hypothesized that diffusible VEGFs synthesized in the VZ of the brain form a concentration gradient to cause angiogenic sprouting from the perineural vascular plexus covering the neural tube (Bar, 1980; Breier et al., 1992). In the neonatal retina, VEGF protein is mostly concentrated in the differentiated areas adjacent to the proliferative zone in which Flk-1 is expressed. It is possible that VEGF in the retina diffuses from the differentiated zone to the progenitor zone and thus serves both as a neurogenic factor for progenitors and newly postmitotic cells and as an angiogenic factor promoting blood vessel network formation (Stone et al., 1995). As the retina matures, expression of VEGF and Flk-1 become colocalized, mostly in the inner nuclear layer of the retina. It is likely that, in the mature retina, Muller glial cells may express both Flk-1 receptor and its ligand to result in an autocrine circuit. The significance of this in Muller glia differentiation and survival is not clear. Alternatively, unidentified ligands for Flk-1 receptor, which mediate distinct downstream events upon binding to the receptor, could exist in the nervous system.
Molecular and cellular markers, especially cell-surface markers, are important tools for evaluating progenitor properties and monitoring the progression of progenitors during development. Functional studies indicate that neural progenitors are not homogenous but exhibit distinctive intrinsic differences at different times in development (Watanabe and Raff, 1990; Altshuler and Cepko, 1992; Lillien and Cepko, 1992; Davis and Temple, 1994; Austin et al., 1995; Williams and Price, 1995). The competence of a progenitor cell to respond to extrinsic cues is likely to be determined by the combined expression of cell-surface receptors, signal transduction components, and transcription factors (Cepko et al., 1996). The Flk-1 receptor clearly qualifies as a progenitor marker in the developing retina and could be responsible for mediating responses to environmental cues. In the retina, the onset of Flk-1 expression in progenitors represents a significant change between the earliest progenitor population, which produces only mitotic progeny, and later progenitor populations, which generate different types of postmitotic neurons and glia. In addition, a steady increase in Flk-1 protein levels between embryonic and early postnatal periods was detected. Because the proportion of progenitor cells expressing Flk-1 at any given stage has not yet been determined, the increases may be caused by increased expression per cell and/or an increased percentage of retinal cells expressing Flk-1. Nonetheless, changes in Flk-1 expression during development may provide a molecular basis for an intrinsic change in the progenitor cell population.
Protein kinase receptors for growth factors, including bFGF, EGF, and neurotrophins, have been implicated in the regulation of CNS progenitor cell proliferation and differentiation (Ghosh and Greenberg, 1995; Lillien, 1995; Vicario-Abejon et al., 1995). However, none of these receptors displays expression patterns resembling that of Flk-1 in the retina, perhaps suggesting a distinct role for this kinase in neurogenesis. Because homozygous Flk-1-deficient embryos die in utero (E8.5–E9.5) before the onset of its expression in neural tissues, perturbing the function of Flk-1 in neural progenitors will require alternative approaches.
Footnotes
This work was supported by a National Research Service Award to X.Y., R01EY09676 to C.L.C., and the Howard Hughes Medical Institute. We thank I. Lemischka for the full-length murine Flk-1 cDNA and C. Brakebusch and A. Ullrich for the anti-Flk-1 antibody. We are grateful to J. Rossant and her colleagues for the Flk-1-deficient mice used in this study. We thank C. P. Austin and J. G. Flanagan for helpful comments on this manuscript.
Correspondence should be addressed to Constance L. Cepko at the above address.
Dr. Yang’s present address: Jules Stein Eye Institute, University of California, Los Angeles, CA 90024.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}